Journal of Cancer Therapy
Vol.2 No.4(2011), Article ID:7801,12 pages DOI:10.4236/jct.2011.24077
Evaluation of the Antitumor and Radiosynthetizing Activity of a Novel Quinoline Sulfonamide Derivative (PIQSA) as a Histone Deacetylase Inhibitor
![]()
1National Centre for Radiation Research and Technology (NCRRT), Atomic Energy Authority (AEA), Cairo, Egypt; 2Department of Zoology, Faculty of Science, El-Azhar University, Cairo, Egypt.
Email: *emnoaman@yahoo.com
Received December 27th, 2010; revised August 11th, 2011; accepted August 20th, 2011.
Keywords: Antitumor, Radiosynthetization, Quinoline Derivative, Sulfonamide Moiety, HDACI, Apoptosis, Cell Cycle, DNA Fragmentation
ABSTRACT
Inhibition of histone deacetylases (HDACs) is emerging as a new strategy in cancer therapy. In the present work a novel pyrimido-quinoline benzene sulfonamide (PIQSA compound) was designed and synthesized postulating its ability to inhibit HDAC enzyme in cancer cells. This study was designed to examine the in vitro anti-tumor efficacy of PIQSA against Ehrlich Ascite carcinoma cells (EAC) and three of the human cancer cell lines (H460), brain (U251) and liver (HepG2). The results of Cytotoxic assays showed that PIQSA exhibited in vitro antitumor activity in a dose dependant manner. The tumor growth delay studies indicating that PIQSA resulted in significant regression in tumor growth, which was more pronounced when PIQSA treatment accompanied with radiation exposure. Also, the efficacy of PIQSA to influence radiation response in Ehrlich solid carcinoma (ESC) tumors was estimated. The results suggest that PIQSA exhibited antitumor activities and strong radioenhancing properties associated with inhibition of HDAC activity, DNA fragmentation followed by apoptotic cell death, preferential cell loss of cells particularly in G1/G0 phase through an apoptotic pathway.
1. Introduction
The major focus research in chemotherapy for cancer includes the identification, characterization and development of new and safe cancer chemo representativeagents [1]. Quinoline derivatives are important biologically active compounds with antibacterial [2] and anticancer activity [3]. On the other hand, a new set of sulfonamide derivatives of potential antitumor agents were developed and synthesized [4-7] and also as HDACIs [4,8]. Acetylation of histones in chromatin is one mechanism involved in the regulation of gene transcription and is tightly controlled by the balance of acetyltransferase (HATs) and deacetylase (HDAC) activities [9]. HDACs mediate changes in nucleosome conformation and are important in the regulation of gene expression and also are involved in cell cycle progression and differentiation [10]. Alterations of HDACs was identified in tumor cells and contributed to the massive perturbations of gene expression in numerous tumors. HDAC inhibition leads to differentiation, cell cycle arrest and apoptosis in tumor cells [11]. The authors stated that HDAC inhibitors (HDACIs) can restore the expression of the tumor suppressor and/or cell cycle regulatory genes in cancer cells and block the cellular proliferation of these cells. They elucidated that Suberoylanilide hydroxamic acid (SAHA) induced either G1 or G2-M arrest as well as apoptosis. They also concluded that HDACIs also down regulate cyclin D1 and D2, and upregulate p53, and p21 [11]. A remarkable increase in the level of acetylated histone associated with the p21 promoter after HDACIs treatment was demonstrated in nude mice and HDACIs significantly inhibited growth in lymphoma cells without major toxic side effects [12], Consequently, efficiently prevent tumor growth both in vitro [13] and in vivo in a variety of preclinical models [11]. Radiotherapy in combination with surgery and chemotherapy has improved treatment for patients with localized and locally advanced cancers. In addition, radiotherapy can be combined with certain chemotherapeutic agents to increase the radiation sensitivity of cancers and to eliminate occult tumor cells located beyond the effective radiation field. Combined-modality therapy has produced moderate improvements in the therapeutic outcome for several cancers [14]. Many investigators revealed the radiosensitizing abilities of various HDACIs both in vitro and in vivo [15-19].
2. Materials and Methods
2.1. Ehrlich Ascites Carcinoma Cell (Mouse Tumour)
A line of Ehrlich ascites carcinoma (EAC) has been used throughout this work. The parent line was supplied through the courtesy of Dr. G. klien, Amsterdam, Holland. EAC cells is originated from breast cancer which modified to grow in female Swiss albino mice and maintained by intraperitoneal inoculation of carcinoma cells in the mice [20].
2.2. Human Tumour Cell Lines
Brain carcinoma cell line (U251), Lung carcinoma cell line (H460), Liver carcinoma cell line (Hepg2) were the three human carcinoma cell lines used in the current study. They were obtained, frozen in liquid nitrogen (–1800C) from the American Type Culture Collection. The tumour cell lines were maintained by serial subculturing at the National Cancer Institute, Cairo, Egypt.
