Journal of Cancer Therapy
Vol.4 No.2(2013), Article ID:30094,26 pages DOI:10.4236/jct.2013.42078
Huh-7 Human Liver Cancer Cells: A Model System to Understand Hepatocellular Carcinoma and Therapy
![]()
1School of Medicine, The University of Notre Dame Australia, Sydney, Australia; 2GenØK—Centre for Biosafety, University of Tromsø, Tromsø, Norway.
Email: George.Mendz@nd.edu.au
Copyright © 2013 Anna C. Krelle et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received December 27th, 2012; revised January 29th, 2013; accepted February 6th, 2013
Keywords: Huh-7 Cells; Hepatoma; Anti-Cancer Therapies
ABSTRACT
In the last decades, the use of in vitro systems in liver research has grown exponentially. Important reasons promoting this work are the high throughput and ease of genetic manipulations afforded by these experiments relative to in vivo experiments. Thousands of investigations of hepatocellular carcinoma have been performed employing the human hepatoma Huh-7 cell line. The extensive body of knowledge produced attests to the importance and value of this in vitro cell system to study the characteristics of hepatomas and the potential of natural and synthetic compounds to prevent and eliminate this liver cancer. The necessarily brief summary provided here attempts to summarise some of the most recent achievements and limitations of investigations with Huh-7 cells and derivatives.
1. Introduction
In general, cirrhosis of any aetiology is the major risk factor for hepatocellular carcinoma (HCC). Major causes of cirrhotic liver are alcohol, hepatitis B virus (HBV) and hepatitis C virus (HCV). About 85% of the global HCC cases are attributable to infections with HBV or HCV (with a small proportion the result of joint infections) [1]. Unlike for HBV, presently there is no vaccine against HCV infection; thus, many current investigations on HCC are focused on understanding the life cycle of HCV in order to elucidate therapeutic and vaccine targets against the disease. Hence, the availability of a suitable cell culture system to enable the study of the replication cycle of HCV is an important prerequisite to understand HCC and to devise strategies for prophylactic and therapeutic interventions. Such model system should mimic adequately the in vivo hepatic environment encountered by the virus, as well as allow full viral replication. The model system also should be suitable for studies, both independent and interactive, of other non-viral aetiologies of HCC, and for evaluating anti-HCC drugs and vaccines.
Hepatitis B and C viruses are poorly propagated in vitro in cell culture systems [2]. Both viruses have limited hosts, infecting only humans and chimpanzees. For this reason, most cell culture models including primary hepatocytes and established cell lines can at best demonstrate infectivity by the virus but are not suitable for studies of HCC requiring complete viral life cycle because of low levels of viral replication. HCV propagation systems have been devised based on transfecting the human hepatoma cell lines Huh-7, HepG2 and IMY-N9 with genomic HCV RNA derived from cloned viral genomes [3,4]; the efficiency of colony-formation in HepG2 and IMY-9 was 10-1000-fold lower compared to that of Huh-7 cells [4-6]. Nonetheless, these systems solve, at least in part, the problem of non HCV replication in cell culture models.
2. Huh-7 Hepatoma Cell Line
Huh-7 is a well-established and differentiated hepatocytederived cellular carcinoma cell line that was originnally taken from a liver tumour in a 57-year old Japanese male in 1982 [7]. Besides Huh-7, other common heaptoma cell lines used in HCC-related studies include HepG2, Huh-6 [8], PLC/PRF/5 [9,10], Hep3B [11], Huh-1 [12], Huh-4 [12], etc. Huh-7 cells can be propagated in a chemically defined medium containing trace amounts of selenium in place of serum [7], unlike other established human heaptoma cell lines with differentiated functions of the liver such as Huh-6, PLC/PRF/5, Hep3B, Huh-1 and Huh-4 that require the addition of serum. It was found that Huh- 7 cells secrete the mitogen heaptoma-derived growth factor that promotes cell growth allowing them not to depend on growth factors found in serum [13]. Accordingly, together with its permissiveness to HCV genomic replication, the ability to propagate Huh-7 cells in a culture medium containing defined components broadens its scope from that of a model for the investigation of oncogenesis to one suitable also for the elucidation of regulatory mechanisms of gene expression; thus, the properties of these hepatoma cells allow systematic studies of in vitro effects of various compounds on their growth and metabolism.
The criteria informing the choice of cell line(s) are highly variable, and there are no final rules to guide the decision on which cell line has relevance for a particular study. The choice of what hepatoma cell line to use depends largely on the aspect of the cancer under investigation. Generally, studies that require the presence of HCV replicons are conducted with Huh-7 cells and HepG2 cells [14]; studies requiring the presence of HBV are more commonly conducted with HepG2 and Hep3B [15]. The latter cell lines were established from liver tumour biopsies obtained during extended lobectomies of a 15- year old Caucasian male from Argentina, and an 18-year old black male from the USA, respectively [11]. The PLC/PRF/5 cell line was derived from a patient with primary liver cancer whose blood had circulating hepatitis B surface antigen [6,9]. In contrast to the PLC/PRF/5 cell line, HepG2 and Hep3B have the capacity to synthesize many human plasma proteins, including both albumin and alpha-fetoprotein [16]. In comparison to HepG2 and Huh-7, Hep3B has been described to express less dioxin-responsive genes, indicating a lower detoxification activity [17]. Epigenetic studies suggested also a more aggressive hepatocellular carcinoma phenotype of Hep3B compared to HepG2 or Huh7 cells [18].
Studies evaluating anti-HCC drug metabolism are commonly conducted in HepG2, although it has recently been proposed that Huh-7 could be an alternative to the limited available primary human hepatocytes and frequently used HepG2 cell line [19]. Huh-6 and Huh-4 both also produce alpha-fetoprotein and albumin in cell culture [12], but unlike the Huh-7, do not allow replication of HCV genomic replicon and, as stated earlier, cannot be propagated in purely chemically defined media, thus limiting their use in HCC related studies. Human hepatoma Huh-7 cells and its derivatives are thus suitable model system to study HCC and other liver diseases, for drug targeting studies, studies of liver metabolism and toxicity of xenobiotics, and for the detection of cytoprotective, anti-genotoxic and co-genotoxic agents [20].
3. Effects of Microbial Carcinogens on Huh-7 Cells
3.1. Hepatitis B Virus
Several viruses have been associated with human cancers. For example, human papilloma virus is a cause of cervical, skin and oropharynx cancers; and Epstein-Barr virus is involved in nasopharyngeal carcinoma, adult cell leukemia, head and neck cancers, Burkitt lymphoma. In the liver, persistent HBV or HCV infection trigger molecular events that can lead to HCC.
Hepatitis B virus is one of the most important aetiological agents implicated in causing HCC. The virus has a unique life cycle that produces large viral loads during active replication but is not directly cytopathic, and liver injury appears to be mostly caused by repeated attempts of the host’s immune responses to control the infection by inducing inflammation, apoptosis and regeneration; thus, fostering the accumulation of genetic and epigenetic alterations. Two of the key events in the life cycle of HBV are the generation of a covalently closed circular (ccc)DNA transcriptional template, and the reverse transcription of the viral pregenomic RNA to form progeny HBV DNA genomes [21]. Notwithstanding its clinical sigificance, there is only a limited understanding of the molecular cellular and environmental mechanisms that in chronic HBV infections lead to chronic hepatitis, cirrhosis and HCC [22]. The unique strategies employed by HBV to replicate and the immunological mechanisms that prevent its eradication allow the virus to persist within infected hepatocytes, and current therapeutic regimens only can suppress efficiently viral replication. Development of more effective therapies to achieve sustained viral control and virus eradication is hindered by the slow progress in understanding specific molecualar events of HBV replication [23].
Hepatocytes have evolved strategies for sensing the presence of infection and for cellular defence, but until recently, knowledge of their innate immune responses was limited. Three hepatoma cell lines, including Huh-7 cells, were employed to characterise gene transcription and signalling of Toll-like receptors (TLR), which are important cellular mediators of microbial recognition. Huh-7 cells express mRNA for all TLR with the exception of TLR8, as well as a limited a repertoire of TLR signalling proteins suggesting that hepatocytes may themselves play an active role in innate immune responses to viruses such as HBV. However, the absence of responses to ligands of specific TLR proteins suggests that not all TLR mRNA may translate into proteins. Nonetheless, the expression of functional TLR2 and selective expression of TLR3 indicated that this cell line is suitable for investigating the interactions between HBV and hepatocyteexpressed pattern recognition receptors [24]. Besides TLR2 receptors, Huh-7 cells express functional interleukin-1 receptors whose stimulation lead to a signalling cascade expressing the pro-inflammatory cytokines tumour necrosis factor-alpha (TNF-α) and IL-8 via a nuclear factor-kappaB (NF-κB)-dependent pathway that inhibits HBV DNA replication [25]. Thus, TNF-α is produced not only by immune cells but also by hepatocytes. It plays a pivotal role in the inflammatory processes of viral infection and hepatocyte death, and many of its biological effects, including ability to induce cell death are mediated by the transcription factor NF-κB.
The synthesis and maintenance of a covalently cccDNA form of the viral genome in the nucleus of host cells is required for establishing an infection with HBV. The Huh-7 cell line was transformed to express high levels of the virus and facilitate the investigation of the synthesis of cccDNA and other aspects of HBV replication [26]. Persistence of HBV infection requires cccDNA formation and amplification. This nuclear form of HBV is synthesised intracellularly by the repair of the viral polymerase-linked relaxed circular (rc) DNA genomes present in infecting virions; additional copies of cccDNA are derived from newly synthesised rcDNA molecules by intracellular amplification. The regulation of the synthesis of cccDNA by the envelope proteins of HBV was investigated in Huh-7 cells; an increase in the level of cccDNA was measured by ablation of the expression of envelope proteins and a decrease in its level by restoration of the expresion of the proteins [27]. Compared to amplification of hepatitis B viruses in other species, the rate of cccDNA formation during HBV infection in vivo is significantly less efficient in humans. A study of the rates of HBV rcDNA to cccDNA conversion in Huh-7 cells found that nuclear import, complete versus partial release from the capsid, and complete versus partial removal of the covalently bound polymerase contribute to limiting cccDNA formation [28]. This lesser efficiency may be due to limitations in the cellular activities promoting the conversion in this cell line, or that inhibitory factors are overexpressed. [28]. Nuclear cccDNA accumulates as a stable episome organized into minichromosomes by histone and nonhistone proteins. The relationships between viral replication and HBV chromatin assembly, transcription, and interaction with viral and cellular regulatory proteins were investigated by transfection of linear HBV DNA into human hepatoma Huh-7 cells. It was found that HBV replication is regulated by the acetylation status of the cccDNA-bound H3/H4 histones. Inhibitors of Class I histone deacetylases induce an increase of both cccDNA-bound acetylated H4 and HBV replication. Histone hypoacetylation and histone deacetylase 1 recruitment onto the cccDNA in liver tissue correlated with low HBV viremia in hepatitis B patients [29].
The limited induction of innate immune responses that follow HBV infection may be due to its ability to evade innate immune recognition. One of the four overlapping open reading frames of the HBV genome encodes the core protein and a non-structural protein known as secreted e-antigen (HBeAg) that shares a significant homology with the core protein. Using Huh-7 cells it was found that HBeAg down-regulates genes involved in cell signalling, RNA transport and processing, cytosol-nuclear trafficking and innate immune responses. The eantigen is not required for viral replication, but displays immune-modulating functions and hence contributes to viral persistence [30].
Another HBV open reading frame, the X gene, encodes the HBx protein, a multifunctional regulator modulating transcription of several genes involved in signal transduction pathways, cell cycle progress, protein degradation, apoptosis, and genetic stability by directly or indirectly interacting with host transcription factors [31]. Isolates of human HCC tissues with integrated HBV DNA possess truncated open reading frames coding for viral polymerase and the core antigen and only encode fully two gene products: the HBx and the surface (HBs) protein. Hence, HBx and HBs proteins represent the two potential candidates involved in HBV hepatocarcinogenesis [32]. Moreover, HBx-defective virus cannot initiate infection in vivo [22], and some mutations in the X gene modulate HBV DNA levels [33]. Although HBx has been extensively investigated on account of its a critical role in neoplastic transformation of hepatocytes in liver infected with HBV, the molecular mechanism(s) involved in HBx-induced HCC are not yet fully understood [22]. Huh-7 cells have been used as an in vitro system to elucidate some of these HBx mechanisms. One of the cell’s earliest responses to HCV infection is the production of interferons (IFN). These cytokines establish an antiviral state in the cell and bridge the innate and adaptive immune systems [34]. HBx protein inhibits activation of interferon-β [35], and also activates NF-κB [36]; considering that this factor regulates many genes that are involved in various cell functions and growth control, its inappropriate activation can lead to inflammation and tumorigenesis. Thus, transactivation of NF- κB by HBx contributes to the pathogenesis of HBV. The potential effects of HBx on the progression of HCC was demonstrated by its up-regulation of pathways that promote proliferation and migration of hepatoma cells [37].
Frequently, reduction of HBV viremia is observed in patients co-infected with HBV and HCV. Double infection leads to more severe liver disease activity and progression, and at the same time to a reciprocal replicative suppression suggesting the existence of viral interferences [23]. To study mechanisms of HBV/HCV viral interference, Huh-7 cell lines inducibly replicating HBV were generated using a tetracycline-regulated gene expression system [38]. Transfection of these cells with HCV replicons or infection with cell culture-derived HCV indicated that both viruses replicated without overt interference suggesting that the reciprocal replicative suppression observed in co-infected patients arises probably from indirect immune-mediate mechanisms [38].
3.2. Hepatitis C Virus
Cellular responses activated by HCV include endoplasmic reticulum (ER) stress/unfolded protein response (UPR), autophagy, apoptosis and cell cycle arrest [39]. These responses are implicated in the pathogenesis of liver disease; persistent HCV infection leads chronicinfected hepatocytes progressively to develop hepatosteatosis, liver cirrhosis, and tumourigenesis of hepatocellular carcinoma.
In vitro and in vivo studies served to characterise HCVregulated ER stress and UPR. The ER is an important organelle for fatty acid synthesis, cholesterol metabolism and protein folding; multiple mechanisms have been proposed for ER stress-induced hepaticsteatosis. Huh-7 cells were employed to establish functional effects of HCV on the ER and UPR; e.g. activation of 5’ mRNA capdependent and cap-independent initiation of translation [40, 41], modulation of intracellular NF-κB signaling [42], supression of antiviral innate response [41], and promotion of viral replication [43,44].
The intracellular process of autophagy targets cytoplasmic components to lysosomes for degradation. It plays a role in the regulation of lipid metabolism and in malignant transformations. Autophagy is reduced in a variety of tumours; inhibition of tumour supressor genes can lead to supression of autophagy, and can promote survival of cancer cells. Cancer cells may exploit the autophagy pathway to counteract a variety of cellular responses and ensure cell survival in the development of tumours [39]. Some viruses, including HCV induce autophagy to benefit their life cycle. Huh-7 cells were used to understand better HCV-activated autophagy in viral replication [43], supression of anti-viral innate immunity [44], protection of host cells from viral infection-induced cell death [45], mitochondria-mediated cytopathic effects [46], lipid metabolism and regulation of lipid storage [47], and dysregulation of glucose homeostasis and induction of insulin resistance [48].
An important strategy of host cells to protect themselves against viral infection is to trigger virus-infected cell death via apoptotic processes. Apoptosis is induced through two major pathways; the mitochondria-mediated intrinsic pathway and the TNF family-induced extrinsic pathway. Mitochondrial apoptosis is initially triggered by various stress signals that are integrated in the mitochondria and produce a release of cytochrome c that results in caspase cascade activation. The extrinsic cell death pathway is triggered by the TNF superfamily of cytokines that activates signaling pathways for cell survival, apoptosis, inflammatory responses and cellular differentiation [39]. HCV activates apoptotic cell death via both pathways [49,50]; at variance with this, some viral proteins inhibit apoptosis in cells [51,52]. Studies with Huh-7 cells showed that some HCV proteins sensitised the cells to apoptosis [53] and promoted apoptosis via the intrinsic pathway [54] or the extrinsic pathway [55]; other HVC components inhibited apoptotic signaling [56] or extrinsic apoptosis [51]. Evidence suggests that apoptosis has a potential role in the pathogenesis of chronic liver disease by inducing liver inflammation, liver injury, and liver fibrosis. Dysregulation of apoptosis may promote hepatocarcinogenesis by altering expression of pro-apoptotic molecules and introducing the cell survival signal, thus promoting unregulated cell proliferation and finally leading to development of hepatocellular carcinoma [39].
