636 R. R. Ji et al. / J. Biomedical Science and Engineering 3 (2010) 633-637
Copyright © 2010 SciRes. JBiSE
Table 3. The regulatory genes of several target genes.
target gene regulatory genes
SWI4 MBP1、CLN3、MCM1、SWI6
SWI5 ACE2、MCM1、MBP1
CLN2 MCM1、SWI4、CLB2、SIC1
CLB2 MCM1、SIC1
dicted by our algorithm which is remained to be verified
further.
3) Discussions
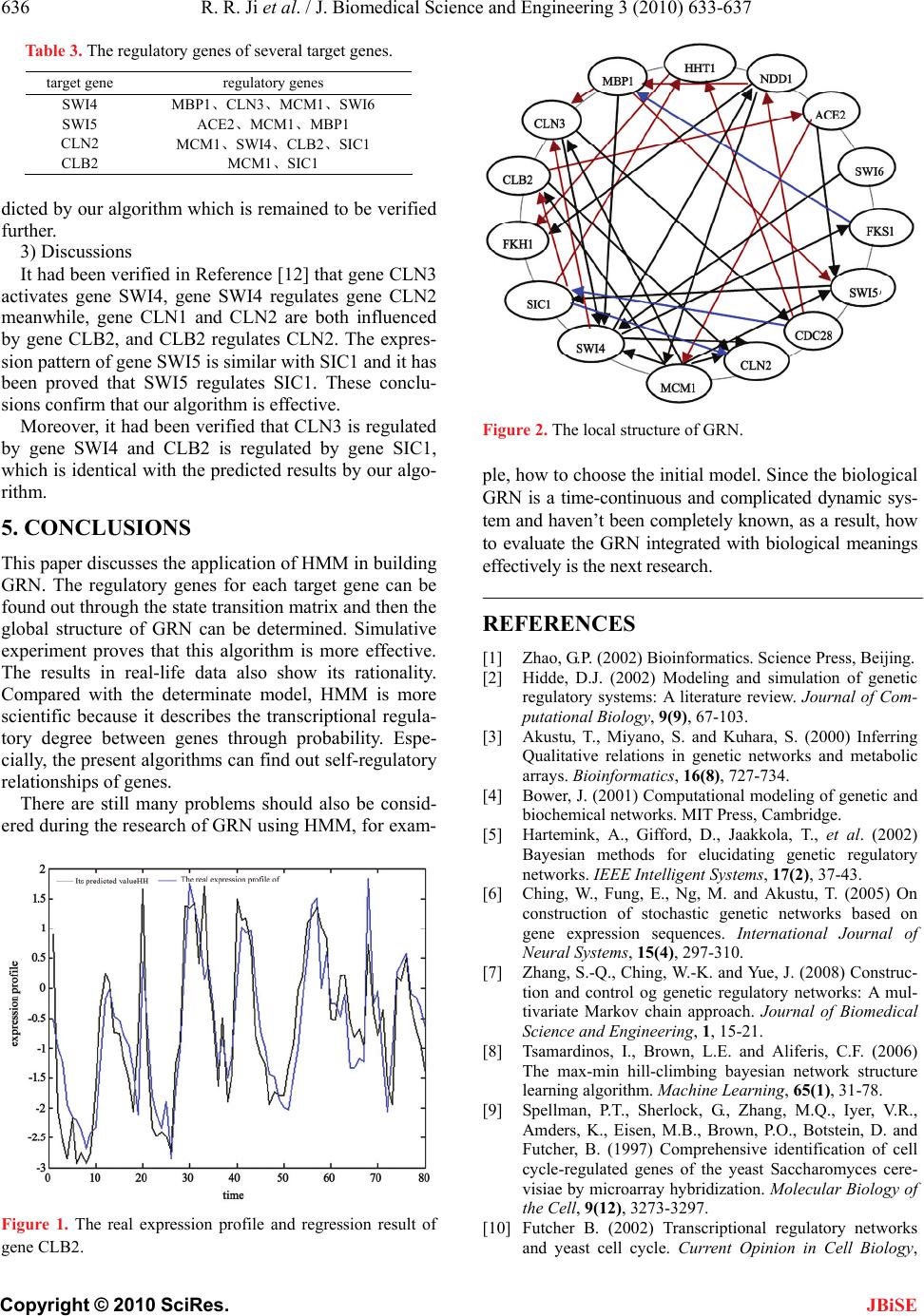
It had been verified in Reference [12] that gene CLN3
activates gene SWI4, gene SWI4 regulates gene CLN2
meanwhile, gene CLN1 and CLN2 are both influenced
by gene CLB2, and CLB2 regulates CLN2. The expres-
sion pattern of gene SWI5 is similar with SIC1 and it has
been proved that SWI5 regulates SIC1. These conclu-
sions confirm that our algorithm is effective.