2.3. Chemicals and Reagents
2.3.1. Chemicals
A novel pyrimido-quinoline compound bearing sulfonamide moiety with molecular formula (C31H29N5O3S2), and chemical structure,4-[4-imino-8,8-dimethyl-6-oxo- 3,5-diphenyl-2-thioxo-1,3,4,5,6,7,8,9-octahydro-2H-pyrimido[4,5-b]quinolin-10-yl]bezenesulfonamide (PIQSA).
Synthesis and analysis identification of the present compound was previously published [6]. RPMI1640: (Roswell park memorial institute) medium with L-glutamine (Cambrex, Belgium), Fatal bovine serum (Sigma, Germany), penicillin and streptomycin (Euroclone, Europe), Dimethyl thiazolyl diphenyl tetrazolium bromide (MTT) dye (American Type Culture Collection (ATCC)), trypan blue & Dimethyl sulfoxide (DMSO) (Sigma, USA).
2.3.2. Gamma-Irradiation Source
Gamma irradiation facility was delivered through NCRRT. Whole body gamma irradiation of animals was performed using Gamma cell 40 (Caesium-137 source). Animals were subjected to three irradiation doses of (2 Gy/dose) at the last week of experimental period. Radiation exposure delivered at a dose rate of 0.85 G/min. All experimental trials have been carried out at the laboratories of NCRRT.
2.4. Methods
The antitumor activity of the novel pyrimido-quinoline benzene sulfonamide (PIQSA) against Ehrlich Asites Carcinoma cells (EAC) was assessed using the trypan blue exclusion test, [20]. The Cytotoxic effect against human cancer cell lines [H460, U251 and Hepg2] cell lines was performed using MTT test [21]. The MTT Cell Proliferation assay is a colorimetric assay system which measures the reduction of a tetrazolium Component (MTT) into an insoluble formazan product by the mitochondria of viable cells. The absorbance was recorded using Elisa reader at 570 nm.
Viable cell% = (A samples – A blank)/(A control – A blank) × 100
3. In-Vivo Study
In the current study, the anti-tumor activity of the novel pyrimido (4,5-b) quinoline sulfonamide compound PIQSA against Ehrlish solid carcinoma ESC tumor in animal models was determined. The mechanisms by which the present compound exerted its anti-cancer effect were also estimated. Beside, the efficacy of the present compound as radiosensitizer induced augmentation of radiationinduced apoptosis in ESC tumor in animal models was also examined.
3.1. Animals
Female Swiss albino mice weighing 20 - 25 g were obtained from the breeding unit at the National Centre for Radiation Research and Technology (NCRRT). The animals were housed (10 animals/cage) and maintained under proper environmental conditions i.e., controlled air, temperature and relative humidity. They were provided with pelleted diet and free access to water. Animal care and handling was done according to the guidelines set by the world health organization, Geneva, Switzerland and approved from the committee for animals care at NCRRT, Atomic Energy Authority (AEA).
3.2. Determination of Acute Toxicity and LD50
Thirty animals were used in this experiment, divided into six groups (five animals per group). Drug was dissolved in DMSO/saline, and injected ip to the six groups of experimental animals (single dose each of 1.6, 1.2, 0.8, 0.6, 0.3, 0.15 mg/kg body weight respectively). Mortality induced due to the toxicity of the drug was noted in each group during the experimental observation period. The median lethal dose (LD50) determined [22].
3.3. Preparation of Tested Compound PIQSA to in Vivo Study
PIQSA compound was freshly prepared by dissolving in DMSO/saline solution and injected ip at a dose of 0.350 mg/kg·b.wt three times/week for three weeks.
3.4. Antitumor Experimental Design
Animals were randomly divided into groups according to the following design: control group, this group left without treatment and served as control. The other groups of animals bearing tumor (80 animals), were injected intramuscular in the thigh of animals with 5 × 106 cells/mouse then left for 10 days until the tumor volume reached to approximately1mm in diameter. Then, they were divided into 4 subgroups each of 20 animals. These groups included, animals bearing tumor left without treatment, animals bearing tumor treated with, ip injection (0.35 mg/kg·b.wt) of PIQSA, three time per weeks for three weeks, animals subjected to Radiation exposure, three doses of (2 Gy/dose) delivered at the last week of experimental, animals exposed to the combined treatment of PIQSA and radiation exposure, each radiation fraction was administered period 20 minutes after PIQSA treatment to maximize growth inhibition.
3.5. Tumor Volume Determination
Animals in each trial were checked daily and checked for any adverse clinical symptoms and deaths were recorded. Through out the entire period of observation, the animals were weighed twice weakly. After 10 days post inoculation with EAC, tumor volume was measured twice weakly using callipers and determined following the published procedure [23].
3.6. Tumor Tissue Preparation
At the end of experiment, Animals were fasted for 16 hrs prior to sampling. As quickly as possible, after animals were sacrificed, the tumors were dissected and a portion of it was used to prepared homogenates in ice-cold phosphate buffer (0.1 M, pH 7.4) using a potter-Elvehjem homogenizer to give a 10% concentration for the determination of DNA fragmentation, and 50% homogenates used for estimation of Histone deacetylase enzyme (HDAC). Portion of tumor was kept in 10% formalin for histochemical examination of apoptosis.