Cyclin-dependent kinase (CDK) complexes exert tight control of progression of the cell cycle. The activities of these kinases are positively regulated by cyclins and negatively regulated by CDK inhibitors (CKI). The role of CKI inhibitors in cell cycle progression is to coordinate internal and external signals and control proliferation at several checkpoints [57]. Although HCV virus infection often leads to cell cycle arrest through altering the activity of some of its regulators [58], it is suggested that the HCV core protein promotes cell proliferation by repressing the expression of tumor suppressor p53 and CKI p21, decreasing the abundance of the retinoblastoma (RB) gene product, and enhancing the transcription factor E2F transactivation of S-phase genes [59]. Investigations with Huh-7 cells helped to elucidate how HCV interferes with RB-mediated cell cycle progression [60], and modulates host gene expression by regulating RB abundance [61]. The tumour suppresor function of RB is exercised by tightly regulating cell proliferation and apoptosis, HCV-induced disruption of the RB-E2F-regulatory pathway in infected cells may promote uncontrolled hepatocellular proliferation, an initial factor leading to development of liver cancer [39]. In addition, Huh- 7 cells were employed also to show that HCV infection induces chromosomal polypolyploidy and impairs mitotic checkpoint [62,63]; this chromosomal instability and defect in mitosis could provide an alternative mechanism to promote hepatocar-cinogenesis. Studies in HCV-infected hepatocytes show that the infection also interferes with host DNA damage/repair responses [64]; inhibition of this pathway may allow infected cells to tolerate massive mutations, leading to tumourigenesis. Overexpression of the HCV protein NS3/4A in Huh-7 cells demonstrated how HCV interferes with DNA repair and potentiates chromosomal instability [65].
3.3. Bacterial Agents
Several non-viral carcinogens have been associated with HCC including Aspergillus aflatoxin B1, haemochromatosis, and fatty liver disease related to diabetes and obesity, but their frequencies of association with the liver cancer are lower than with HBV or HCV. Many of the chronic carriers of HBV or HCV do not develop cirrhotic liver, and only a subset of patients suffering from viralinduced liver cirrhosis eventually progress to HCC. These data suggest that in the presence of HBV or HCV, some co-factors may contribute to hepatocarcinogenesis.
Increasing evidence shows that the bacterium Helicobacter pylori, a group 1 carcinogen [66], together with other Helicobacter bacteria could play a role in human HCC [67]. Associations between various of species of this genus and HCC have been proposed because their nucleic acids have been found in tissue biopsies of HCV-induced HCC patients [67], as well as in patients without other risk factors [68]. Hence, a number of studies have been conducted employing the Huh-7 in vitro model system to elucidate the mechanisms by which Helicobacter species may cause or potentiate the events leading to neoplasia. Several strains of H. pylori and one strain of H. hepaticus form podosomes in Huh-7 cells [69]. Podosomes are actin-based protrusive organelles arranged into peculiar dot-like structures, and consist of a densely packed F-actin core and actin-regulatory proteins such as vinculins, integrins and signaling proteins [70]. They have been described in responses of cells to environmental stresses such as ischemia or nutrient deprivation, where they are likely to manifest a defense mechanism aimed at reducing mechanical or metabolic stress [71]. It was suggested that the formation of podosomes by Huh-7 cells might mirror loss of tissue integrity and bacterial infection in diseased livers [69]. Similar results were obtained in primary hepatocyte cultures indicating that Huh-7 effectively mirrored the behavior of heaptocytes, in this in vitro context, mimicking a pathological state [69].
To elucidate potential mechanisms leading to the development of HCC in the presence of Helicobacter spp., a global proteomic approach was employed to investigate the differential protein expression in co-cultures of Helicobacter bilis with Huh-7 cells harbouring the HCV sub-genomic replicon, and replicon cured-Huh-7 cells [72]. The analyses revealed the effects of the bacterium on the expression of Huh-7 proteins in the presence of HCV replicon and when the replicon had been removed from the cells through treatment with IFN-α. The hummingbird morphology was observed in both the transfectedand cured-Huh-7 cells co-cultured with the bacterium, and no significant decline in cell proliferation was observed between them, suggesting that neither the presence of the HCV-replicon nor its inactivation by IFN-α treatment affected differently the morphology and growthresponse of the liver cells. At the molecular level, modulation of several proteins that participate in differrent biological processes were observed including stress response proteins, metabolic enzymes, protein translation, modification and degradation, cell proliferation and structure, as well as tumor-related proteins [72]. Several metabolic enzymes expressed in the transfected-Huh-7 cells in response to H. bilis were not expressed in the curedHuh-7 cells from which the replicon has been eliminated, or in the parental Huh-7 cells not transfected with the replicon [73]. This finding suggested an interaction between the effects of the replicon and H. bilis on metabolic pathways of the hepatoma cells. The differences in the expression of metabolic proteins between the parental and cured Huh-7 cells suggests an effect of IFN-α treatment of the cured cells and/or a permanent effect of the virus on the cells. The latter would explain the different modulation of protein expression in response to H. bilis of cured and parental Huh-7 cells. These findings are in agreement with the results of a study showing variations in protein expression between parental Huh-7 cells, transfected with an HCV replicon and cured cells [74].
4. Natural Products as Chemopre-Ventive Agents against HCC
Current treatments for hepatocellular carcinoma, including chemotherapy, hepatectomy and liver transplant, do not always offer optimal outcomes for patients. Liver cancer is often not detected until late in the progression of the disease, and thus chemotherapy can prove a poor treatment option and can induce severe side effects. Likewise, hepatectomy and liver transplantation can be curative, but they have high recurrence rates and poor subsequent survival rates [75].
Natural products are non-toxic natural extracts or compounds that compared with synthetic materials, generally produce less side effects [75]; thus, potentially they provide an alternative or adjunct therapeutic option for patients with cancer. Many natural compounds have been the focus of research to determine their anti-cancer properties and mechanisms of action. From these studies, new chemopreventive agents have been found that may be developed for the treatment of HCC.
4.1. Apoptosis-Inducing Compounds
Apoptosis, also known as “programmed cell death”, is the method of many anti-cancer treatments; apoptosis signal transduction can occur via extrinsic or intrinsic pathways. A significant proportion of recent research into natural products as chemopreventive agents has focused on natural products that exhibit their anticancer effects through the induction of apoptosis.
4.1.1. Flavonoids
Flavonoids are a group of natural products that have been investigated as agents for the prevention and treatment of HCC. Commonly, they are found in fruits and vegetables, and are known to possess anti-allergic, anti-inflammatory, antioxidant and anticancer properties [75]. Like many other natural compounds, flavonoids do not cause significant toxicity or side effects, a property that enhances their potential as therapeutic agents against cancers, including HCC [75]. Flavonoids can act as anticancer agents through the induction of apoptosis. However, little is known about the exact mode of action inducing their apoptotic activity. Results of recent studies using the Huh-7 cell line provided a deeper understanding of the exact mechanisms of these compounds in inducing apoptosis (Table 1).
Apigenin (APG) is a flavonoid found in orange, tea, chamomile, onion and wheat sprouts, that exhibits antioxidant and anti-inflammatory effects [76]. The effects of APG on Huh-7 cells, were investigated in a recent study of APG-induced apoptosis which focused on the changes in the levels of the intracellular protein vimentin and type I collagen; vimentin has a central role in maintaining cell shape and supporting and anchoring the position of the organelles in the cytosol. They were chosen in the study of APG owing to its significant involvement in cell migration, adhesion and signalling, and apoptotic processes [76]. In particular, vimentin methylation has been proposed as biomarker of upper gastrointestinal neoplasias [77]. APG-treated Huh-7 cells had decreased expression of vimentin, consistent with increased apoptosis of these cells. Also, the levels of the angiogenic proteins vascular endothelial growth factor (VEGF) and matrix metalloproteinase-8 (MMP-8) were decreased. These results on Huh-7 cells indicate that a central effect of APG is to lower expression of proteins essential for cell structural integrity and function such as vimentin and type I collagen, as well as of proteins that promote vascularisation and tumour growth.
Studies with the flavonoid Aloe-emodin (AE), a hydroxyanthraquinone that can be extracted from Aloe vera leaves, established that DNA damage and apoptosis were induced in Huh-7 cells treated with it. Specifically, two apoptotic pathway signalling proteins calpain-2 (CAPN2) and ubiquitin-protein ligase E3A (UBE3A), were downregulated. Like for APG, the mechanism of apoptosis for AE has been identified, and could serve as the basis for further research into its development as a chemopreventive drug for the treatment of HCC [78].
The flavonoid luteolin is found in fruits and vegetables, and also in tea. It possesses anti-inflammatory, antioxidant and anti-proliferative properties. This natural product exhibits anticancer apoptotic effects on Huh-7 cells through production of reactive oxygen species (ROS) and changes in the expression of levels of the proteins peroxiredoxin 6 (PRDX6) and prohibitin (PHB) [79]. The conclusion of this work was that PRDX6 and PHB are key targets of luteolin, and that the flavonoid induced apoptosis in Huh-7 cells through effects involving intracellular ROS [79].
Fisetin is a flavonoid found in strawberries, apples and grapes; it exhibits anti-inflammatory, anti-platelet, antioxidant, and anticancer properties. Specifically, fisetin increased intracellular levels of ROS, contributing to induction of apoptosis in cells via the caspase cascade through the release of cytochrome c. This flavonoid has effects also on the expression of proteins involved in apoptosis; in Huh-7 cells it causes down-regulation of the baculoviral IAP repeat-containing protein 8 (BIRC8) and the apoptosis regulator Bcl-W (Bcl2L2) [80]. Thus, the mode of action of fisetin in Huh-7 cell line is by inducing apoptosis through the dysregulation of apoptotic proteins and increasing intracellular levels of ROS. These properties make it a key flavonoid to be further studied for its potential as a chemopreventive agent in HCC.
A natural product extract from the stem bark of the African tree Pycnanthus angolensis contains several isoflavones: irilone, tectorigenine, formononetin, genistein, 2-hydroxybiochanin A, a mixture of biochanin A and prunetin, and 4’,7-dihydroxy-2’-methoxyisoflavan, as well as the flavonone liquiritigenin. The study was undertaken to determine whether these compounds possess cytotoxic properties on Huh-7 cells; the results indicated that the flavonoids had apoptotic effects on the hepatoma cells through induction of caspase-3 activity [81]. Further studies will serve to determine in greater detail the specific mechanism of action of each flavonoid and its potential as chemopreventive agent.
The flavonoids described in this section induce apoptosis via diverse pathways. Recent research has made clearer the cytotoxic mechanisms of many of these compounds, but further investigations will help to achieve a complete understanding of their mode of action. Notwithstanding the knowledge gap, current data provide evidence for their potential as chemopreventive agents in the treatment of HCC.
4.1.2. Non-Flavonoid Compounds
In addition to flavonoids, recently, other natural compounds have been investigated for their potential as chemopreventive agents in the treatment of HCC. A study of Pulsatilla koreana extract (PKE), a traditional
Table 1. Natural products with cytotoxic properties for Huh-7 hepatocytes.

Korean herbal medicine, found that it has apoptotic effects on Huh-7 cells by increasing the expression of caspase-3 and poly(ADP-ribose) polymerase (PARP) [82]. To exploit the potential of PKE in chemotherapy, investtigations are needed to determine its specific anti-cancer components.
Squamocin extracted from the seeds of the plant Annona squamosa has cytotoxic effects on Huh-7 cells. Specifically, it arrests the cell cycle at the G1 phase, and activates both intrinsic and extrinsic pathways to apoptosis. Squamocin induces apoptosis through modulation of histone H3 phosphorylation together with down-regulation of the expression of aurora B kinase and mitogen-and-stress-activated protein kinase-1 MSK1 [83]. Interestingly, squamocin anti-tumour effects occur via epigenetic changes resulting from modulation of histone phosphorylation.
Resveratrol is a stilbenoid natural phenol that inhibits Huh-7 cell proliferation and induces cell cycle arrest and apoptosis. Apoptosis is achieved through up-regulation of cyclin-dependent kinase inhibitor 1 (p21/WAF1) and down-regulation of cyclin E, cyclin A, and cyclin-dependent kinase 2. The study showed that resveratrol increases the ratio of pro-apoptotic/anti-apoptotic proteins, is associated with the mitochondrial membrane depolarization and the increase in caspase activity, and increases expression of the autophagy-related proteins Atg5, Atg7, Atg9, and Atg12 [84].
Monoterpene and β-carboline indole alkaloids are isolated from the methanol extract of leaves of Tabernaemontana elegans. Huh-7 cells treated with these alkaloids showed increased caspase-3 activity and subsequent induction of apoptosis. The work identified the corynanthe-type monoterpene alkaloids tabernaemontanine and vobasine to have the best potential as chemopreventive agents against HCC [85].
The triterpenoids saponins gypenosides derived from Gynostemma pentaphyllum are widely employed in traditional Chinese medicine. They have cytotoxic properties and a study undertaken to understand their effects on Huh-7 cells found that they induce apoptosis through a mitochondria-dependent caspase-9 activation cascade [86]. A more detailed investigation of the mechanisms leading to apoptosis in gypenoside-treated cells showed that the saponins induced the generation of ROS, disruption of the mito-chondrial membrane potential, inactivation of ERK, increased miotchondrial Bax and decreased mitochondrial Bcl-2 levels, and cause a rapid rise in intracellular Ca2+ levels. It was concluded that the increase in Ca2+ levels acts as a trigger for apoptosis of these cells [87].
Ginsenosides are pharmacologically active steroidal saponins of ginseng, natural herbal extract of the root of the medicinal plant Panax ginseng. The 20S-ginsenoside Rg(3) promoted apoptosis of Huh-7 cells via the tumour necrosis factor-related apoptosis-inducing ligand (TRAIL) pathway. Rg(3) up-regulates tumour necrosis factor receptor member 10b (TNFRSF10B, DR5) expression at the transcriptional level that is mediated bythe transcription factor CCAAT-enhance-binding protein (C/EBP) homologous protein (CHOP) an endoplasmic reticulum stress response protein. Since Rg(3) is well tolerated, its properties support its further devlopment as novel anticancer therapeutic agent in combined therapy with TRAIL [88].
The properties of the natural herbs Morarah and Khaltita were studied to determine their effects on Huh-7 cells. Extracts of these herbs induced apoptosis in the cells in culture showing anticancer effects in vitro and potential as chemopreventive agents against HCC. Further investigations need to take place in order to establish their mechanism of action in induction of apoptosis and possible side effects [89].
Artemisinin is a natural sesquiterpene lactone from the plant Artemesia annua that traditionally has been used in the treatment of malaria. A recent study showed that some derivatives, including dihydroartemisinin, artemether and artesunate, have also anticancer capabilities [90]. These artemisinin derivatives exert anticancer properties through induction of apoptosis. A recent study employing Huh-7 cells was undertaken to better understand the mode of action of these products in their induction of apoptosis, It established that they cause cell-cycle arrest at the G1-phase; they decrease the levels of cyclin D1, cyclin E, cyclin-dependent kinase 2, cyclin-dependent kinase 4, and E2F1; and they increase the levels of cyclin-dependent kinase inhibitors 1 (Cip1/p21) and 7 (Kip1/p27). In addition, the derivatives cause an increase the Bax/Bcl-2 ratio and up-regulation of caspase-3, an elevation of PARP levels, and down-regulation of the double minute oncogene MDM2 [90]. The PARP family of proteins is found in the nucleus, and the proteins are involved in DNA repair and programmed cell death; they are activated in cells experiencing stress and/or DNA damage. PARP stimulate the release of mitochondrial apoptosis-inducing factor that initiates caspase-independent apoptosis and is an intrinsic regulator of apoptosis [91]. All these effects contribute to their cell death effects on Huh-7 cells and support their potential use as chemopreventive agents against HCC.
The naturally occurring triterpenoid oleanolic acid (OA) and pentacyclic triterpene ursolic acid (UA) commonly found in plants and herbs posses hepatoprotective, anti-inflammatory and anti-cancer activities. OA and UA inhibit the growth of Huh-7 cells by stopping cell-cycle progression at the G1 phase. Treatment of this hepatoma cell line produced a loss of mitochondria membrane potential, and changed the ratio of expression levels of proand anti-apoptotic proteins. In the treated Huh-7 cells mitochondrial cytochrome c is released to the cytosol, caspase-3 and caspase-9 are activated followed by PARP cleavage. In addition, treated cell suppressed the activity of NF-κB and modulated the expression of X-linked apoptotic protein (XIAP). The data indicated that OA and UA induced apoptosis of Huh-7 cells via a mitochondria-mediated pathway and down-regulation of XIAP [92].