Moreover, it had been verified that CLN3 is regulated
by gene SWI4 and CLB2 is regulated by gene SIC1,
which is identical with the predicted results by our algo-
rithm.
5. CONCLUSIONS
This paper discusses the application of HMM in building
GRN. The regulatory genes for each target gene can be
found out through the state transition matrix and then the
global structure of GRN can be determined. Simulative
experiment proves that this algorithm is more effective.
The results in real-life data also show its rationality.
Compared with the determinate model, HMM is more
scientific because it describes the transcriptional regula-
tory degree between genes through probability. Espe-
cially, the present algorithms can find out self-regulatory
relationships of genes.
There are still many problems should also be consid-
ered during the research of GRN using HMM, for exam-
Figure 1. The real expression profile and regression result of
gene CLB2.
Figure 2. The local structure of GRN.
ple, how to choose the initial model. Since the biological
GRN is a time-continuous and complicated dynamic sys-
tem and haven’t been completely known, as a result, how
to evaluate the GRN integrated with biological meanings
effectively is the next research.
REFERENCES
[1] Zhao, G.P. (2002) Bioinformatics. Science Press, Beijing.
[2] Hidde, D.J. (2002) Modeling and simulation of genetic
regulatory systems: A literature review. Journal of Com-
putational Biology, 9(9), 67-103.
[3] Akustu, T., Miyano, S. and Kuhara, S. (2000) Inferring
Qualitative relations in genetic networks and metabolic
arrays. Bioinformatics, 16(8), 727-734.
[4] Bower, J. (2001) Computational modeling of genetic and
biochemical networks. MIT Press, Cambridge.
[5] Hartemink, A., Gifford, D., Jaakkola, T., et al. (2002)
Bayesian methods for elucidating genetic regulatory
networks. IEEE Intelligent Systems, 17(2), 37-43.
[6] Ching, W., Fung, E., Ng, M. and Akustu, T. (2005) On
construction of stochastic genetic networks based on
gene expression sequences. International Journal of
Neural Systems, 15(4), 297-310.
[7] Zhang, S.-Q., Ching, W.-K. and Yue, J. (2008) Construc-
tion and control og genetic regulatory networks: A mul-
tivariate Markov chain approach. Journal of Biomedical
Science and Engineering, 1, 15-21.
[8] Tsamardinos, I., Brown, L.E. and Aliferis, C.F. (2006)
The max-min hill-climbing bayesian network structure
learning algorithm. Machine Learning, 65(1), 31-78.
[9] Spellman, P.T., Sherlock, G., Zhang, M.Q., Iyer, V.R.,
Amders, K., Eisen, M.B., Brown, P.O., Botstein, D. and
Futcher, B. (1997) Comprehensive identification of cell
cycle-regulated genes of the yeast Saccharomyces cere-
visiae by microarray hybridization. Molecular Biology of
the Cell, 9(12), 3273-3297.
[10] Futcher B. (2002) Transcriptional regulatory networks
and yeast cell cycle. Current Opinion in Cell Biology,