3.7. Histone Deacetylase (HDAC) Activity Assay
HDAC extraction was carried out according to the method previously described [24]. HDAC activity was evaluated using colorimetric Histone deacetylase (HDAC) activity assay Kit of Biovision, the Catalogue (k331-100) [25]. Briefly, 10 μg of nuclear extract was incubated with HDAC assay buffer and HDAC assay substrate buffer at 370c for 1 hour. A total 40 μg of quenching solution was added to stop the activator solution at room temperature for 15 min. Activity was measured using ELISA micro plate reader at 405 nm.
3.8. DNA Fragmentation Assay
A characteristic event of apoptosis is DNA fragmentation and release of nucleosome into the cytoplasm. These can be detected by an ELISA assay [26]. The absorbance was determined using ELISA microplate reader at 600 ml. The percentage of fragmented DNA can be calculated using the formula:
% of DNA fragmented = OD supernatant/(OD pellet + OD supernatant).
3.9. Histochemical Detection of Apoptosis
Apoptosis was quantitated by visualizing distinctive nuclear and cytoplasmic fluorescence, chromatin condensation and formation of multiple apoptotic bodies following staining of cells with acridine orange and ethidium bromide depending on Fluorescence Imaging [27].
3.10. Flow Cytometric Cell Cycle Analysis
Cell cycle analysis was carried out according the method previously described [28]. Cells were trypsinized, washed with PBS, and fixed in 75% ethanol at 4˚C for 30 min. Prior analyses, suspended in cold propidium iodide (PI) solution, and incubated at room temperature in the dark for 30 min. Flow cytometry analyses were performed using a MoFlo flow cytometer (MoFlo, Dako Cytomation).
4. Statistical Analysis
The results were expressed as the mean value ± standard deviation (SD).Statistical analysis was performed using analysis of variance (ANOVA) for testing the significance between treated groups [29].
5. Results
5.1. Cytotoxic Effect of PIQSA on ESC Cancer Cells and Different Human Cell Lines
The tested compound PIQSA exhibited very cytotoxic effect on the cell viability in a dose dependant manner, the results also, pointed to that its IC50 is 0.065 mM/ml (Table 1).The Cytotoxic effect of PIQSA on different human cancer cell lines illustrated in (Table 2), showed a Cytotoxic effect on lung cancer cells (H460), brain cancer cells (U250), and also on liver carcinoma cells (HepG2) with IC50 = 50 μM/ml, 55 μM/ml, and 50 μM/ml respectively.
5.2. The in Vivo Cytotoxic Effect of PIQSA
The in vivo acute cytotoxic effect of the PIQSA compound was performed and the LD50 found to be 0.670 mg/kg body weight.
Treatment with PIQSA at dose of 0.350 mg/kg resulted in significant delay in tumor growth with (p < 0.01) for tumor weight and (p < 0.05) for tumor size. Three doses of γ-IR (2Gy/dose) induced highly significant delay in tumor growth with (p < 0.001) for tumor weight and tumor volume. The results also revealed significantly

Table 1. Effect of different concentrations of PIQSA on the viability of EAC cells in vitro by trypan blue exclusion test indicating the effect of different doses in μM/ml and the percentage of nonviable.
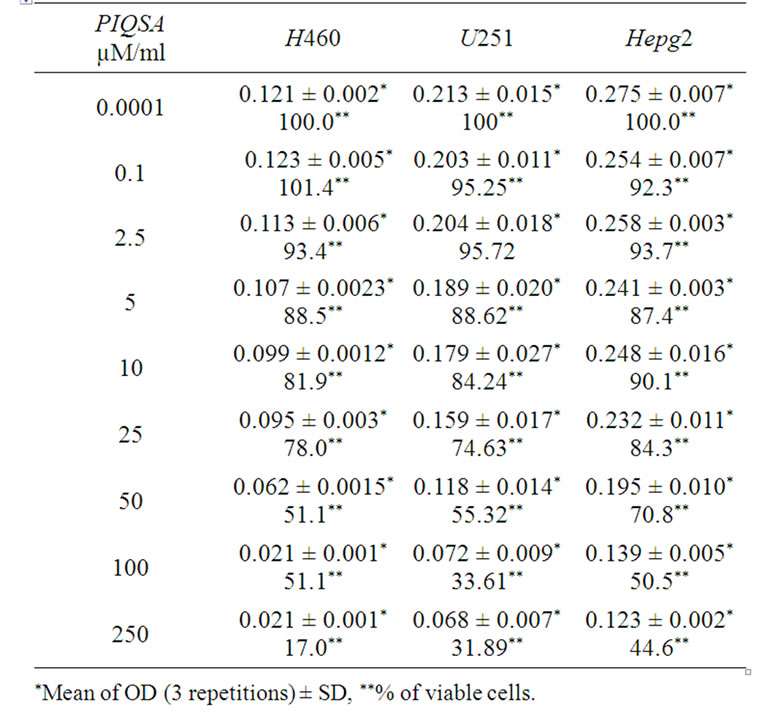
Table 2. Cytotoxic effect of different concentrations of PIQSA on the viability of three human cancers Cell lines in vitro using MTT cell proliferation assay.
smaller tumor size in animal group received combination treatment (p < 0.01) with –22.73% decrease from the control. The tumor weight also recorded highly significant decrease (p < 0.001) by –62.26% from the control, –38.52% decrease from PIQSA treatment alone and –33.07% decrease from irradiation treatment alone, indicating a significant effect of PIQSA in combined treatment on the tumor growth compared with each other treatment modality alone as shown in (Table 3).