4.2. Compounds Affecting Non-Apoptotic Pathways
Apoptosis is a common mechanism of action of many natural products, but many others exert their effects through various cellular pathways. Geranylgeranoic acid (GGA) is a polyprenoic natural product found in the Chinese herb Schisandra chinensis [93]. A recent study of the effects of GGA and its derivatives showed that it causes an initiation of autophagy on Huh-7 cells, but blocks maturation of autolysomes and consequently the late stages of autophagy [94]. Another study with these hepatoma cells showed in GGA-treated cells suppression of cyclin D1 protein, subsequent decrease in the phosphorylation and nuclear translocation of retino-blastoma protein (RB), and nuclear accumulation of RB. Such activity disrupts cell growth and cell-cycle progression, making it a potential agent for the treatment of HCC [95].
Another compound that disrupts the cell cycle is Dallose, which has been the focus of a recent study of its effects on various cancer cells. In particular on Huh-7 cells GCA causes G1 cell-cycle arrest, as well as upregulation of gene expression of the thioredoxin interacting protein (TXNIP) and stabilisation of p27kip1, that cause also cell-cycle arrest [96].
Prostaglandins (PG) are mediators of inflammation and have an important role in hepatocarcinogenesis [97]. The are derived from arachidonic acid, an omega-6 polyunsaturated fatty acids (ω-6 PUFA), by the action of cyclooxygenase enzymes (COX). Elevated levels of PG and increased expression of the inducible cyclooxygenase-2 (COX-2) have been measured in HCC. The Wnt/beta-catenin (β-catenin) pathway ialso is implicated in hepatic carcino-genesis [98], and elevated levels of β-catenin are found in hepatomas. In contrast, there is considerable evidence that omega-3 poly-unsaturated fatty acids (ω-3 PUFA) prevent carcinogenesis. A study of the effects on Huh-7 cells of the ω-3 PUFA docosahexaenoic acid (DHA) and eicosapentaenoic acid (EPA) showed that they inhibit COX-2 and β-catenin. Treatment of Huh-7 cell cultures with DHA and EPA causes cleavage of poly(ADP-ribose) polymerase, and activation of caspase-3 and caspase-9. These effects suggest some ω-3 PUFA acids as potential agents for the treatment of HCC [99].
Isofraxidin (7-hydroxy-6,8-dimethoxy-2H-1-benzopyran- 2-one), is a coumarin component extracted from the stem bark of the Chinese herb Acanthopanax senticosus. Recent studies of its effect on Huh-7 cells show that it has anti-cancer effects and is a potential future therapeutic agent for HCC. Specifically, isofraxidin inhibits metalloproteinase-7 (MMP-7) expression, and also the phosphorylation of extracellular-signal-regulated kinases 1 and 2 (ERK1/2); the functional outcome is to inhibit in vitro cell invasion [100].
Cellular senescence is a process that limits the proliferation (growth) of normal human cells in culture. It is an essentially irreversible growth arrest [101]. Penta-1,2, 3,4,6-O-galloyl-beta-D-glucose (PGG) is a naturally occurring gallotannin polyphenolic compound found in herbs such as Rhus chinensis and Paeonia suffructicosa capable of inducing S-phase and G1 cell-cycle arrests [102,103]. PGG-induced senescence-like S-phase arrest in Huh-7 human hepatoma cells; the senescence-like response was mediated by intracellular ROS generation. These results suggest a novel mechanism of PGG action to induce an atypical cellular senescence in hepatoma cells, adding to its potential as a potential chemopreventive agent [104]. Figure 1 indicates the cellular loci where these natural compounds act.
5. Systemic Therapies in the Treatment of HCC
Systemic therapies of HCC include chemotherapy, hormone therapy and immunotherapy. These treatments have been used in the past, but the evidence indicates that they are not effective in advanced or unresectable presentations of the disease, shown by low response rates and marginal survival benefits [105]. In addition, systemic cytotoxic chemotherapy can have significant toxicity in patients with underlying liver dysfunction [106]. Several characteristics of HCC contribute to the ineffectiveness of systemic therapies; hepatomas tend to be chemotherapy-resistant and neoplastic hepatocytes express the multidrug-resistant gene MDR, also, HCC are heterogeneous tumours that have different pathways of carcinogenesis [105]. The discovery of molecularly targeted agents and their success in treating various types of cancers has renewed interest in the development of new active agents that increase the chemosensitivity of HCC to systemic therapies. This section reviews a number of single agents that have employed the Huh-7 cell line to increase understanding of the tumour biology of HCC and improve systemic chemotherapies.
Anthracyclines are antibiotic compounds produced by the bacterium Streptomyces peucetius whose derivatives have been used as anti-neoplastic drugs; doxorubicin (DOX) or adriamycin is an anthracycline produced by genetically modified Streptomyces strains. The cytotoxic

Figure 1. Cellular loci where natural compounds described inthis section act. CAS, caspase cascade, NUC, nucleus; MIT, mitochondrion; CTS, cytoskeleton; ER, endoplasmic reticulum.
activity of DOX stems from inhibition of macromolecular biosynthesis at the transcription level through intercalation with DNA and stopping topoisomerase II. It has been used commonly to treat HCC, but as a single agent has not proved effective owing to tumour resistance, and its toxicity to normal cells. Thus, investigations were directed at discovering new therapeutic agents that can enhance the effectiveness of doxorubicin treatment. A reason for HCC resistance to DOX is the ability of neoplastic cells to undergo epithelial-mesenchymal transition (EMT) during tumour progression; a process by which epithelial cells down-modulate cell-adhesion, change their polarity and cytoskeleton, become isolated and motile and develop resistance to programmed cell death; in hepatoma cells, activation of signal transducer and activator of transcription 3 (STAT3) contributes to the process of EMT [107]. A study of the novel STAT3 inhibitor NSC 74859 using Huh-7 cells showed that co-administered with DXR it has synergistic anti-tumour effects enhancing DOX cytotoxicity [108].
Cancer stem cells (CSC) are defined as a minor population that possess a prominent ability to generate new tumours, and are capable to self-renew and generate differentiated progenies like normal stem cells [109]; they are key factors in tumour progression and growth. A side population of Huh-7 cells considered representative of HCC stem cells was used to identify molecular signalling pathways in liver CSC, and to suggest new therapeutic strategies based on the results [110]. The study established that in the CSC side population of Huh-7 cells hypoxic conditions promoted cell proliferation and growth owing to overexpression of cytochrome P450 2C9 (CYP2- C9), a protein localised in the endoplasmic reticulum and that metabolises many xenobiotics. Also, in the CSC subpopulation STAT3 was activated by interleukin-6 was more significantly than in non-CSC cells, and plays a role in resistance to DOX. Treatment with sulfaphenazole, an inhibitor of CYP2C9 inhibitor, or AG490, an inhibitor of STAT3, sensitised the CSC population to DOX, suggesting that co-administration of the inhibitors with DOX may be a potential new therapeutic strategy [110].
Nutlin-3 is cis-imidazole analogue that inhibits interactions between the tumour suppressors p53 and p73 and HDM2, the human analogue of the murine double minute (MDM2) oncoprotein. Recent investigations using Huh-7 cells showed that Nutlin-3 enhances the chemosensitivity to DOX through inhibition of p73-HDM2 binding with subsequent activation of the apoptotic pathway. Such result suggests that Nutlin-3 may be a potential HCC therapy when co-administered with DOX [111].
The effects of some chemotherapeutic agents are enhanced by co-administration with natural products. A study investigated the benefits caffeine-assisted chemotherapy in the treatment of HCC with cis-diamminedichloroplatinum-II (cisplatin). Caffeine could increase the anti-tumour effect of cisplatin or other DNA-damaging agents because it inhibits DNA repair. Cisplatin is an agent used in the treatment of HCC, but its administration alone is not optimal [200]. Huh-7 cells were treated with caffeine and cisplatin and the co-administration enhances the anti-tumour effects of cisplatin [112]. Expression of the MDR gene is mechanism by which HCC cells become resistant to many of the available chemotherapy drugs. Recent work found that coadministration of Kampo drugs, traditional Japanese medicines, has potential to overcoming drug resistance of HCC to paclitaxel (PTX), a mitotic inhibitor synthesised by fungi in the bark of Taxus brevifolia, the Pacific yew tree. Using the Huh-7 paclitaxel-resistant variant Huh-7/PTX that expresses high levels of the multidrug resistance permeability glycoprotein 1 (MDR-1), 26 kinds Kampo medicines were tested and two of them, takushato and goreisan, increased the sensitivity of HuH-7/PTX cells to paclitaxel. These compounds contain the triterpenes Alisol A, Alisol B, and Alisol B acetate that increase resistance to paclitaxel by preventing drug efflux by MDR-1 without affecting its expression levels. This study identified Kampo drugs as potentially effective when coadministered with the chemotherapy drug paclitaxel [113].
The uptake of chemotherapy drugs and their subsequent therapeutic effects can be enhanced by the use of nanocarriers. A nanoparticle from an amphiphilic block copolymer composed of conventional monomethoxy (polyethylene glycol)-poly (D,L-lactide-co-glycolide)-poly (L-lysine) (mPEG-PLGA-b-PLL) was used for delivery of DOX or of small interfering RNA-negative (siRNA) into Huh-7 cells. The study shows increased cellular uptake of the drugs when they were delivered into the cell via the nanoparticle carrier. The findings suggest the potential of these carriers to deliver chemotherapy agents to HCC with subsequent enhanced cellular uptake and therapeutic results [114].
Another study investigated the delivery to Huh-7 cells of mitoxantrone (MX), an anthracenedione anti-neoplastic agent that disrupts DNA synthesis and DNA repair. Dual-functional liposomes (LPG) with the synthetic polymeric nano-biomaterial (Gal-P123) that targets cancer cells and reverses multidrug resistance were loaded with MX. Delivery via LPG enhanced uptake of MX by Huh- 7 cells, and inhibited drug efflux, thus improving significantly the cytotoxic therapeutic efficacy of MX [115].
To increase the effectiveness of cisplatin treatment, further knowledge has been required of its pro-apoptotic mechanisms. The mitogen-activated protein kinase (MAPK) pathway plays a key role in cell response to cisplatin. A recent study determined the individual contribution of each kinase of this pathway on cisplatininduced death. ERK1 has a greater role than ERK2 in cisplatin-induced pro-apoptotic signal in HCC cells and it was up-regulated by ERK2 inhibition. The results of this study provide the basis for more targeted cisplatin therapy suggesting that its efficacy of could be increased by targeting ERK2 [116].
Recent studies show that gene therapy can enhance the effects of some chemotherapeutic agents. A study demonstrated that transfection of the sodium iodine symporter (NIS) or the mutant Herpes-simplex virus type1 sr39 thymidine kinase (HSV1-sr39tk) gene into Huh-7 cells enhances intracellular accumulation of therapeutic radionuclides and guanosine nucleoside analogue prodrugs. The work demonstrates the potential of combination gene therapy using NIS and HSV1-sr39tk followed by radioiodine treatment and chemotherapy to treat human hepatocellular carcinoma cells [117].
6. Targeted Therapies in the Treatment of HCC
Dysfunction of many different signaling pathways occurs in the pathogenesis of hepatocellular carcinoma [118]. Also, the etiology of hepatocellular carcinoma is complex and diverse; hence, improved understanding of these pathways is required [119]. Unlike systemic therapies, targeted therapies aim at specific aberrant pathways [120]. Thus, a better of understanding of the biology of neoplastic hepatocytes will facilitate the development of new, targeted therapeutic agents that overcome drug resistance and minimise side effects, with the ultimate aim of improving patient outcomes [119], for this reason targeted therapy for HCC is currently under intensive investigation [120]. The Huh-7 cell line is extensively used in current investigations to develop of therapies for HCC, and this section attempts to provide an overview of this research. Table 2 sumamrises the targeted therapies discussed in this section.
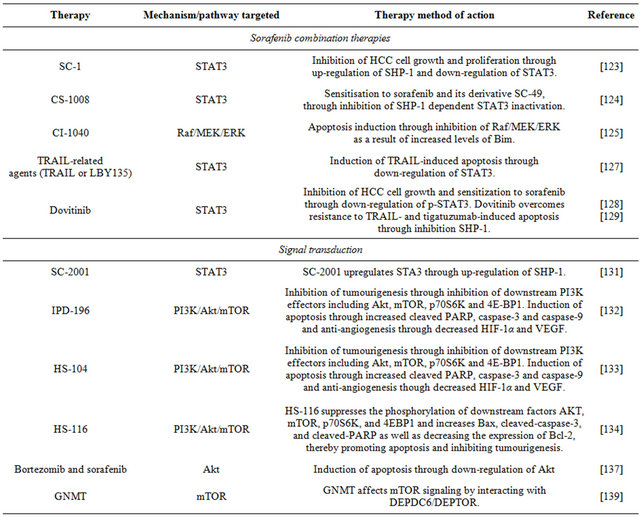
6.1. Sorafenib Combination Therapies
Presently, the mainstay of targeted therapy against HCC is sorafenib, a bi-aryl urea which interferes with tyrosine protein kinases; it inhibits cancer cell growth signaling and angiogenesis and promotes apoptosis [121]. The story of this drug has proven the concept that targeted therapy brings benefits to patients with HCC, although at present these remain limited [120] because sorafenib like other therapeutic agents has a number of limitations, including the development of resistance against it and side effects [122]. A number of investigations have been undertaken to identify sorafenib combination therapies that would improve its efficacy.
To investigate the role of the signal transducers and activators of transcription 3 (STAT3) in sorafenib inhibittion of HCC, Huh-7 cells were treated with this drug or SC-1, one of its derivatives that is structurally similar but without kinase inhibition activity; the treated heaptoma cells showed growth inhibition and apoptosis. The STAT3 signaling pathway regulates proteins such as myeloid cell leukemia sequence 1 (Mcl-1), survivin and cyclin D1 that are involved in cell growth, survival, differentiation and anti-apoptosis; STAT3 is down-regulated by the protein tyrosine phosphatase SHP-1. The results showed that SC-1 and sorafenib up-regulated SHP-1 activity and down-regulated STAT3, thereby inhibiting HCC cell growth and proliferation. This work suggests that for sorafenib-treated patients, STAT3 could potentially be used as a biomarker for determining HCC prognosis [123]. In a study with Huh-7 cells, it was determined that sorafenib or SC-49, one of its derivatives devoid of kinase inhibition activity, in combination with CS-1008, a novel anti-human death receptor 5 antibody that induces apoptosis, down-regulate the phosphorylation of STAT3 and subsequently reduce the protein levels of STAT3-regulated proteins. The mechanism of action of the combination therapy was STAT3 inactivation through increasing the activity of the modulator of signaling cascades SHP-1 [124].
The effects of co-administration of sorafenib and CI- 1040, an inhibitor of mitogen-activated protein kinase/ extracellular signal-regulated kinase (ERK) kinase (MEK) were investigated in Huh-7 cells. Combination of sorafenib and CI-1040 synergistically inhibited ERK phosphorylation and cell growth and induced apoptosis. Vertical blockade of signaling through the protooncogene serine/threonine-protein kinase (Raf)/MEK/ERK was achieved by the co-administration of these agents, improving the anti-tumour effects of sorafenib in HCC [125].
Programmed cell death can be stimulated by tumour necrosis factor (TNF)-related apoptosis-inducing ligand (TRAIL) [126]; hence, TRAIL is a potential target for anti-cancer drugs. However, hepatomas often display resistance to TRAIL-induced apoptosis; specifically, Huh-7 cells show significant resistance to TRAIL-induced apoptosis. These hepatoma cells were treated with sorafenib and/or LBY135, an agonistic death receptor 5 (DR5) antibody, and the cell responses were investigated in terms of signal transduction and apoptosis. Besides downregulating phospho-STAT3, and subsequently reducing the expression levels of STAT3-related proteins, sorafenib in combination with LBY135 significantly supressed xenograft tumour growth, suggesting that it sensitises hepatoma cells resistant to TRAIL-induced apoptosis [127]. This study provides a positive foundation for future applications of sorafenib as an agent to help overcome TRAIL resistance in HCC [127].
Table 2. Targeted therapies for HCC that have been investigated using Huh7-cells.
Continued
Dovitinib is a multiple kinase inhibitor currently undergoing clinical investigation for its use in the treatment of HCC. It shows anti-tumour activity in Huh-7 and other liver cell lines. Its mechanism of action is down-regulation of phospho-STAT3 (p-STAT3), subsequently reducing the levels of expression of STAT-3 related proteins, and inducing apoptosis. An inhibitor of SHP-1 reversed the down-regulation of p-STAT3 and the apoptosis induced by dovitinib, and this inhibitor up-regulates the activity of SHP-1 via direct interactions. In addition, dovitinib induced apoptosis in two sorafenib-resistant cell lines through inhibition of STAT3, and sorafenib-resistant cells showed significant activation of STAT3, suggesting that targeting STAT3 may be a useful approach to overcome drug resistance in HCC [128] Dovitinib and tigatuzumab, a novel humanized anti-human DR5 agonistic antibody, are currently undergoing clinical investigation for their co-administration in the treatment of HCC. Besides showing significant resistance to TRAIL-induced apoptosis, Huh-7 cells are resistant also to tigatuzumabinduced apoptosis. The mechanism of action of the antitumour effects of dovitinib in resistant Huh-7 cells was through inhibition of phospho-STAT3. The combination of dovitinib and tigatuzumab increased the activity of SHP-1 and restored the sensitivity of the hepatoma cells to TRAILand tigatuzumab-induced apoptosis [129].