5.3. Effects of Different Treatments on Histone Deacetylase Activity and DNA Fragmentation
Treatment with PIQSA alone caused inhibition of HDAC activity (p < 0.01), also very high significant increase in DNA fragmentation was noticed (p < 0.001) by 77.46% increase from the control. Radiation treatment alone increased the % of DNA fragmentation by 377% (p < 0.001) where there was no significant change in HDAC activity from the control. Combination of PIQSA and radiation induced very high significant change in both HDAC activity and DNA fragmentation as compared to the control and also compared with each other treatment modality alone (p < 0.001) indicating a significant effect of the combined treatment on the enzyme activity and DNA fragmentation compared with each other treatment alone (Table 4).
5.4. Effects of Different Treatments on Induction of Cell Death
5.4.1. Biochemical Analysis
Table 5 showed the biochemical analysis of ESC cell death after different treatments. There was high significant increase in necrotic cell death (p < 0.001) and apoptotic cell death (p < 0.01). Treatment with radiation alone induced very high significant increase in both necrotic and apoptotic cell death (p < 0.001). Combined PIQSA and radiation increased the % of apoptotic cell death as compared to the control and each modality treatment alone indicating that pre-treatment with PIQSA synthesize ESC cells to radiation dose to commit apoptosis than to necrosis.
5.4.2.Histochemical Analysis of ESC Cell Death
Figure 1, showing sections of ESC tumors in control group, in PIQSA treated group and in radiation treated group. (a,b) indicating viable cells with bright green nuclei in peripheral and core region of tumor sections from the vehicle control group. (c,d) revealed aggregation of apoptotic cells with orange nuclei in the peripheral a number of apoptotic vacuoles scattered in the inner region after PIQSA treatment alone. (e,f) tumor sections
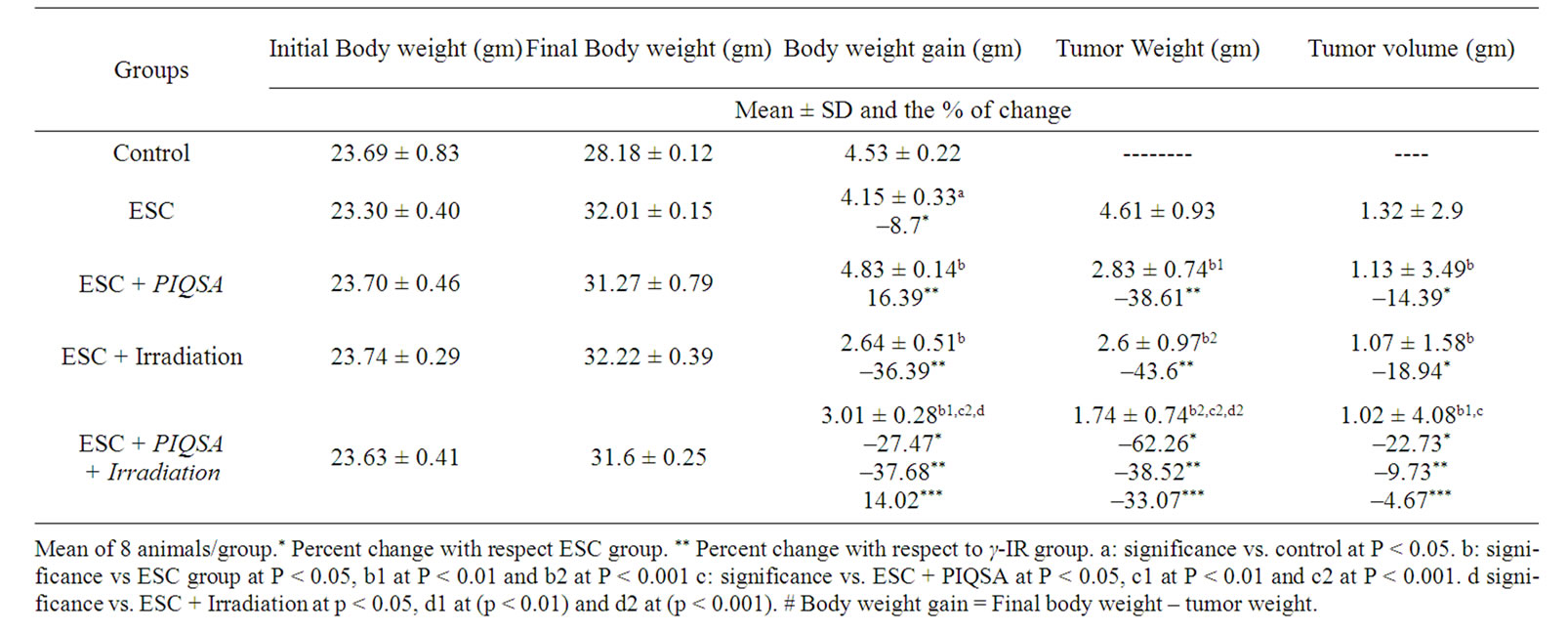
Table 3. Effect of different treatments on body weight gain, tumor weight and tumor volume in treated mice.
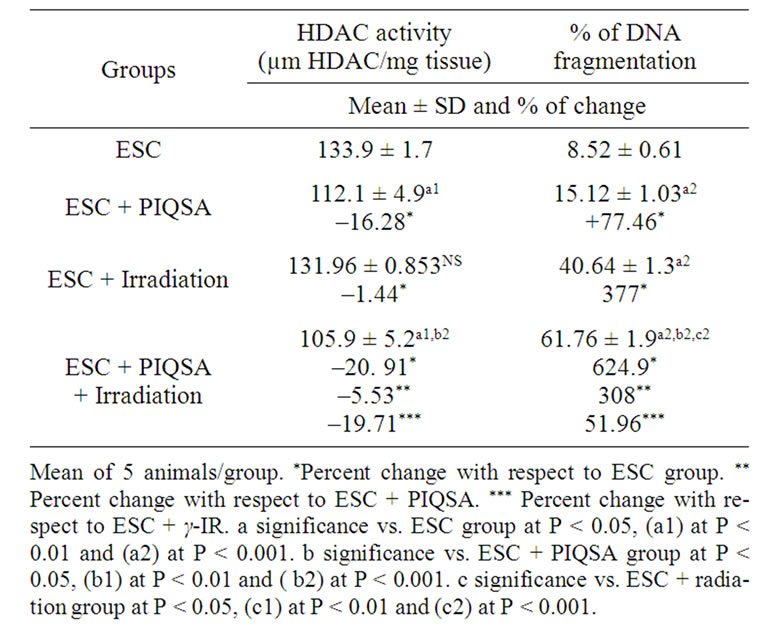
Table 4. The effect of different treatments of mice on histone deacetylase activity and DNA fragmentation.
showed the effect of radiation alone indicating increase of apoptotic cells in both peripheral and core region with the presence of apoptotic strips and colonies in the outer and inner regions of the tumor sections. (g,h) tumor sections of animals after combined of PIQSA and radiation, there are greater increase in apoptotic cells as compared to control and also as compared to PIQSA alone and radiation alone.
5.5. Effect of Different Treatment on Distribution of Cell Cycle Phases
A significant change in distribution of cells in different phases of cell cycle where G1/G0 phase (P < 0.01) and S phase, and (p < 0.001) in G2/M phase in control group
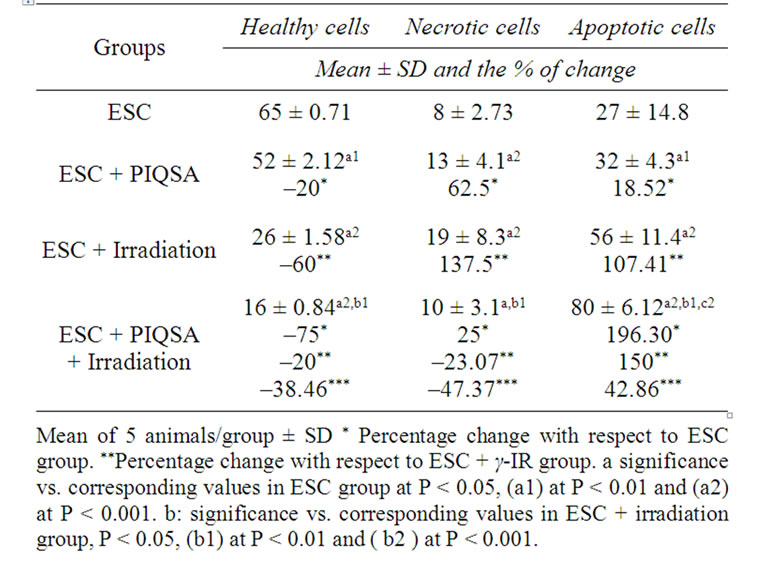
Table 5. Biochemical analysis of apoptotic cell death after different treatments.
compared with normal animals. PIQSA treatment resulted in redistribution of cells in the different phases where a significant change was recorded in G1/G0 phase and G2/M phase (p < 0.05), also, S phase (p < 0.01) with percent changes amounted by 23.52%; –31.9%; and 15.69% respectively, compared to control. Treatment with radiation alone resulted in increase of cells arrested in G2/M (p < 0.001) amounted by 90.77% associated with a proportional decrease of cells at the S-phase (–73.26%) and also at G1/G0 phase (–5.55%). Combined treatment increase in cell population at G1/G0 phase (p < 0.001) accompanied with highly significant decrease in S phase and G2/M phase (p < 0.001) as compared to either the control vehicle or animal received PIQSA alone and radiation alone Table 6.