6.2. Signal Transduction
The anti-apoptotic proteins of the BCL-2 family are overexpressed and dysregulated in various cancers, including HCC. In particular, Bcl-xL and Mcl-1 have significant cytoprotective roles in HCC. A common strategy in the design of Bcl-2 protein inhibitors is based on mimicking the actions of endogenous inhibitors that bind anti-apoptotic Bcl-2 proteins via the Bcl-2 homology 3 (BH3) domains (BH3 mimetics) [130]. Obatoclax is a pan-Bcl-2 inhibitor that acts as a BH3-mimetic to disrupt the interactions of anti-apoptotic and proapoptotic proteins, such as Mcl-1 and Bak. It uniquely displaces BH3 domains by activation of the pocket of Mcl-1 followed by a triggering of apoptosis mediated by oligomerization of Bak and release of cytochrome c. Important anti-apoptotic members of the Bcl-2 family, such as Bcl-xL and Mcl-1, can be regulated by oncogenic transcription factors, such as STAT3 which is constitutively activated in HCC. SC-2001 is a novel compound structurally related to obatoclax; similarly, it inhibits in Huh-7 cells protein-protein interactions between Mcl-1 and Bak, and, in addition, it down-regulates Mcl-1 protein levels by reducing its transcription. SC-2001 down-regulates the phosphorylation of STAT3 subsequently inhibiting its transcriptional activities, and repressing also survivin and cyclin D1. The inhibitor upregulated expression of SHP1 inducing apoptosis in HCC and reducing tumour growth [131].
Normal cell function, including cell survival, cell proliferation and cell growth, involves the phosphatidy-linositol 3-kinase/protein kinase B/mammalian target of rapamycin (PI3K/Akt/ mTOR) pathway. This pathway is initiated by the enzyme PI3-kinase which causes phosphorylation of cell membrane components. Subsequentlyphosphorylated components bind to Akt and activate this protein kinase, which initiates the activation of several pathways that promote cell survival, proliferation and growth. Akt activation is followed in the PI3K/Akt/ mTOR pathway by activation of mTOR; this kinase plays a role in protein synthesis that ultimately supports cell proliferation and growth, and also is involved in nutrient uptake and angiogenesis. Impairment of any of the components of this pathway can result in tumourigenesis, and human cancers frequently have overactive PI3K/Akt/ mTOR pathways that reduce apoptosis and allow unregulated proliferation. In particular, this pathway is dysregulated in HCC; for this reason, in recent cancer research, this pathway and its components are the focus for finding targets target for therapeutic intervention.
The Huh-7 cell line has been employed in studies targeting the PI3K/Akt/mTOR pathway in HCC. A focus has been the development of PI3K inhibitors specific to HCC. The compound IPD-196 [ethyl 6-(5-(2,4-di-fluorophenylsulfonamido)pyridine-3-yl)imidazo[1,2-a] pyridine-3-carboxylate] is a PI3K inhibitor that is effective decreasing phosphorylation of downstream PI3K effecttors, including Akt, mTOR, the p70S6 ribosomal protein kinase and the translator repressor 4E-BP1, and it has more potent anti-proliferative effects that sorafenib. IPD- 196 caused cell cycle arrest, induction of apoptosis, and inhibition of angiogenesis in human hepatoma cells, suggesting that it may be a potential candidate drug for targeted HCC therapy [132].
Another PI3K inhibitor is the compound HS-104 [N- (5-(3-(3-methyl-1,2,4-oxadiazol-3-yl)imidazo [1,2-a] pyridin-6-yl)pyridin-3-yl)benzenesulfonamide)], recently studied in relation to its potential as an anticancer treatment for HCC. In Huh-7 cells, HS-104 inhibits cell growth, and its apoptotic effects include increased cleaved caspase-3 and PARP, as well as DNA fragmentation. These results together with those from experiments in animal xenografts suggest that HS-104 is considered a novel drug candidate for the treatment of HCC [133].
The compound HS-116 [N-(5-(3-(3-cyanophenyl)-1Hpyrrolo[2,3-b]pyri-din-5-yl)pyridin-3-yl)ben-zene-sulfonamide (HS-116)] show effects on Huh-7 cells similar to those of other PI3K inhibitors. Specifically, HS-116 suppresses the phosphorylation of downstream factors Akt, mTOR, p70S6K, and 4E-BP1 and promotes apoptosis by increasing Bax levels, cleaved-caspase-3 and PARP, and decreasing the expression of Bcl-2 [134].
Bortezomib is a proteasome inhibitor that binds with high affinity to the catalytic site of the 26S proteasome; inhibition of these protein complexes may prevent degradation of pro-apoptotic factors, thus permitting activation of programmed cell death in neoplastic cells dependent upon suppression of pro-apoptotic pathways. A major molecular determinant of bortezomib-induced apoptosis in Huh-7 cells is down-regulation of phosphoAkt; in cells able to develop resistance against bortezomib, phospho-Akt is not down-regulated [135]. This inhibitor overcomes TRAIL-induced apoptosis in Huh7- cells in part through inhibition of the (PI3K/Akt/mTOR) pathway [136]. It was found that in Huh-7 cells treated with sorafenib and/or bortezomib each inhibitor causes increased levels of apoptotic cell death, and they show synergy in combination by through Akt inactivation [137].
Mammalian mTOR resides in two distinct multiprotein complexes referred to as mTOR complex 1 (mTORC1) and 2 (mTORC2). Complex 1 controls cell growth in part by phosphorylating key regulators of protein synthesis, and mTORC2 modulates cell survival in response to growth factors by phosphorylating downstream effectors such as Akt. In addition to activating Akt by mTORC2, mTORC1 regulates negatively Akt by suppressing the growth factor-driven pathways upstream of it [138]. DEPTOR as an mTOR binding protein that normally functions to inhibit the mTORC1 and mTORC2 pathways, but overexpression of DEPTOR inhibits mTORC1, and this leads to the activation of Akt via the inhibition of a negative feedback loop to PI3K [138]. Loss of DEPTOR in Huh-7 cells reduced Akt activation and cell growth. It was revealed that GNMT affects mTOR signaling by interacting with DEPTOR, and that GNMT can sensitise HuH-7 cells to rapamycin both in vitro and in vivo. These results make GNMT and DEPTOR potential targets for future therapeutic agents for the treatment of HCC [139].
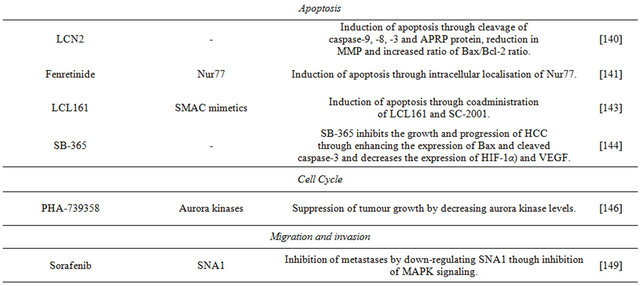
6.3. Apoptosis
Besides the pro-apoptotic effects of sorafenib, dovitinib and their derivatives, other compounds capable of promoting apoptosis in HCC have been investigated.
Abnormal expression of the secreted iron-binding glycoprotein lipocalin 2 (LCN2) is a feature of some human cancers. LCN2 overexpression in Huh-7 cells dramatically inhibited cell viability, induced cell-cycle arrest in sub-G1 phase, DNA fragmentation, and condensation of chromatin. LCN2 induces apoptosis in Huh-7 cells through the cleavage of caspase-9, -8, -3, and PARP protein, and a reduction in the mitochondrial membrane potential; it also increases the Bax/Bcl-2 ratio. The study improved the understanding of the role of LCN2 on HCC progression; by demonstrating that overexpression of LCN2 actively induces apoptosis of hepatoma cells, it suggests that the glycoprotein could be considered as therapeutic agent against HCC [140].
The mechanism of apoptosis induction by the synthetic retinoid fenretinide has been unknown; consequently, a recent study examined the mechanisms fenretinide-induced apoptosis in HCC cells. Apoptosis was correlated with the induction and cytoplasmic distribution of the nerve growth factor IB (Nur77). In Huh-7 cells, the sensitivity to fenretinide is related to the translocation of Nurr77 from the nucleus and targeting to mitochondria. The results of the study show that fenretinide-induced apoptosis is dependent on Nur77 and more specifically, of its the intracellular localisation [141].
The endogenous pro-apoptotic protein second mitochondria-derived activator caspases (SMAC) is normally localised in mitochondria. Apoptopic stimuli make SMAC translocate to the cytosol, bind inhibitor of apoptosis proteins (IAP) and release caspases. Disturbing this mechanism by immobilization of SMAC in the mitochondria or overexpressing of IAPs is linked to evasion from apoptosis by tumour cells. Strategies to mimic the action of SMAC are called SMAC mimetics. In immunogenic tumour killing, death-inducing agents also instigate a potent anti-tumour immune response [142]. LCL161 is an orally bioavailable SMAC mimetic and inhibitor of IAP family. The endogenous levels of Bcl-2 sensitivity of hepatoma cell lines cells to LCL161. Huh-7 cells have high Bcl-2 levels of expression an d proved resistant to LCL161. Down-regulation of Bcl-2 expression by siRNA or treatment with the inhibitor SC-2001 results in Huh-7 cells overcoming resistance to LCL161 and undergoing apoptosis, and indicates synergistic effects between the SMAC mimetic and teh BCL-2 inhibitor [143]. The results show that these combined therapeutic agents may be useful for future treatments of HCC.
Saponins exhibit cytostatic and cytotoxic activity against various cancer cells. Saponin D (SB365), an active component isolated from Pulsatilla koreana, suppresses the growth of Huh-7 cells and promotes apoptosis through induction of expression of Bax and cleaved caspase-3. This saponin has anti-angiogenic activity by decreasing the expression of HIF-1α and VEGF. The mechanism of action of SB365 is to suppress the phosphorylation of PI3K downstream factors, such as Akt, mTOR and p70S6K both in vitro and in vivo. The results suggest that saponin D may be a candidate for anti-HCC chemotherapy [144].
6.4. Cell Cycle
Aurora kinases (AURK) are key regulators of the cell cycle; their uncontrolled expression promotes aneuploidy and tumour cells often overexpress AURK. The AURK family includes aurora-A, aurora-B, and aurora-C enzymes [145]. Aurora-A kinase expression is detected in human HCC samples, and the levels of aurora-B correlate with the proliferation index of hepatomas. The Huh- 7 cell line expresses aurora-A and -B, and reveals phosphorylation of histone H3 at a locus indicative of auroraB kinase activity. Investigation of the effects of the AURK inhibitor PHA-739358 on Huh-7 cells demonstrated that in vitro the inhibitor suppresses completely cell proliferation and decreases strongly histone H3 phosphorylation. The results show that PHA-739358 suppresses tumour growth by decreasing aurora kinase activiity. Additive effects are observed in combination with sorafenib, and following the failure of sorafenib therapy, treatment with PHA-739358 reduced tumour growth. This investigation suggests that targeting AURK with PHA-739358 is a promising new approach for treatment of HCC, either administered alone or in combination with sorafenib [146].
6.5. Migration and Invasion
Hepatocellular carcinoma has the potential to metastasise to the lungs, lymph nodes, adrenal gland and bones and if such metastases occur, prognosis is poor. Epithelialmesenchymal transition (EMT) is a key development in the ability of HCC to metastasise [147]. The zinc-finger SNAI1 protein can initiate EMT and is involved in the formation and maintenance of embryonic mesoderm. Ecadherin is a calcium-dependent cell-cell adhesion molecule with pivotal roles in epithelial cell behaviour, tissue formation, and suppression of cancer [148]. In invasive tumour cell lines, there is loss of the “invasionsuppressor” E-cadherin and up-regulation of N-cadherin, another adhesion, Eto N-cadherin switching in epithelial carcinomas impacts on their metastatic progression. Hepatocyte growth factor (HGF) induces in Huh-7 cells EMT-like morphologic changes, up-regulates expression of the SNAI1 protein and N-cadherin, and down-regulates E-cadherin. Sorafenib and the MEK inhibitor U0126 inhibit HGF morphologic changes in hepatoma cells, the up-regulation of SNAI1 and cadhering switching. Thus, sorafenib inhibits EMT in these cells, and the study provides insight into the anti-EMT effect of tyrosine kinases in HCC cells [149].
The cell death regulatory protein retinoid-interferoninduced mortality 19 (GRIM-19) has been identified as a tumour suppressor associated with apoptosis and growth inhibition; it is thought to be involved in regulating hepatoma invasiveness, since GRIM-19 levels are significantly attenuated in patients with invasive potential HCC. Using Huh-7 cell lines that had been depleted of GRIM-19, a recent study identified that these cells show EMT-like morphology coupled with loss of contact inhibition. As a result, GRIM-19 is proposed as a new potential target for adjuvant treatment of aggressive and invasive HCC [150].
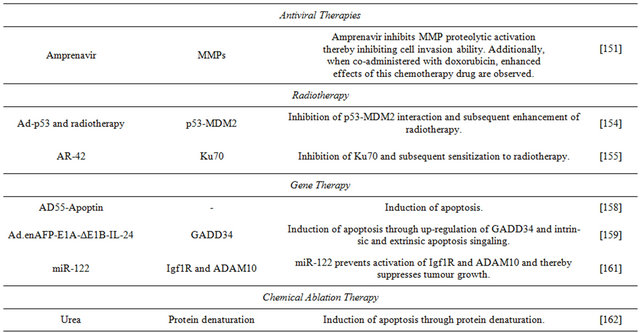
6.6. Antiviral Therapies
Amprenavir is a protease inhibitor used for treatment of patients with HIV. A recent study evaluated its anti-tumour activity on HCC and potential synergistic effects in combination with standard HCC chemoterapeutic drugs, such as DOX. In vitro, Amprenavir has direct inhibitory effects on matrix metalloproteinase proteolytic activation in Huh-7 cells. In vivo, has a synergistic effect with DOX and promotes regression of hepatocarcinoma growth by anti-angiogenesis and overall anti-tumor activities. The novel anti-neoplastic action of Amprenavir on liver cancer shows the possibility of novel combination therapies [151].
6.7. Radiotherapy
Radiotherapy has been used as a treatment for HCC, but less so than other treatments including chemotherapy and liver resection. It is often used in cases in which HCC is untreatable. The minimal use of radiotherapy for HCC therapy is largely due to low whole liver tolerance, as challenges exist in delivering high-dose radiotherapy to the tumour while trying to spare the healthy parts of the liver [152]. HDM2 plays a key role in the effects of radiotherapy, it is an important negative regulator of the p53 tumour suppressor, and the amount of HDM2 expression determines the level of p53 induction by radiation, and decreased levels of this protein sensitise cells to ionising radiation [153]. Since inhibition of HDM2 means increased expression of p53, the regulator has been the focus of recent studies investigating its potential as a target for improving the efficacy of radiotherapy in the treatment of HCC. Work using different HDM2 cellular levels performed adenovirusmediated modified p53 (Ad-p53) gene transfer in Huh-7 cells, and investigated its effectiveness in enhancing radiotherapy cell death, and also whether the p53-HDM2 interaction would be disrupted thereby enhancing the response to radiotherapy. It found that in Huh-7 cells (in both in vitro and in vivo xenografts), gene therapy combined with radiotherapy caused decreased cell viability, making this a future potentially beneficial therapeutic regime for treatment of HCC [154].
In cancer cells, histone deacetylase (HDAC) can be dysregulated and cause tumour growth and repression of tumour suppressor genes. Inhibitors of HDAC target acetylation of the protein Ku70 involved in DNA double strand break repairs and interfere with cellular DNA repair mechanisms. The effects a combination of AR-42, a phenylbutyrate-derived HDAC inhibitor, with radiotherapy were investigated in Huh-7 cells. This study shows that AR-42 causes inhibition of Ku70 activity and sensitised cells to radiotherapy. Thus, AR-42 is another potential agent for future co-administration with radiotherapy to increase its effectiveness against HCC [155].