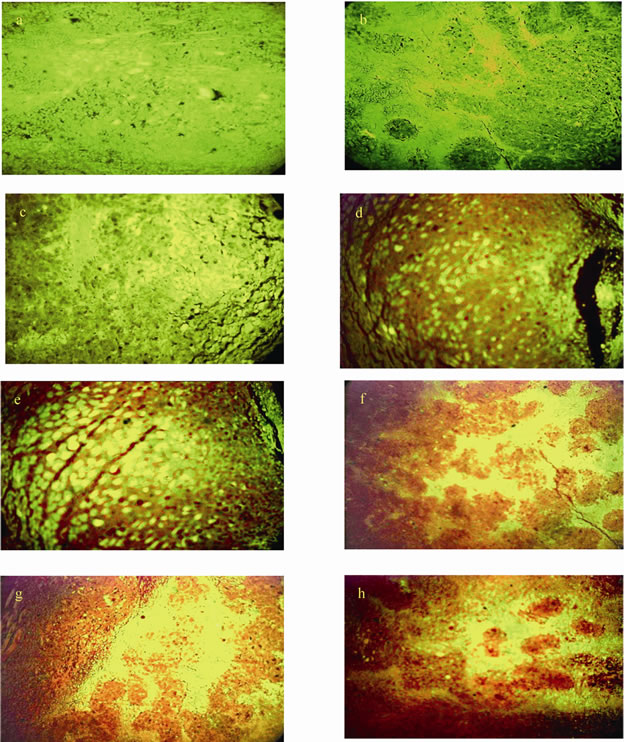
Figure 1. Analyses of tumor sections from mice bearing ESC tumor and treated with vehicle control PIQSA alone, radiation alone, or combined PIQSA and radiation. (a,b) showing tumor sections in control animals. (Acridine orange & Ethidium bromide ×100). (c,d) tumor sections after PIQSA treatment alone. (Acridine orange & Ethidium bromide ×100). (e,f) TUMOR sections after radiation treatment alone. (Acridine orange & Ethidium bromide × 100). (g,h) tumor sections after combined PIQSA and radiation. (Acridine orange &Ethidium bromide × 100).
6. Discussion
Our results confirmed that the pyrimido(4,5-b)quinoline sulfonamide (PIQSA compound) has a broad spectrum of antitumor activity in vitro against EAC cell lines and the three human cell lines,(H460), (U251) and (HepG2) used in the present work and agreed with other published HDAC inhibitors, including trichostatin A [30], LAQ- 824 [31], MS-275 [32], PXD101 [33], cyclic hydroxamic acid containing peptide 31 [34], Valproate [35]. The growth inhibition in human cell lines was recorded with accompanied by accumulation in both acetylated histones and acetylated tubulin [36] and tumor suppressor protein p21Cip1/WAF1, and generation of cleaved PARP fragment and phosphorylated H2AX (H2AX), two known markers of apoptosis [36]. Previously [37] it was suggested that, p21 could be involved in the inhibition of proliferation and induction of differentiation in human cancer cell lines induced by treatment with HDACIs. Others [38] reported that, HDAC inhibitors exert their antitumor activity via induction of cell cycle arrest in G1 or G2-M, followed by differentiation or apoptosis of various tumor cell types with little or no toxicity against normal cells. In vivo studies, the results revealed that (PIQSA) exhibited antitumor activity against ESC tumor in mice, presented by the decrease in tumor weight and
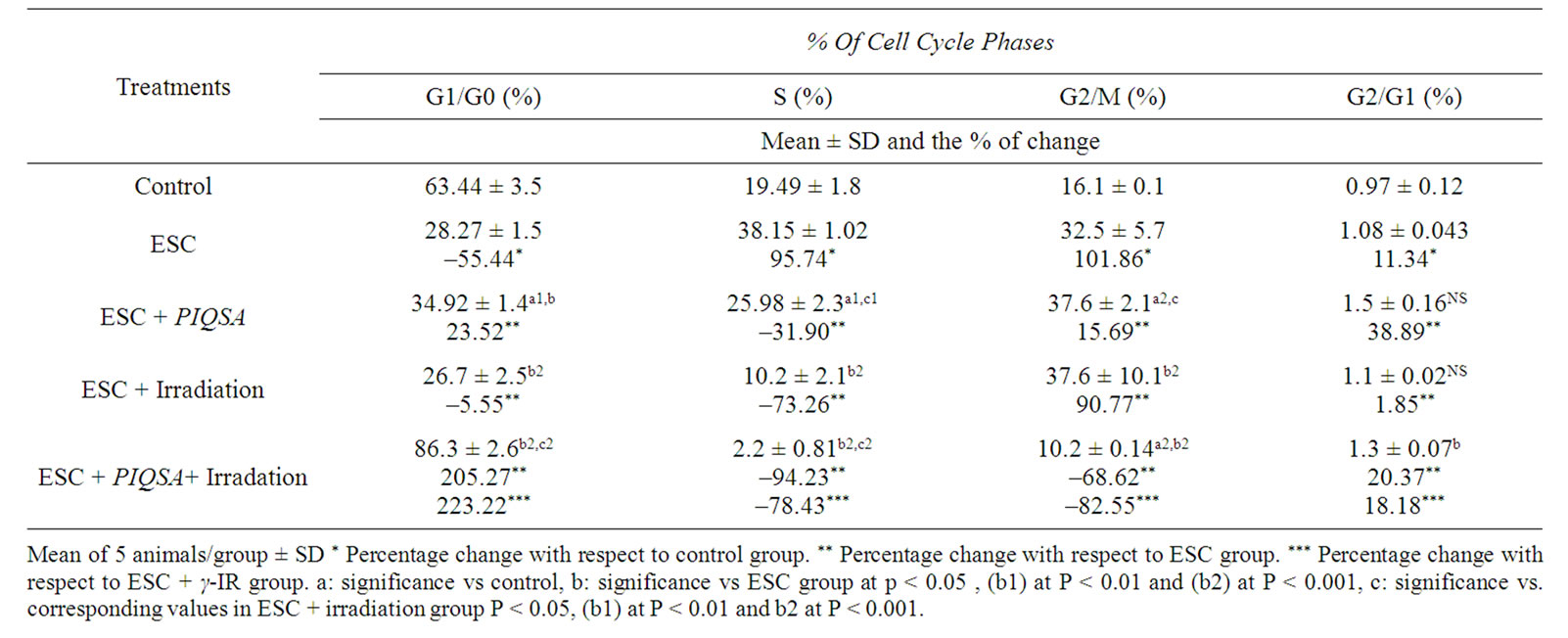
Table 6. Effect of different treatments on distribution of cell cycle Phases in mice bearing tumors.