6.8. Gene Therapy
Gene and viral therapies for cancer have yielded therapeutic effects, but it has been determined that the new strategy Cancer Targeting Gene-Virotherapy (CTGVT) is more effective than either gene or viral therapy alone. This method inserts anti-tumour genes into an oncolytic virus; once inside the tumour cells the virus replicates and causes amplification of the therapeutic gene [156]. The strategy was employed using the double-regulated oncolytic adenovirus AD55 and the cancer therapeutic gene Apoptin to create the recombinant virus AD55- Apoptin. This particular virus was chosen for its ability to induce apoptosis in tumour cells, while preserving normal cells [157]. The combination AD55-Apoptin induced apoptosis in Huh-7 cells, and the effects were greater than those achieved by either AD55 or the oncolytic genetically engineered ONYX-015 virus alone, suggesting its potential as a therapeutic agent for treatment of HCC [158].
Another study using the CTGVT strategy investigated the use of IL-24 inserted into an oncolytic adenovirus to form the construct Ad.enAFP-E1A-ΔE1B-IL-24. The construct has a potent anti-tumour effect on liver cancer cell lines in vitro, but little or no effect on normal cell lines. The mechanism of the anti-tumour effect of Ad. enAFP-E1A-ΔE1B-IL-24 is to up-regulate GADD34, a member of the protein family whose expression is upregulated by growth arrest and DNA damage, and intrinsic and extrinsic apoptotic signaling. In vivo, intratumour injection Ad.enAFP-E1A-ΔE1B-IL-24 resulted in complete elimination of Huh-7 liver cancer in nude mice [159].
MicroRNA-122 (miR-122) is a liver-specific microRNA. It is abundantly expressed in normal hepatocytes but is absent in many hepatocellular carcinoma cells and plays a pivotal role in the liver [160]. Its target is the insulin-like growth factor receptor Igf1R that is involved in cell growth, survival, differentiation and proliferation, tumour metastases and resistance to therapy. The impact miR-122 on tumour growth and progression was examined in Huh-7 cells; anti-miR-122 transfected into the cells increased cell growth, replication, migration and clonogenic survival relative to controls. The study establishes that in Huh-7 cells, Igf1R is negatively regulated by miR-122, as is the distintegrin and metalloprotease family 10 ADAM10 protein that plays a role in tumour survival. These results help to understand the mechanism of anti-tumour activity of miR-122, and will serve in future development of it as a therapeutic agent against HCC [161].
6.9. Chemical Ablation
Chemical ablation methods have been used to treat HCC, and systemic toxicity can result from such treatments. To remedy the currently limited understanding of the mode of action of this therapy, a study was performed in chemical ablation of Huh-7 using urea, which has little systemic toxicity for cells. The work showed induction of cell death in Huh-7 cells as a result of protein denaturation, making such a method a potential for future chemical ablation treatment of solid tumours such as HCC [162].
7. Conclusions
Human hepatoma cell lines have been used extensively to study the biology of HCC, to understand the mechanisms of action of various therapeutic agents and to model the development of acquired resistance to drugs.
An advantage of investigations using in vitro systems is that they can be conducted significantly faster and cheaper than in vivo studies. Cell culture systems offer unique possibilities to perform analyses under wellcontrolled experimental conditions and at a single cell level, thus potentially helping to understand in greater detail the subtleties of cancer formation. An important consideration to keep in mind is that cells in culture may respond differently to various stimuli than cells in the intact liver, for this reason discrepancies between data obtained in vitro and in vivo could be expected, and the biological and clinical relevance of results from in vitro work should be critically evaluated. In addition, the genomic heterogeneity of the patient population is refletecd in tumour-derived cell lines, and their variable response to therapeutic agents.
To establish the genetic basis for drug sensitivity, tissue-specific panels of cell cancer lines have shown remarkable similarity to the primary tumours from which they originated, validating their genetic surrogacy of tumours in vivo. To evaluate combinations of candidate anti-cancer agents, technological advances in cell linebased screening platforms could be employed in a highthroughput fashion to probe the combinatorial space of drugs [163].
Tumour cell 3D cultures such as multilayer cell systems, cells embedded in matrices or hollow fibres, and multicellular tumour spheroids, present opportunities to mimic more closely the environment of cells growing in vivo and uncover characteristics of tumour biology as yet unknown [163].
Notwithstanding the limitations of cell culture studies, this review shows that the specific characteristics of the invasive Huh-7 cell line and its derivatives have been exploited to obtain detailed molecular information of multiple processes in liver malignant tumours, e.g. the biology of neoplastic hepatocytes; dysregulation of pleiotropic growth factors, receptors and their down-stream signaling pathway components; identification of tumour markers; efficacy of anti-cancer drugs and cellular responses; the progression of HCC to a metastatic state, etc. Thus, research with this cell line has made a significant contribution to the knowledge of hepatomas and potential therapies, and future investigations are expected to continue advancing the knowledge of liver cancer.
REFERENCES
- D. M. Parkin, “The Global Health Burden of InfectionAssociated Cancers in the Year 2002,” International Journal of Cancer, Vol. 118, No. 12, 2006, pp. 3030- 3044. doi:10.1002/ijc.21731
- R. Bartenschlager and V. Lohmann, “Replication of Hepatitis C Virus,” Journal of General Virology, Vol. 81, No. 7, 2000, pp. 1631-1648.
- V. Lohmann, F. Korner, J. Koch, U. Herian, L. Theilmann, et al., “Replication of Subgenomic Hepatitis C Virus RNAs in a Hepatoma Cell Line,” Science, Vol. 285, No. 5424, 1999, pp. 110-113. doi:10.1126/science.285.5424.110
- T. Date, T. Kato, M. Miyamoto, Z. Zhao, K. Yasui, et al., “Genotype 2a Hepatitis C Virus Subgenomic Replicon can Replicate in HepG2 and IMY-N9 cells,” Journal of Biological Chemistry, Vol. 279, No. 21, 2004, pp. 22371- 22376. doi:10.1074/jbc.M311120200
- K. S. Chang, Z. Cai, C. Zhang, G. C. Sen, B. R. Williams, et al., “Replication of Hepatitis C Virus (HCV) RNA in Mouse Embryonic Fibroblasts: Protein Kinase R (PKR)- Dependent and PKR-Independent Mechanisms for Controlling HCV RNA Replication and Mediating Interferon Activities,” Journal of Virology, Vol. 80, No. 15, 2006, pp. 7364-7374. doi:10.1128/JVI.00586-06
- T. Kato, T. Date, M. Miyamoto, Z. Zhao, M. Mizokami, et al., “Nonhepatic Cell Lines HeLa and 293 Support Efficient Replication of the Hepatitis C Virus Genotype 2a Subgenomic Replicon,” Journal of Virology, Vol. 79, No. 1, 2005, pp. 592-596. doi:10.1128/JVI.79.1.592-596.2005
- H. Nakabayashi, , K. Taketa, K. Miyano, T. Yamane and J. Sato, “Growth of Human Hepatoma Cells Lines with Differentiated Functions in Chemically Defined Medium,” Cancer Research, Vol. 42, No. 9, 1982, pp. 3858- 3863.
- I. Doi, “Establishment of a Cell Line and Its Clonal Sublines from a Patient with Hepatoblastoma,” Gann, Vol. 67, No. 1, 1976, pp. 1-10.
- J. J. Alexander, E. M. Bey, E. W. Geddes and G. Lecatsas, “Establishment of a Continuously Growing Cell Line from Primary Carcinoma of the Liver,” Sout African Medical Journal, Vol. 50, No. 54, 1976, pp. 2124-2128.
- G. M. MacNab, J. J. Alexander, G. Lecatsas, E. M. Bey and J. M. Urbanowicz, “Hepatitis B Surface Antigen Produced by a Human Hepatoma Cell Line,” British Journal of Cancer, Vol. 34, No. 5, 1976, pp. 509-515. doi:10.1038/bjc.1976.205
- D. P. Aden, A. Fogel, S. Plotkin, I. Damjanov and B. B. Knowles, “Controlled Synthesis of HBsAg in a Differentiated Human Liver Carcinoma-Derived Cell Line,” Nature, Vol. 282, No. 5739, 1979, pp. 615-616. doi:10.1038/282615a0
- N. Huh, and T. Utakoji, “Production of HBs-Antigen by Two New Human Hepatoma Cell Lines and Its Enhancement by Dexamethasone,” Gann, Vol. 72, No. 1, 1981, pp. 178-179.
- H. Nakamura, Y. Izumoto, H. Kambe, T. Kuroda, T. Mori, et al., “Molecular Cloning of Complementary DNA for a Novel Human Hepatoma-Derived Growth Factor. Its Homology with High Mobility Group-1 Protein,” Journal of Biological Chemistry, Vol. 269, No. 40, 1994, pp. 25143-25149.
- M. Triyatni, E. A. Berger and B. Saunier, “A New Model to Produce Infectious Hepatitis C Virus without the Replication Requirement,” PLoS Pathogens, Vol. 7, No. 4, 2011, p. e1001333. doi:10.1371/journal.ppat.1001333
- H. Tang, L. Liu L, F. J. Liu, E. Q. Chen, S. Murakami, et al., “Establishment of Cell Lines Using a DoxycyclineInducible Gene Expression System to Regulate Expression of Hepatitis B Virus X Protein,” Archives of Virology, Vol. 154, No. 7, 2009, pp. 1021-1026. doi:10.1007/s00705-009-0402-0
- B. B. Knowles, C. C. Howe and D. P. Aden, “Human Hepatocellular Carcinoma Cell Lines Secrete the Major Plasma Proteins and Hepatitis B Surface Antigen,” Science, Vol. 209, 1980, pp. 497-499.
- W. K. Kim, Y. J. In, J. H. Kim, H. J. Cho, J. H. Kim, et al., “Quantitative Relationship of Dioxin-Responsive Gene Expression to Dioxin Response Element in Hep3B and HepG2 Human Hepatocarcinoma Cell Lines,” Toxicology Letters, Vol. 165, No. 2, 2006, pp. 174-181. doi:10.1016/j.toxlet.2006.03.007
- C. Jiang, B. Zhou, K. Fan, E. Heung, L. Xue, et al., “A Sequential Treatment of Depsipeptide followed by 5-Azacytidine Enhances Gadd45beta Expression in Hepatocellular Carcinoma Cells,” Anticancer Research, Vol. 27, No. 6B, 2007, pp. 3783-3789.
- J. Lin, L. Schyschka, R. Muhl-Benninghaus, J. Neumann, L. Hao, et al., “Comparative Analysis of Phase I and II Enzyme Activities in 5 Hepatic Cell Lines Identifies Huh-7 and HCC-T Cells with the Highest Potential to Study Drug metabolism,” Archives of Toxicology, Vol. 86, No. 1, 2012, pp. 87-95. doi:10.1007/s00204-011-0733-y
- V. Mersch-Sundermann, S. Knasmuller, X. J. Wu, F. Darroudi and F. Kassie, “Use of a Human-Derived Liver Cell Line for the Detection of Cytoprotective, Antigenotoxic and Cogenotoxic agents,” Toxicology, Vol. 198, No. 1-3, 2004, pp. 329-340. doi:10.1016/j.tox.2004.02.009
- S. Locarnini and F. Zoulim, “Molecular Genetics of HBV Infection,” Antiviral Therapy, Vol. 15, Suppl. 3, 2010, pp. 3-14. doi:10.3851/IMP1619
- M. Bharadwaj, G. Roy, K. Dutta K, M. Misbah, M. Husain M and S. Hussain, “Tackling Hepatitis B VirusAssociated Hepatocellular Carcinoma—The Future is Now,” Cancer and Metastasis Reviews, 2012. doi:10.1007/s10555-012-9412-6
- M. Dandri, M. Lütgehetmann and J. Petersen, “Experimental Models and Therapeutic Approaches for HBV,” Seminars in Immunopathology, Vol. 35, No. 1, 2012, pp. 7-21. doi:10.1007/s00281-012-0335-7
- S. Preiss, A. Thompson, X. Chen, S. Rodgers, V. Markovska, et al., “Characterization of the Innate Immune Signalling Pathways in Hepatocyte Cell Lines,” Journal of Viral Hepatitis, Vol. 15, No. 12, 2008, pp. 888-900. doi:10.1111/j.1365-2893.2008.01001.x
- A. J. Thompson, D. Colledge, S. Rodgers, R. Wilson, P. Revill , et al., “Stimulation of the Interleukin-1 Receptor and Toll-Like Receptor 2 Inhibits Hepatitis B Virus Replication in Hepatoma Cell Lines in Vitro,” Antiviral Therapy, Vol. 14, No. 6, 2009, pp. 797-808.
- T. B. Lentz and D. D. Loeb, “Development of Cell Cultures that Express Hepatitis B Virus to High Levels and Accumulate cccDNA,” Journal of Virological Methods, Vol. 169, No. 1, 2010, pp. 52-60. doi:10.1016/j.jviromet.2010.06.015
- T. B. Lentz and D. D. Loeb, “Roles of the Envelope Proteins in the Amplification of Covalently Closed Circular DNA and Completion of Synthesis of the Plus-Strand DNA in Hepatitis B Virus,” Journal of Virology, Vol. 85, No. 22, 2011, pp. 11916-11927. doi:10.1128/JVI.05373-11
- J. Köck, C. Rösler, J. J. Zhang, H. E. Blum, M. Nassal and C. Thoma, “Generation of Covalently Closed Circular DNA of Hepatitis B Viruses via Intracellular Recycling Is Regulated in a Virus Specific Manner,” PLoS Pathogens, Vol. 6, No. 9, 2010, p. e1001082. doi:10.1371/journal.ppat.1001082
- T. Pollicino, L. Belloni, G. Raffa, N. Pediconi, G. Squadrito, et al., “Hepatitis B Virus Replication Is Regulated by the Acetylation Status of Hepatitis B Virus cccDNABound H3 and H4 Histones,” Gastroenterology, Vol. 130, No. 3, 2006, pp. 823-837.
- S. Locarnini, T. Shaw, J. Dean, D. Colledge, A. Thompson, et al., “Cellular Response to Conditional Expression of the Hepatitis B Virus Precore and Core Proteins in Cultured Hepatoma (Huh-7) cells,” Journal of Clinical Virology, Vol. 32, No. 2, 2005, pp. 113-121.
- X. Zhang, H. Zhang and L. Ye, “Effects of Hepatitis B Virus X Protein on the Development of Liver Cancer,” Journal of Laboratory and Clinical Medicine, Vol. 147, No. 2, 2006, pp. 58-66.
- H. C. Wang, W. Huang, M. D. Lai and I. J. Su, “Hepatitis B Virus Pre-S Mutants, Endoplasmic Reticulum Stress and Hepatocarcinogenesis,” Cancer Science, Vol. 97, No. 8, 2006, pp. 683-688. doi:10.1111/j.1349-7006.2006.00235.x
- H. Yu, R. Zhu, Y. Z. Zhu, Q. Chen and H. G. Zhu, “Effects of Mutations in the X Gene of Hepatitis B Virus on the Virus Replication,” Acta Virologica, Vol. 56, No. 2, 2012, pp. 101-110. doi:10.4149/av_2012_02_101
- A. Le Bon, G. Schiavoni, G. D’Agostino, I. Gresser, F. Belardelli, et al., “Type I Interferons Potently Enhance Humoral Immunity and can Promote Isotype Switching by Stimulating Dendritic Cells in Vivo,” Immunity, Vol. 14, No. 4, 2001, pp. 461-470.