tumor volume (Table 3). The reduction in body weight gain and the decrease in tumor volume are the criteria for judging on the volume of any antitumor activity drugs [39]. Our results appear to be in consistent with others [20,36],whose concluded that, HDACIs may interfere with cancer cell proliferation and/or survival through the modulation of cell-cycle traverse, induction of differentiation, or by promoting cell death (i.e., apoptosis) [39]. Acetylation and deacetylation of histones play an important role in the regulation of gene transcription and in the modulation of chromatin structure [40]. The equilibrium of steady-state acetylation is tightly controlled by the antagonistic effects of histone acetyltransferases (HATs) and HDACs. Aberrant gene expression resulting in functional inactivation of HAT activity or overexpression of HDACs can mediate tumor cell proliferation [41] and deregulation of HDAC recruitment to promoters contributes to tumorigenesis [42].
Our results revealed that PIQSA resulted in inhibition of HDAC activity.
The inhibition of HDAC enzymes by HDACIs shifts the balance between the deacetylation activity of HDAC enzymes and the acetylation activity of histone acetyltransferases, resulting in hyperacetylation of core histone [43]. HDAC inhibitors have been shown to affect the growth and survival of tumor cells in vitro and in vivo [44]. These compounds have polar end which binds zinc ion of HDAC catalytic pocket structure, other part produce effect through block passage of active site. Mechanism of action of some of these compounds was interaction with catalytic site of HDAC, which blocks substrate approach active zinc ion of enzymes [45]. Other HDAC inhibitors bind to HDAC active site powerfully and the effect of inhibit HDAC is stronger [43].
DNA fragmentation is a hallmark of apoptosis [46]. It was recognized that apoptotic DNA fragmentation is carried out by a heterodimeric protein complex called DNA fragmentation factor (DFF) [47] or caspase activated DNase (CAD) [48]. When apoptosis is initiated, the activated caspase cleaves DFF45/ICAD, and DFF40/ CAD is released into the nucleus to carry out DNA fragmentation. The genes encoding the nuclease that are responsible for the fragmentation of DNA during apoptosis, are aberrantly expressed in many tumor types [49] .In addition, the abnormalities in this gene are associated with poor prognosis in cancer patients. Most significantly, tumor-specific DFF45 gene mutations or deletions were identified in human germ cell tumor and neuroblastoma tumors from patients [50] indicating the involvement of this gene in tumor development. Our results revealed PIQSA alone as well as IR alone (Table 4) resulted in very highly significant DNA fragmentation. The production of DNA damage is the basis of cancer treatments such as chemoand radiotherapy [51]. Such treatments induce mitotic catastrophe, a form of cell death resulting from abnormal mitosis and leading to the formation of interphase cells with multiple micronuclei. The sequential generation of large chromatin fragments followed by internucleosomal fragmentation due to the effect of γ-IR or chemical agents is a biochemical hallmark of apoptosis [52]. This genomic DNA fragmentation is correlated with chromatin condensation and nuclear breakdown. It was concluded that treatment of human tumor cell lines with HDACIs suppress cell growth with decreasing DNA synthesis and stimulate apoptosis and that was associated with mechanisms responsible for these effects including increased histone acetylation as well as enhanced expression of p21 and gelsolin [53]. Others [54] demonstrated that ionizing radiation can induce numerous effects in cells including DNA degradation. Which was attributed to chromatin dysfunction such as singleand double-strand DNA fragmentation due to oxidative stress that leading to cell death through apoptosis or necrosis [55]. The results in the present study revealed that treatment with PIQSA caused significant induction of apoptosis. The results being in consistent with previous investigators [56-59]. Inhibition of HDACs will activate these silenced genes, contributing to growth arrest, differentiation, and/or apoptosis of transformed cells. Treatment with HDAC inhibitors triggers both the intrinsic and sensitizes tumor cells to the death ligands that initiate the extrinsic pathway of apoptosis [56]. Several HDAC inhibitors induce mitochondrial permeability transition, in which pro-apoptotic molecules, such as cytochrome c, are released into the cytosol, resulting in eventual activation of caspase-dependent apoptotic cascades [57,58]. Reactive oxygen species have recently been identified as a major cell death mechanism of several HDAC inhibitors [59]. Others mentioned that, histone deacetylase inhibitors cause accumulation of reactive oxygen species (ROS), activation of a family of cysteine proteases known as caspases and cell death of transformed but not normal cells. In addition, another intracellular system exist which plays an important role in response to stress stimuli and cause an increase in ROS is the thioredoxin (Trx) reduction-oxidation system. Trx protein can function as a hydrogen donor for many protein targets and an active scavenger of ROS [60].