- M. Kumar, S. Y. Jung, A. J. Hodgson, C. R. Madden, J. Qin and B. L. Slagle, “Hepatitis B Virus Regulatory HBx Protein Binds to Adaptor Protein IPS-1 and Inhibits the Activation of Beta-Interferon,” Journal of Virology, Vol. 85, No. 2, 2011, pp. 987-995. doi:10.1128/JVI.01825-10
- B. Y. Jiao, W. S. Lin, F. F. She, W. N. Chen and X. Lin, “Hepatitis B Virus X Protein Enhances Activation of Nuclear Factor κB through Interaction with Valosin-Containing Protein,” Archives of Virology, Vol. 156, No. 11, 2011, pp. 2015-2021. doi:10.1007/s00705-011-1099-4
- R. Tang, F. Kong, L. Hu, H. You, P. Zhang, et al., “Role of Hepatitis B Virus X Protein in Regulating LIM and SH3 Protein 1 (LASP-1) Expression to Mediate Proliferation and Migration of Hepatoma Cells,” Virology Journal, Vol. 16, No. 9, 2012, p. 163. doi:10.1186/1743-422X-9-163
- P. Bellecave, J. Gouttenoire, M. Gajer, V. Brass, G. Koutsoudakis, et al., “Hepatitis B and C Virus Coinfection: A Novel Model System Reveals the Absence of Direct Viral Interference,” Hepatology, Vol. 50, No. 1, 2009, pp. 46-55. doi:10.1002/hep.22951
- P.-Y. Ke and S. S.-L. Chen, “Hepatitis C Virus and Cellular Stress Response: Implications to Molecular Pathogenesis of Liver Diseases,” Viruses, Vol. 4, No. 10, 2012, pp. 2251-2290. doi:10.3390/v4102251
- K. D. Tardif, K. Mori and A. Siddiqui, “Hepatitis C Virus Subgenomic Replicons Induce Endoplasmic Reticulum Stress Activating an Intracellular Signaling Pathway,” Journal Virology, Vol. 76, No. 15, 2002, pp. 7453-7459. doi:10.1128/JVI.76.15.7453-7459.2002
- A. von dem Bussche, R. Machida, K. Li, G. Loevinsohn, A. Khander, J. Wang, et al., “Hepatitis C Virus NS2 Protein Triggers Endoplasmic Reticulum Stress and Suppresses its Own Viral leplication,” Journal of Hepatology, Vol. 53, No. 5, 2010, pp. 797-804. doi:10.1016/j.jhep.2010.05.022
- S. Li, L. Ye, X. Yu, B. Xu, K. Li, et al., “Hepatitis C Virus NS4B Induces Unfolded Protein Response and Endoplasmic Reticulum Overload Response-Dependent NF-Kappab Activation” Virology, Vol. 391, No. 2, 2009, pp. 257-264.
- D. Sir, W. L. Chen, J. Choi, T. Wakita, T. S. Yen and J. H. Ou, “Induction of Incomplete Autophagic Response by Hepatitis C Virus via the Unfolded Protein Response,” Hepatology, Vol. 48, No. 4, 2008, pp. 1054-1061. doi:10.1002/hep.22464
- P. Y. Ke and S. S. Chen, “Activation of the Unfolded Protein Response and Autophagy after Hepatitis C Virus Infection Suppresses Innate Antiviral Immunity in Vitro,” Journal of Clinical Investigations, Vol. 121, No. 1, 2011, pp. 37-56. doi:10.1172/JCI41474
- S. Taguwa, H. Kambara, N. Fujita, T. Noda, T. Yoshimori, et al., “Dysfunction of Autophagy Participates in Vacuole Formation and Cell Death in Cells Replicating Hepatitis C Virus,” Journal of Virology, Vol. 85, No. 24, 2011, pp. 13185-13194. doi:10.1128/JVI.06099-11
- V. C. Chu, S. Bhattacharya, A. Nomoto, J. Lin, S. K. Zaidi, et al., “Persistent Expression of Hepatitis C Virus Non-Structural Proteins Leads to Increased Autophagy and Mitochondrial Injury in Human Hepatoma Cells,” PLoS ONE, Vol. 6, 2011, p. 28551. doi:10.1371/journal.pone.0028551
- T. Vescovo, A. Romagnoli, A. B. Perdomo, M. Corazzari, F. Ciccosanti, et al., “Autophagy Protects Cells from HCV-Induced Defects in Lipid Metabolism,” Gastroenterology, Vol. 142, No. 3, 2012, pp. 644-653.
- G. C. Das and F. B. Hollinger, “Molecular Pathways for Glucose Homeostasis, Insulin Signaling and Autophagy in Hepatitis C Virus Induced Insulin Resistance in a Cellular Model,” Virology, Vol. 434, No. 1, 2012, pp. 5-17. doi:10.1016/j.virol.2012.07.003
- P. Zhao, T. Han, J. J. Guo, S. L. Zhu, J. Wang, et al., “HCV NS4B Induces Apoptosis through the Mitochondrial Death Pathway,” Virus Research, Vol. 169, No. 1, 2012, pp. 1-7. doi:10.1016/j.virusres.2012.04.006
- E. A. Prikhod’ko, G. G. Prikhod’ko, R. M. Siegel, P. Thompson, M. E. Major and J. I. Cohen, “The NS3 Protein of Hepatitis C Virus Induces Caspase-8-Mediated Apoptosis Independent of Its Protease or Helicase Activities,” Virology, Vol. 329, No. 1, 2004, pp. 53-67. doi:10.1016/j.virol.2004.08.012
- S. H. Lee, Y. K. Kim, C. S. Kim, S. K. Seol, J. Kim, et al., “E2 of Hepatitis C Virus Inhibits Apoptosis,” The Journal of Immunology, Vol. 175, No. 12, 2005, pp. 8226- 8235.
- M. Tanaka, M. Nagano-Fujii, L. Deng, S. Ishido, et al., “Single-Point Mutations of Hepatitis C Virus NS3 that Impair p53 Interaction and Anti-Apoptotic Activity of NS3,” Biochemical and Biophysical Research Communications, Vol. 340, No. 3, 2006, pp. 792-799. doi:10.1016/j.bbrc.2005.12.076
- H. L. Chiou, Y. S. Hsieh, M. R. Hsieh and T. Y. Chen, “HCV E2 May Induce Apoptosis of Huh-7 Cells via a Mitochondrial-Related Caspase Pathway,” Biochemical and Biophysical Research Communications, Vol. 345, No. 1, 2006, pp. 453-458. doi:10.1016/j.bbrc.2006.04.118
- Y. Nomura-Takigawa, M. Nagano-Fujii, L. Deng, S. Kitazawa, S. Ishido, et al., “Non-Structural Protein 4A of Hepatitis C Virus Accumulates on Mitochondria and Renders the Cells Prone to Undergoing MitochondriaMediated Apoptosis,” Journal of General Virology, Vol. 87, No. 7, 2006, pp. 1935-1945. doi:10.1099/vir.0.81701-0
- H. Zhu, H Dong, E. Eksioglu, A. Hemming, M. Cao, et al., “Hepatitis C Virus Triggers Apoptosis of a Newly Developed Hepatoma Cell Line through Antiviral Defense System,” Gastroenterology, Vol. 133, No. 5, 2007, pp. 1649-1659. doi:10.1053/j.gastro.2007.09.017
- K. H. Lan, M. L. Sheu, S. J. Hwang, S. H. Yen, S. Y. Chen, et al., “HCV NS5A Interacts with P53 and Inhibits P53-Mediated Apoptosis,” Oncogene, Vol. 21, No. 31, 2002, pp. 4801-4811. doi:10.1038/sj.onc.1205589
- D. G. Johnson and C. L. Walker, “Cyclins and Cell Cycle Checkpoints,” Annual Review of Pharmacology and Toxicology, Vol. 39, No. 1, 1999, pp. 39:295-312.
- V. Sanchez and D. H. Spector, “Subversion of Cell Cycle Regulatory Pathways,” Current Topics in Microbiology and Immunology, Vol. 325, 2008, pp. 243-262.
- M. Hassan, H. Ghozlan and O. Abdel-Kader, “Activation of RB/E2F Signaling Pathway Is Required for the Modulation of Hepatitis C Virus Core Protein-Induced Cell Growth in Liver and Non-Liver Cells,” Cellular Signalling, Vol. 16, No. 12, 2004, pp. 1375-1385. doi:10.1016/j.cellsig.2004.04.005
- [61] T. Munakata, Y. Liang, S. Kim, D. R. McGivern, J. Huibregtse, et al., “Hepatitis C Virus Induces E6AP-Dependent Degradation of the Retinoblastoma Protein,” PLoS Pathogens, Vol. 3, No. 9, 2007, pp. 1335-1347. doi:10.1371/journal.ppat.0030139
- [62] D. R. McGivern, R. A. Villanueva, S. Chinnaswamy, C. C. Kao and S. M. Lemon, “Impaired Replication of Hepatitis C Virus Containing Mutations in a Conserved NS5B Retinoblastoma Protein-Binding Motif,” Journal of Virology, Vol. 83, No. 15, 2009, pp. 7422-7433. doi:10.1128/JVI.00262-09
- [63] K. Machida, J. C. Liu, G. McNamara, A. Levine, L. Duan and M. M. Lai, “Hepatitis C Virus Causes Uncoupling of Mitotic Checkpoint and Chromosomal Polyploidy through the Rb Pathway,” Journal of Virology, Vol. 83, No. 23, 2009, pp. 12590-12600. doi:10.1128/JVI.02643-08
- [64] R. P. Kannan, L. L. Hensley, L. E. Evers, S. M. Lemon and D. R. McGivern, “Hepatitis C Virus Infection Causes Cell Cycle Arrest at the Level of Initiation of Mitosis,” Journal of Virology, Vol. 85, No. 16, 2011, pp. 7989-8001. doi:10.1128/JVI.00280-11
- [65] K. Machida, G. McNamara, K. T. Cheng, J. Huang, C. H. Wang, et al., “Hepatitis C Virus Inhibits DNA Damage Repair Through Reactive Oxygen and Nitrogen Species and by Interfering with the ATM-NBS1/-Mre11/ Rad50 DNA Repair Pathway in Monocytes and Hepatocytes,” The Journal of Immunology, Vol. 185, No. 11, 2010, pp. 6985-6998. doi:10.4049/jimmunol.1000618
- [66] C. K. Lai, K. S. Jeng, K. Machida, Y. S. Cheng and M. M. Lai, “Hepatitis C Virus NS3/4A Protein Interacts with ATM, Impairs DNA Repair and Enhances Sensitivity to Ionizing Radiation,” Virology, Vol. 370, No. 2, 2008, pp. 295-309.
- [67] IARC, “Agents Classified by the IARC Monographs,” 2011. http://monographs.iarc.fr/ENG/Classification/ClassificationsGroupOrder.pdf
- [68] Q. V. Tu, A. S. Okoli, Z. Kovach and G. L. Mendz, “Hepatocellular Carcinoma: Prevalence and Molecular Pathogenesis of Helicobacter spp,” Future Microbiology, Vol. 4, No. 10, 2009, pp. 1283-1301. doi:10.2217/fmb.09.90
- [69] P. Avenaud, B. Le Bail, K. Mayo, A. Marais, R. Fawaz, et al., “Natural History of Helicobacter hepaticus Infection in Conventional A/J mice, with Special Reference to Liver Involvement,” Infection and Immunity, Vol. 71, No. 6, 2003, pp. 3667-3672. doi:10.1128/IAI.71.6.3667-3672.2003
- [70] E. Le Roux-Goglin, C. Varon, P. Spuul, C. Asencio, F. Megraud, et al., “Helicobacter Infection Induces Podosome Assembly in Primary Hepatocytes in Vitro,” European Journal of Cell Biology, Vol. 91, No. 3, 2012, pp. 161-170. doi:10.1016/j.ejcb.2011.11.003
- [71] S. Linder and M. Aepfelbacher, “Podosomes: Adhesion Hot-Spots of Invasive Cells,” Trends in Cell Biology, Vol. 13, No. 7, 2003, pp. 376-385. doi:10.1016/S0962-8924(03)00128-4
- [72] W. B. VanWinkle, M. Snuggs and L. M. Buja, “Hypoxiainduced Alterations in Cytoskeleton Coincide with Collagenase Expression in Cultured Neonatal Rat Cardiomyocytes,” Journal of Molecular and Cellular Cardiology, Vol. 27, No. 12, 1995, pp. 2531-2542. doi:10.1006/jmcc.1995.0040
- [73] A. S. Okoli, M. J. Raftery and G. L. Mendz, “Comparison of Helicobacter bilis—Associated Protein Expression in Huh7 Cells Harbouring HCV Replicon and in RepliconCured Cells,” International Journal of Hepatology, Vol. 12, 2012, pp. 501-671.
- [74] A. S. Okoli, “Molecular Studies of the Response of Helicobacter hepaticus to Bile, and the Effect of Helicobacter bilis on Human Hepatoma Cells,” PhD Thesis, University of New South Wales, Sydney, 2009.
- [75] Y. Nishimura-Sakurai, N. Sakamoto, K. Mogushi, S. Nagaie, M. Nakagawa, et al., “Comparison of HCV-associated Gene Expression and Cell Signaling Pathways in Cells with or without HCV Replicon and in RepliconCured Cells,” Journal of Gastroenterology, Vol. 45, No. 5, 2010, pp. 523-536.
- [76] E. Genoux, E. Nicolle and A. Boumendjel, “Flavonoids as Anticancer Agents: Recent Progress and State of the Art?” Current Organic Chemistry, Vol. 15, No. 15, 2011, pp. 2608-2615.
- [77] B. R. Kim, Y. K. Jeon and M. J. Nam, “A Mechanism of Apigenin-Induced Apoptosis is Potentially Related to Anti-Angiogenesis and Anti-Migration in Human Hepatocellular Carcinoma Cells,” Food and Chemical Toxicology, Vol. 49, No. 7, 2011, pp. 1626-1632. doi:10.1016/j.fct.2011.04.015
- [78] H. Moinova, “Aberrant Vimentin Methylation Is Characteristic of Upper GI Pathologies,” Cancer Epidemiology Biomarkers & Prevention, Vol. 21, No. 4, 2012, pp. 594- 600. doi:10.1158/1055-9965.EPI-11-1060
- [79] W. Jeon, Y. K. Heon and M. J. Nam, “Apoptosis by Aloe-Emodin Is Mediated through Down-Regulation of Calpain-2 and Ubiquitin-Protein Ligase E3A in Human Hepatoma Huh-7 Cells,” Cell Biology International, Vol. 36, No. 2, 2012, pp. 163-167. doi:10.1042/CBI20100723
- [80] D. R. Yoo, Y. H. Jang, Y. K. Jeon, J. Y. Kim, W, Jeon, et al., “Proteomic Identification of Anti-Cancer Proteins in Luteolin-Treated Human Hepatoma Huh-7 Cells,” Cancer Letters, Vol. 282, No. 1, 2009, pp. 48-54. doi:10.1016/j.canlet.2009.02.051
- [81] J. Y. Kim, Y. K. Jeon, W. Jeon and M. J. Nam, “Fisetin Induces Apoptosis in Huh-7 Cells via Downregulation of BIRC8 and Bcl2L2,” Food and Chemical Toxicology, Vol. 48, No. 8-9, 2010, pp. 2259-2264. doi:10.1016/j.fct.2010.05.058
- [82] T. A. Mansoor, R. M. Ramalho, X. Luo, C. Ramalhete, C. M. P. Rodrigues and M. J. U. Ferreira, “Isoflavones as Apoptosis Inducers in Human Hepatoma HuH-7 Cells,” Physiotherapy Research, Vol. 25, No. 12, 2011, pp. 1819- 1824. doi:10.1002/ptr.3498
- [83] S. Hong, K. H. Jung, H. Lee, M. Choi, H. Zheng, M. K. Son, G. Lee and S. Hong, “Apoptotic and Anti-Angiogenic Effects of Pulsatilla koreana Extract on Hepatocellular Carcinoma,” Internal Journal of Oncology, Vol. 40, No. 2, 2011, pp. 452-460. doi:10.3892/ijo.2011.1204
- [84] C-C. Lee, Y-H. Lin, W-H. Chang, P-C. Lin, Y-C. Wu and J-G. Chang, “Squamocin Modulates Histone H3 Phosphorylation Levels and Induces G1 Phase Arrest and Apoptosis in Cancer Cells,” BMC Cancer, Vol. 11, No. 58, 2011. doi:10.1186/1471-2407-11-58
- [85] P. C. Liao, L. T. Ng, L. T. Lin, C. D. Richardson, G. H. Wang and C. C. Lin, “Resveratrol Arrests Cell Cycle and Induces Apoptosis in Human Hepatocellular Carcinoma Huh-7 Cells,” Journal of Medicinal Food, Vol. 13, No. 6, 2010, pp. 1415-1423. doi:10.1089/jmf.2010.1126
- [86] T. A. Mansoor, R. M. Ramalho, S. Mulhovo, C. M. Rodrigues and M. J. Ferreira, “Induction of Apoptosis in Huh-7 Cancer Cells by Monoterpene and Beta-Carboline Indole Alkaloids Isolated from the Leaves of Tabernaemontana elegans,” Bioorganic & Medicinal Chemistry Letters, Vol. 19, No. 15, 2009, pp. 4255-4258. doi:10.1016/j.bmcl.2009.05.104
- [87] Q. F. Wang, J. C. Chen, S. J. Hsieh, C. C. Cheng and S. L. Hsiu, “Regulation of Bcl-2 family molecules and Activation of Caspase Cascade Involved in GypenosidesInduced Apoptosis in Human Hepatoma Cells,” Cancer Letters, Vol. 183, No. 2, 2002, pp. 169-178. doi:10.1016/S0304-3835(01)00828-X
- [88] Q. F. Wang, C. W. Chiang, C. C. Wu, C. C. Cheng, S. J. Hsieh, et al., “Gypenosides Induce Apoptosis in Human Hepatoma Huh-7 Cells through a Calcium/Reactive Oxygen Species-Dependent Mitochondrial Pathway,” Planta Medica, Vol. 73, No. 6, 2007, pp. 535-544. doi:10.1055/s-2007-967200
- [89] J. Y. Lee, K. H. Jung, M. J. Morgan, Y. R. Kang and H. S. Lee, et al., “Sensitization of TRAIL-Induced Cell Death by 20S-Ginsenoside Rg3 via CHOP-Mediated DR5 UpRegulation in Human Hepatocellular Carcinoma Cells,” Molecular Cancer Therapeutics, 10 October 2012.
- [90] S. Baig and M. Alamgir, “Cell Death Induced by Morarah and Khaltita in Hepatoma Cancer Cells (Huh-7),” Journal of the College of Physicians and Surgeons Pakistan, Vol. 19, No. 10, 2009, pp. 644-648.
- [91] J. Hou, D. Wang, R. Zhang and H. Wang, “Experimental Therapy of Hepatoma with Artemisinin and its Derivatives: In Vitro and in Vivo Activity, Chemosensitization, and Mechanisms of Action,” Clinical Cancer Research, Vol. 14, No. 17, 2007, pp. 5519-5530.
- [92] D. Davar, J. H. Beumer, L. Hamieh and H. Tawbi, “Role of PARP Inhibitors in Cancer Biology and Therapy,” Current Medicinal Chemistry, Vol. 19, No. 23, 2012, pp. 3907-3921. doi:10.2174/092986712802002464
- [93] M. H. Shu, T. C. Ko and G. C. Yen, “Oleanoic Acid and Ursolic Acid Induce Apoptosis in Huh-7 Human Hepatocellular Carcinoma Cells through a Mitochondrial-Dependent Pathway and Down-Regulation of XIAP,” Journal of Agricultural and Food Chemistry, Vol. 58, No. 10, 2010, pp. 6110-6118. doi:10.1021/jf100574j
- [94] Y. Shidoji and H. Ogawa. “Natural Occurrence of CancerPreventive Geranyl-Geranoic Acid in Medicinal Herbs,” Journal of Lipid Research, Vol. 45, No. 6, 2004, pp. 1092-1103. doi:10.1194/jlr.M300502-JLR200
- [95] K. Okamoto, Y. Sakimoto, K. Imai, H. Senoo and Y. Shidoji, “Induction of an Incomplete Autophagic Response by Cancer-Preventive Geranlygeranoic Acid in a Human Hepatoma-Derived Cell Line,” Biochemical Journal, Vol. 440, No. 1, 2011, pp. 63-71. doi:10.1042/BJ20110610
- [96] S. Shimonishi, T. Muraguchi, M. Mitake, C. Sakane, K. Okamoto and Y. Shidoji, “Rapid Downregulation of Cyclin D1 Induced by Geranylgeranoic Acid in Human Hepatoma Cells,” Nutrition and Cancer, Vol. 64, No. 3, 2012, pp. 473-480.
- [97] F. Yamaguchi, M. Takata, K. Kamitori, M. Nonaka, Y. Dong, et al., “Rare Sugar D-Allose Induces Specific UpRegulation of TXNIP and Subsequent G1 Cell Cycle Arrest in Hepatocellular Carcinoma Cells by Stabilization of P27kip1,” International Journal of Oncology, Vol. 32, No. 2, 2008, pp. 377-385.
- [98] T. Wu, “Cyclooxygenase-2 in Hepatocellular Carcinoma,” Cancer Treatment Reviews, Vol. 32, No. 1, 2006, pp. 28- 44. doi:10.1016/j.ctrv.2005.10.004
- [99] S. C. Larsoo, M. Kumlin, M. Ingelman-Sundberg and A. Wolk, “Dietary Long-chain n-3 Fatty Acids for the Prevention of Cancer: A Review of Potential Mechanisms,” American Journal of Clinical Nutrition, Vol. 79, No. 6, 2004, pp. 935-945.
- [100] K. Lim, C. Han, Y. Dai, M. Shen and T. Wu, “Omega-3 Polyunsaturated Fatty Acids Inhibit Hepatocellular Carcinoma Cell Growth through Blocking Beta-Catenin and Cyclooxygenase-2,” Molecular Cancer Therapeutics, Vol. 8, No. 11, 2009, pp. 3046-3055. doi:10.1158/1535-7163.MCT-09-0551
- [101] T. Yamazaki and T. Tokiwa, “Isofraxidin, a Coumarin Component from Acanthopanax senticosus, Inhibits Matrix Metalloproteinase-7 Expression and Cell Invasion of Human Hepatoma Cells,” Biological and Pharmaceutical Bulletin, Vol. 33, No. 10, 2010, pp. 1716-1722.
- [102] F. Rodier and J Campisi, “Four Faces of Cellular Senescence,” The Journal of Cell Biology, Vol. 192, No. 4, 2011, pp. 547-556. doi:10.1083/jcb.201009094
- [103] G. S. Oh, H. O. Pae, H. Oh, S. G Hong, I. K. Kim, K. Y. Chai, Y. G. Yun, T. O. Kwon and H. T. Chung. “In vitro Anti-proliferative Effect of 1,2,3,4,6-Penta-O-galloylbeta-D-glucose on Human Hepatocellular Carcinoma Cell Line, SK-HEP-1 Cells,” Cancer Letters, Vol. 174, No. 1, 2001, pp. 17-24. doi:10.1016/S0304-3835(01)00680-2
- [104] J. E. Huh, E. O. Lee, M. S. Kim, K. S. Kang, C. H. Kim, et al., “Penta-O-galloyl-b-D-glucose Suppresses Tumor Growth via Inhibition of Angiogenesis and Stimulation of Apoptosis: Roles of Cyclooxygenase-2 and Mitogen-Activated Protein Kinase Pathways,” Carcinogenesis, Vol. 26, No. 8, 2005, pp. 1436-1445. doi:10.1093/carcin/bgi097
- [105] S. Yin, Y. Dong, J. Li, J. Lü and H Hu. “Penta-1,2,3,4,6- O-galloyl-beta-D-glucose Induces Senescence-Like Terminal S-Phase Arrest in Human Hepatoma and Breast Cancer Cells,” Molecular Carcinogenesis, Vol. 50, No. 8, 2011, pp. 592-600. doi:10.1002/mc.20743
- [106] A. X. Zhu, “Systemic Therapy of Advanced Hepatocellular Carcinoma: How Hopeful Should We Be?” The Oncologist, Vol. 11, No. 7, 2006, pp. 790-800. doi:10.1634/theoncologist.11-7-790
- [107] M. B. Thomas, “Systemic Therapy for Hepatocellular Carcinoma,” Cancer Journal, Vol. 14, No. 2, 2008, pp. 123-127. doi:10.1097/PPO.0b013e31816a6058
- [108] F. van Zijl, G. Zulehner, M. Petz, D. Schneller and D. C. Kornauth, et al., “Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma,” Future Oncology, Vol. 5, No. 8, 2009, pp. 1169-1179. doi:10.2217/fon.09.91
- [109] Q. D. Hu, W. Chen, T. L Yan, T. Ma, C. L. Chen, et al., “NSC 74859 Enhances Doxorubicin Cytotoxicity via Inhibition of Epithelial-Mesenchymal Transition in Hepatocellular Carcinoma Cells,” Cancer Letters, Vol. 325, No. 2, 2012, pp. 207-213. doi:10.1016/j.canlet.2012.07.003
- [110] T. Reya, S. J. Morrison, M. F. Clarke and I. L. Weissman, “Stem Cells, Cancer, and Cancer Stem Cells,” Nature, Vol. 414, No. 1, 2001, pp. 105-111. doi:10.1038/35102167
- [111] S. J. Myung, J. H. Yoon and S. J Yu, “STAT3 & Cytochrome P450 2C9: A Novel Signaling Pathway in Liver Cancer Stem Cells,” Biomedicine & Pharmacotherapy, Vol. 66, No. 8, 2012, pp. 612-616. doi:10.1016/j.biopha.2012.08.011
- [112] T. Zheng, J. Wang, X. Song, X. Meng, S. Pan, H. Jiang and L. Liu, “Nutlin-3 Cooperates with Doxorubicin to Induce Apoptosis of Human Hepatocellular Carcinoma Cells through P53 or P73 Signaling Pathways,” Journal of Cancer Research and Clinical Oncology, Vol. 136, No. 10, 2010, pp. 1597-1604. doi:10.1007/s00432-010-0817-8
- [113] Y. Kawano, M. Nagata, T. Kohno, A. Ichimiya, T. Iwakiri, M. Okumura and K. Arimori, “Caffeine Increases the Antitumor Effect of Cisplatin in Human Hepatocellular Carcinoma Cells,” Biological and Pharmaceutical Bulletin, Vol. 35, No. 3, 2012, pp. 400-407. doi:10.1248/bpb.35.400
- [114] S. Hyuga, M. Shiraishi, A. Hori, M. Hyuga and T. Hanawa, “Effects of Kampo Medicines on MDR-1-Mediated Multidrug Resistance in Human Hepatocellular Carcinoma Huh-7/PTX Cells,” Biological and Pharmaceutical Bulletin, Vol. 35, No. 10, 2012, pp. 1729-1739. doi:10.1248/bpb.b12-00371
- [115] P. Liu, H. Yu, Y. Sun, M. Zhu and Y. Duan, “A mPEGPLGA-b-PLL Copolymer Carrier for Adriamycin and siRNA delivery,” Biomaterials, Vol. 33, No. 17, 2012, pp. 4403-4412. doi:10.1016/j.biomaterials.2012.02.041
- [116] X. Zhang, S. Guo, R. Fan, M. Yu, F. Li, et al., “DualFunctional Liposome for Tumor Targeting and Overcoming Multidrug Resistance in Hepatocellular Carcinoma Cells,” Biomaterials, Vol. 33, No. 29, 2012, pp. 7103-7114. doi:10.1016/j.biomaterials.2012.06.048
- [117] J. Guegan, F. Ezan, N. Theret, S. Langouet and G. Baffet, “MAPK Signaling in Cisplatin-Induced Death: Predominant Role of ERK1 over ERK2 in Human Hepatocellular Carcinoma Cells,” Carcinogenesis, Vol. 34, No. 1, 2013, pp. 38-47.
- [118] Y. L. Lee, Y. J. Lee, S. J. Ahn, T. H. Choi, B. S. Moon, et al., “Combined Radionuclide-Chemotherapy and in Vivo Imaging of Hepatocellular Carcinoma Cells after Transfection of a Triple-Gene Contruct, NIS, HSV1-sr39tk, and EGFP,” Cancer Letters, Vol. 290, No. 1, 2010, pp. 129- 138. doi:10.1016/j.canlet.2009.09.004
- [119] N. Mendez-Sanchez, F. Vasquez-Fernandez, D. ZamoraValdes and M. Uribe, “Sorafenib, a Systemic Therapy for Hepatocellular Carcinoma,” Annals of Hepatology, Vol. 7, No. 1, 2008, pp. 46-51.
- [120] M. B. Thomas and J. L. Abbruzzese, “Opportunities for Targeted Therapies in Hepatocellular Carcinoma,” Journal of Clinical Oncology, Vol. 23, No. 31, 2005, pp. 8093- 8108. doi:10.1200/JCO.2004.00.1537
- [121] S. Chan and W. Yeo, “Targeted Therapy of Hepatocellular Carcinoma: Present and Future,” Journal of Gastroenterology and Hepatology, Vol. 27, No. 5, 2012, pp. 862- 872. doi:10.1111/j.1440-1746.2012.07096.x
- [122] NCI, “Drug Directory 2012,” National Cancer Institute, December 2012. http://www.cancer.gov/drugdictionary?cdrid=299013
- [123] C. Frenette and R. Gish, “Targeted Systemic Therapies for Hepatocellular Carcinoma: Clinical Perspectives, Challenges and Implications,” World Journal of Gastroenterology, Vol. 18, No. 6. 2012, pp. 498-506. doi:10.3748/wjg.v18.i6.498
- [124] W. T. Tai, A. L. Cheng, C. W. Shiau, H. P. Huang, J. W. Huang, et al., “Signal Transducer and Activator of Transcription 3 Is a Major Kinase-Independent Target of Sorafenib in Hepatocellular Carcinoma,” Journal of Hepatology, Vol. 55, No. 5, 2011, pp. 1041-1048. doi:10.1016/j.jhep.2011.01.047
- [125] K. F. Chen, C. W. Shiau, C. Y. Liu, P. Y. Chu, W. T. Tai, et al., “Sorafenib and Its Derivative SC-49 Sensitize Hepatocellular Carcinoma Cells to CS-1008, a Humanized Anti-DR5 Antibody,” British Journal of Pharmacology, Vol. 168, No. 3, 2013, pp. 658-672. doi:10.1111/j.1476-5381.2012.02212.x
- [126] D. L. Ou, Y. C. Shen, J. D. Liang, J. Y. Liou, S. L. Yu, et al., “Induction of Bim Expression Contributes to the Antitumor Synergy between Sorafenib and MitogenActivated Protein Kinase/Extracellular Signal-Regulated Kinase Kinase Inhibitor CI-1040 in Hepatocellular Carcinoma,” Clinical Cancer Research, Vol. 15, No. 18, 2009, pp. 5820-5828. doi:10.1158/1078-0432.CCR-08-3294
- [127] R. W. Johnstone, A. J. Frew and M. J. Smyth, “The TRAIL Apoptotic Pathway in Cancer Onset, Progression and Therapy,” Nature Reviews Cancer, Vol. 8, No. 10, 2008, pp. 782-798. doi:10.1038/nrc2465
- [128] K. F. Chen, W. T. Tai, T. H. Liu, H. P. Huang, Y. C. Lin, et al., “Sorafenib Overcomes TRAIL Resistance of Hepatocellular Carcinoma Cells through the Inhibition of STAT3,” Clinical Cancer Research, Vol. 16, No. 21, 2010, pp. 5189-5199. doi:10.1158/1078-0432.CCR-09-3389
- [129] W. T. Tai, A. L. Cheng, C. W. Shiau, C. Y. Liu, C. H. Ko, et al., “Dovitinib Induces Apoptosis and Overcomes Sorafenib Resistance in Hepatocellular Carcinoma through SHP-1: Mediated Inhibition of STAT3,” Molecular Cancer Therapeutics, Vol. 11, No. 2, 2012, pp. 452-463. doi:10.1158/1535-7163.MCT-11-0412
- [130] K. F. Chen, H. L. Chen, C. Y. Liu, W. T. Tai, K. Ichikawa, et al., “Dovitinib Sensitizes Hepatocellular Carcinoma Cells to TRAIL and Tigatuzumab, a Novel Anti-DR5 Antibody, through SHP-1-Dependent Inhibition of STAT3,” Biochemical Pharmacology, Vol. 83, No. 6, 2012, pp. 769- 777. doi:10.1016/j.bcp.2011.12.035
- [131] B. Fleischer, H. Schulze-Bergkamen, M. Schuchmann, A. Weber, S. Biesterfeld, et al., “Mcl-1 Is an AntiApoptotic Factor for Human Hepatocellular Carcinoma,” International Journal of Oncology, Vol. 28, No. 1, 2006, pp. 25-32.
- [132] K. F. Chen, J. C. Su, C. Y. Liu, J. W. Huang, K. C. Chen, W. L. Chen, W. T. Tai and C. W. Shiau, “A Novel Obatoclax Derivative, SC-2001, Induces Apoptosis in Hepatocellular Carcinoma Cells through SHP-1-Dependent STAT3 Inactivation,” Cancer Letters, Vol. 321, No. 1, 2012, pp. 27-35. doi:10.1016/j.canlet.2012.03.023
- [133] J. H. Lee, H. Lee, S. M. Yun, K. H. Jung, Y. Jeong, et al., “IPD-196, a Novel Phosphatidylinositol 3-Kinase Inhibitor with Potent Anticancer Activity against Hepatocellular Carcinoma,” Cancer Letters, Vol. 322, No. 1, 2013. pp. 99-108.
- [134] K. H. Jung, H. M. Zheng, Y. Jeong, M. J. Choi, H. Lee, S.W. Hong, H. S. Lee, M. K. Son, S. Lee, S. Hong and S. S. Hong, “Suppression of Tumor Proliferation and Angiogenesis of Hepatocellular Carcinoma by HS-104, a Novel Phosphoinositide 3-Kinase Inhibitor,” Cancer Letters, Vol. 328, No. 1, 2013, pp. 176-187. doi:10.1016/j.canlet.2012.08.005
- [135] K. H. Jung, M. J. Choi, S. Hong, H. Lee, S. W. Hong, H. M. Zheng, H. S. Lee, S. Hong and S. S. Hong, “HS-116, a Novel Phosphatidylinositol 3-Kinase Inhibitor Induces Apoptosis and Suppresses Angiogenesis of Hepatocellular Carcinoma Through Inhibition of the PI3K/AKT/mTOR Pathway,” Cancer Letters, Vol. 316, No. 2, 2011, pp. 187- 195. doi:10.1016/j.canlet.2011.10.037
- [136] K. F. Chen, P. Y. Yeh, K. H. Yeh, Y. S. Lu, S. Y. Huang and A. L. Cheng, “Down-Regulation of Phospho-Akt Is a Major Molecular Determinant of Bortezomib-Induced Apoptosis in Hepatocellular Carcinoma Cells,” Cancer Research, Vol. 68, No. 16, 2008, pp. 6698-6707. doi:10.1158/0008-5472.CAN-08-0257
- [137] K. F. Chen, P. Y. Yeh, C. Hsu, C. H. Hsu, Y. S. Lu, et al., “Bortezomib Overcomes Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand Resistance in Hepatocellular Carcinoma Cells in Part through the Inhibition of the Phosphatidylinositol 3-Kinase/Akt Pathway,” Journal of Biological Chemistry, Vol. 284, No. 17, 2009, pp. 11121- 11133. doi:10.1074/jbc.M806268200
- [138] K. F. Chen, H. C. Yu, T. H. Liu, S. S. Lee, P. J. Chen and A. L. Cheng, “Synergistic Interactions between Sorafenib and Bortezomib in Hepatocellular Carcinoma Involve PP2A-Dependent Akt Inactivation,” Journal of Hepatology, Vol. 52, No. 1, 2009, pp. 88-95. doi:10.1016/j.jhep.2009.10.011
- [139] T. R. Peterson, M. Laplante, C. C. Thoreen, Y. Sancak, S. A. Kang, et al., “DEPTOR Is an mTOR Inhibitor Frequently Overexpressed in Multiple Myeloma Cells and Required for Their Survival,” Cell, Vol. 137, No. 5, 2009, pp. 873-886. doi:10.1016/j.cell.2009.03.046
- [140] C. H. Yen, Y. C. Lu, C. H. Li, C. M. Lee, C. Y. Chen, et al., “Functional Characterization of Glycine N-Methyl-Transferase and Its Interactive Protein DEPDC6/-DEPTOR in Hepatocellular Carcinoma,” Molecular Medicine, Vol. 18, No. 1, 2012, pp. 286-296. doi:10.2119/molmed.2011.00331
- [141] M. H. Chien, T. H. Ying, S. F. Yang, J. K. Yu, C. W. Hsu, S. C. Hsieh and Y. H. Hsieh, “Lipocalin-2 Induces Apoptosis in Human Hepatocellular Carcinoma Cells through Activation of Mitochondria Pathways,” Cell Biochemistry and Biophysics, Vol. 63, No. 3, 2012, pp. 177-186. doi:10.1007/s12013-012-9370-1
- [142] H. Yang, N. Bushue, P. Bu and Y. J. Wan, “Induction and Intracellular Localization of Nur77 Dictate FenretinideInduced Apoptosis of Human Liver Cancer Cells,” Biochemical Pharmacology, Vol. 79, No. 7, 2009, pp. 948- 954. doi:10.1016/j.bcp.2009.11.004
- [143] P. U. Emeagi, K. Thielemans and K. Breckpot, “The Role of SMAC Mimetics in Regulation of Tumor Cell Death and Immunity,” OncoImmunology, Vol. 1, No. 6, 2012, pp. 965-967. doi:10.4161/onci.20369
- [144] K. Chen, J. Lin, C. Shiau, W. Tai, C. Liu, H. Yu, P. Chen and A. Cheng, “Inhibition of Bcl-2 Improves Effect of LCL161, a SMAC Mimetic, in Hepatocellular Carcinoma Cells,” Biochemical Pharmacology, Vol. 84, No. 3, 2012, pp. 268-277. doi:10.1016/j.bcp.2012.04.023
- [145] S. W. Hong, K. H. Jung, H. S. Lee, M. J. Choi, M. K. Son, H. M. Zheng and S. S. Hong, “SB365 Inhibits Angiogenesis and Induces Apoptosis of Hepatocellular Carcinoma through Modulation of PI3K/Akt/mTOR Signaling Pathway,” Cancer Science, Vol. 103, No. 11, 2012, pp. 1929-1937. doi:10.1111/j.1349-7006.2012.02409.x
- [146] R. Hrabakova, M. Kollareddy, J. Tyleckova, P. Halada, M. Hajduch, S. J. Gadher and H. Kovarova, “Cancer Cell Resistance to Aurora Kinase Inhibitors: Identification of Novel Targets for Cancer Therapy,” Journal of Proteome Research, Vol. 12, No. 1, 2012, pp. 455-469. doi:10.1021/pr300819m
- [147] D. Benten, G. Keller, A. Quaas, J. Schrader, A. Gontarewicz, S. Balabanov, M. Braig, H. Wege, J. Moll, A. W. Lohse and T. H. Brummendorf, “Aurora Kinase Inhibitor PHA-739358 Suppresses Growth of Hepatocellular Carcinoma in Vitro and in a Xenograft Mouse Model,” Neoplasia, Vol. 11, No. 9, 2009, pp. 934-944.
- [148] S. Kummar and N. Q. Shafi, “Metastatic Hepatocellular Carcinoma,” Clinical Oncology, Vol. 15, No. 5, 2003, pp. 288-294. doi:10.1016/S0936-6555(03)00067-0
- [149] F. van Roy and G. Berx, “The Cell-Cell Adhesion Molecule E-Cadherin,” Cellular and Molecular Life Sciences, Vol. 65, No. 23, 2008, pp. 3756-3788. doi:10.1007/s00018-008-8281-1
- [150] T. Nagai, T. Arao, K. Faruta, K. Sakai, K. Kudo, et al., “Sorafenib Inhibits the Hepatocyte Growth Factor-Mediated Epithelial Mesenchymal Transition in Hepatocellular Carcinoma,” Molecular Cancer Therapeutics, Vol. 10, No. 1, 2011, pp. 169-177. doi:10.1158/1535-7163.MCT-10-0544
- [151] H. Hao, J. Liu, G. Liu, D. Guan, Y. Yang, X. Zhang, X. Cao and Q. Liu, “Depletion of GRIM-19 Accelerates Hepatocellular Carcinoma Invasion via Inducing EMT and Loss of Contact Inhibition,” Journal of Cellular Physiology, Vol. 227, No. 3, 2012, pp. 1212-1219. doi:10.1002/jcp.24025
- [152] V. Esposito, A. Verdina, L. Manente, E. P. Spugnini, R. Vigietti, et al., “Amprenavir Inhibits the Migration in Human Hepatocarcinoma Cell in Vitro and the Growth Xenografts in Vivo,” Journal of Cellular Physiology, Vol. 228, No. 3, 2013, pp. 640-645. doi:10.1002/jcp.24173
- [153] M. A. Hawkins and L. A. Dawson, “Radiation Therapy for Hepatocellular Carcinoma: From Palliation to Cure,” Cancer, Vol. 106, No. 8, 2006, pp. 1653-1663. doi:10.1002/cncr.21811
- [154] M. E. Perry, “Mdm2 in the Response to Radiation,” Molecular Cancer Research, Vol. 2, No. 1, 2004, pp. 9-19.
- [155] W. S. Koom, S. Y. Park, W. Kim, M. Kim, J. S. Kim, H. Kim, I. K. Choi, C. O. Yun and J. Seong, “Combination of Radiotherapy and Adenovirus-Mediated P53 Gene Therapy for MDM2-Overexpressing Hepatocellular Carcinoma,” Journal of Radiation Research, Vol. 53, No. 2, 2012, pp. 202-210.
- [156] Y. S. Lu, C. H. Chou, K. Y. Tzen, M. Gao, A. L. Cheng, S. K. Kulp and J. C. Cheng, “Radiosensitizing Effect of a Phenylbutyrate-Derived Histone Deacetylase Inhibitor in Hepatocellular Carcinoma,” International Journal of Randiation Oncology, Vol. 83, No. 2, 2012, pp. 181-189. doi:10.1016/j.ijrobp.2011.12.022
- [157] X. Y. Liu, “Targeting Gene-Virotherapy of Cancer and Its Prosperity,” Cell Research, Vol. 16, 2006, pp. 879-886. doi:10.1038/sj.cr.7310108
- [158] A. A. A. M. Danen-Van Oorschot, D. F. Fischer, J. M. Grimbergen, B. Klein, S.-M. Zhang, et al., “Apoptin Induces Apoptosis in Human Transformed and Malignant Cells But Not in Normal
 Cells,” Proceedings of the National Academy of Science of the United States of America, Vol. 94, No. 11, 1997, pp. 5843-5847. doi:10.1073/pnas.94.11.5843
Cells,” Proceedings of the National Academy of Science of the United States of America, Vol. 94, No. 11, 1997, pp. 5843-5847. doi:10.1073/pnas.94.11.5843 - [159] K. J. Zhang, J. Qian, S.B. Wang and Y. Yang, “Targeting Gene-Viro-Therapy with AFP Driving Apoptin Gene Shows Potent Antitumor Effect in Hepatocarcinoma,” Journal of Biomedical Science, Vol. 19, 2012, p. 20. doi:10.1186/1423-0127-19-20
- [160] K. J. Zhang, J. Zhang, Y. M. Wu, J. Qian, X. J. Liu, et al., “Complete Eradication of Hepatomas Using an Oncolytic Adenovirus Containing AFP Promoter Controlling E1A and an E1B Deletion to Drive IL-24 Expression,” Cancer Gene Therapy, Vol 19, No. 9, 2012, pp. 619-629. doi:10.1038/cgt.2012.40
- [161] W. C. Tsai, S. D. Hsu, C. S. Hsu, T. C. Lai, S. J. Chen, et al., “MicroRNA-122 Plays a Critical Role in Liver Homeostasis and Hepatocarcinogenesis,” Journal of Clinical Investigations, Vol. 122, No. 8, 2012, pp. 2884-2897. doi:10.1172/JCI63455
- [162] S. Bai, M. W. Nasser, B. Wang, S. H. Hsu, J. Datta, et al., “MicroRNA-122 Inhibits Tumorigenic Properties of Hepatocellular Carcinoma Cells and Sensitizes These Cells to Sorafenib,” The Journal of Biological Chemistry, Vol. 284, No. 46, 2009, pp. 32015-32027. doi:10.1074/jbc.M109.016774
- [163] R. N. Aravalli, J. Choi, S. Mori, D. Mehra, J. Dong, et al., “Spectroscopic and Calorimetric Evaluation of Chemically Induced Protein Denaturation in HuH-7 Liver cancer Cells and Impact on Cell Survival,” Technology in Cancer Research and Treatment, Vol. 11, No. 5, 2012, pp. 409-527. doi:10.7785/tcrt.2012.500271
- [164] S. V. Sharma, D. A. Haber and J. Settleman, “Cell LineBased Platforms to Evaluate the Therapeutic Efficacy of Candidate Anticancer Agents,” Nature Reviews Cancer, Vol. 10, No. 4, 2010, pp. 241-253. doi:10.1038/nrc2820
Abbreviations
AD55: adenovirus 55 ADAM10: ADAM metallopeptidase domain 10 AE: Aloe-emodin AG490: EGFR, ErbB2 and JAK2 inhibitor APG: Apigenin AR-42: Ku70 inhibitor Atg5, 7, 9, 12: autophagy protein 5, 7, 9, 12 AURK: aurora kinases Bax: Bcl-2-associated X protein Bcl-2, -W, -xL: antiapoptotic B-cell lymphoma 2, -W, and -extra large proteins BH3: proapoptotic interacting-domain death agonist protein BIRC8: baculoviral IAP repeat-containing protein 8 CAPN2: calpain-2 cccDNA: covalently closed circular DNA CDK: cyclin-dependent kinase C/EBP: CCAAT-enhancer-binding transcription factor protein CHOP: C/EBP homology protein transcription factor Cip1/p21: CDK inhibitor 1 Cisplatin: cis-diamminedichloroplatinum-II CKI: CDK inhibitors COX: cyclooxygenase COX-2: cyclooxygenase-2 CSC: cancer stem cells CS-1008: humanized agonistic monoclonal antibody directed against human TRAIL receptor 2 CTGVT: cancer targeting gene-virotherapy CYP2C9: cytochrome P450 2C9 DEPTOR: domain-containing mTOR-interacting protein DHA: docosahexaenoic acid DOX: doxorubicin DR5: death receptor 5 4E-BP1: 4E-binding protein 1 E2F: transcription factor involved in the G1/S transition of the cell cycle E2F1: E2F transcription factor 1 EMT: epithelial-mesenchymal transition EPA: eicosapentaenoic acid ER: endoplasmic reticulum ERK, ERK1, ERK2: extracellular signal-regulated kinases, 1, 2 GADD34: growth arrest and DNA damage-inducible protein 34 Gal-P123: polymeric nano-biomaterial GGA: geranylgeranoic acid GNMT: glycine N-methyltransferase GRIM-19: cell death regulatory protein retinoid-interferon-induced mortality 19 H3, H4: histone H3 protein, H4 protein HBeAg: hepatitis B “e” antigen HBs: hepatitis B surface protein HBV: hepatitis B virus HBx: hepatitis B protein X HCC: hepatocellular carcinoma HCV: hepatitis C virus HDAC: histone deacetylase HDM2: human double minute 2 homolog protein HGF: hepatocyte growth factor HIF-1: hypoxia-inducible transcription factor 1 HSV1-sr39tk: Herpes-simplex virus type1 sr39 thymidine kinase IAP: inhibitor of apoptosis protein family IFN, IFN-α: interferon, interferon-alpha Igf1R: insulin-like growth factor receptor IL-8: interleukin 8 Kip1/p27: CDK inhibitor 7 Ku70: DNA double-strand break-end binding protein LBY135: monoclonal antibody agonist of DR5 LCL161: inhibitor of IAP LCN2: lipocalin 2 LPG: linear polyglycerol MAPK: mitogen-activated protein kinase Mcl-1: myeloid cell leukemia sequence 1 MDM2: mouse double minute 2 homolog MDR: multidrug-resistant gene MDR-1: multidrug resistance (MDR) 1 MEK: MAP kinase kinase miR-122: microRNA-122 MMP-7: meta-lloproteinase-7 MMP-8: matrix metalloproteinase-8 MSK1: mitogen-and-stress-activated protein kinase-1 mTOR, mTOR 1, mTOR 2: mammalian target of rapamycin protein, complex 1, complex 2 MX: mitoxantrone NF-κB: nuclear factor-kappaB NIS: sodium iodine symporter NSC 74859: STAT3 inhibitor NS3/4A: HCV serine protease Nur77: nerve growth factor IB OA: oleanolic acid ONYX-015: oncovirus created by genetically engineering an adenovirus PARP: poly(ADP-ribose) polymerase PG: prostaglandins PGG: Penta-1,2,3,4,6-O-galloyl-beta-D-glucose PHA-739358: aurora kinase inhibitor PHB: prohibitin PI3K: phosphatidylinositide 3-kinases PI3K/Akt/ mTOR: phosphatidylinositol 3-kinase/protein kinase B/mammalian target of rapamycin pathway PKE: Pulsatilla koreana extract PRDX6: peroxiredoxin 6 p70S6: p70s6k-p70 S6 kinase p70S6K: 70 kDa ribosomal protein S6 kinase p-STAT3: phospho-Stat3 PTX: paclitaxel
ω-3 PUFA: omega-3 poly-unsaturated fatty acids
ω-6 PUFA: omega-6 polyunsaturated fatty acids p21/WAF1: cyclin-dependent kinase inhibitor 1 RB: retinoblastoma tumour suppressor protein RB-E2F: retinoblastoma tumour suppressor transcription factor pathway rcDNA: relaxed circular deoxyribonucleic acid ROS: reactive oxygen species SB365: saponin D SC-1, -49: sorafenib derivatives SHP-1: Src homology region 2 domain-containing phosphatase-1 SMAC: second mitochondria-derived activator caspases SNAI1: snail homolog 1 STAT3: signal transducer and activator of transcription 3 TLR, TLR2, 3, 8: toll-like receptor, toll-like receptor 2, 3, 8 TNF: tumour necrosis factor TNF-α: tumour necrosis factor-alpha TNFRSF10B: tumour necrosis factor receptor member 10b TRAIL: tumour necrosis factor-related apoptosis-inducing ligand TXNIP: thioredoxin interacting protein UA: ursolic acid UBE3A: ubiquitin-protein ligase E3A UPR: unfolded protein response U0126: MEK inhibitor VEGF: vascular endothelial growth factor XIAP: X-linked apoptotic protein