Results in Table 6 indicated that PIQSA caused significant increase of cells arrested in G1/G0 phase of the cell cycle. This result is in Harmony with previous investigators [61,62]. HDACIs-mediated growth inhibition is associated with a marked decrease in cyclin B1 mRNA levels. The decrease in cyclin B1 occurred, is completely blocked by concomitant treatment with protein synthesis inhibitors, and appears to be dependent on changes in transcription. The mechanism of cyclin B1 repression by a HDACI requires prolonged histone hyperacetylation and is at least partly dependent on p21expression [61]. Also, HDACIs blocked two colon cancer cell lines mainly in the G1 phase. In these two human colon cancer cell lines, HDAC inhibitors increased the p21 (WAF1) gene expression by selectively increasing the degree of acetylation of the gene-associated histones, and induced a G1 cell cycle arrest [62]. The results also indicated that γ-IR caused cell accumulation at G2/G1 phase. The results are in agreement with others [63]. They observed a constant accumulation of cells in G2/M phase with the increased level of apoptosis. They concluded that the block of cells in G2/M phase suggests accumulation of sublethal damages and hence increased radiosensitivity manifested by higher level of apoptosis and debris. Our results also, showed that in combined treatment; pretreatment with PIQSA enhanced the ESC tumor due to the exposure dose of IR. As the combined treatment of mice augment the inhibition of HDAC enzyme, increased DNA fragmentation, and also increased cell arrest at G1/G0 phase, consequently induced apoptotic cell death. The results in the current work agreed with previous investigators who reported that HDAC have been shown synergistic effects with cytotoxic cancer therapy [64]. This synergy is related to the inhibition of DNA repair [18] and synthesis [19]. Others stated that different HDAC classes may have differential effects for radiosensitization, which in some cell lines may be influenced by p53 expression [65]. They suggested that the presence of p53 protein may further augment the radiosensitization of cancer cells by HDACIs. HDACs have previously been linked to the regulation of p53, a key molecule in cellular response to DNA damage [66,67]. They also reported that HDAIs suppressed the growth of Wild Type (WT) p53–containing cells more efficiently by increasing G2-M arrest whereas cells without WT p53 accumulated mainly in G1 phase of the cell cycle. Apoptosis was also considerably reduced in the absence of p53 [68] clarifying the precise mechanisms by which p53 potentiate radiosensitization by HDIs inhibiting class I and II HDACs. The enhanced response to radiation was accompanied by inhibition of cellular proliferation and by increased phosphorylation of H2AX; implicating DNA double-strand breaks in the antineoplastic effects of the combined treatment modality and suggesting a decrease in the repair of radiation-induced DNA double-strand breaks (DSBs) [15].The formation of γ-H2AX is necessary for the recruitment of many factors involved in DNA repair, which regulate cell cycle checkpoints [69]. DNA DSBs induced by IR activate checkpoint pathways that inhibit progression of cells through G1 and G2 phases and delay the progression through S phase [69]. Previous reports have shown the ability of HDAC inhibitors to prolong the duration of γ-H2AX in irradiated cancer cell lines [18, 66]. These reports attribute the radiosensitization ability of HDAC inhibitors to the inhibition of DNA repair. Many reports have shown that altered gene expression leads to radiosensitization [70-73].
7. Conclusions
Depending on the findings obtained in the present work, the novel quinoline sulfonamide compound (PIQSA) showed in vitro antitumor efficacy, as it inhibited the viability of EAC cell and other three human cancer cells (lung, brain and liver). Also, it seems that PIQSA compound induced in vivo antitumor and radiosensitizing activity against ESC tumor transplanted in the experimental animals. It is possible to conclude that PIQSA exerted it’s in vivo antitumor and its radiosensitizing activity through different mechanisms including inhibition of histone Deacetylase activity, increasing DNA fragmentation, arresting cell cycle particularly at G1/G2 phase which led to apoptosis of cancer cells.
8. Acknowledgements
The authors would like to thank Prof. Dr. M.M Ghorab; Prof. of applied organic chemistry, National Centre for Radiation Research and technology (NCRRT), Atomic Energy Authority, Cairo, Egypt, for providing the chemical material used as a chemical drug and radiothenthetizing agent during this work.
REFERENCES
- G. J. Kellof, “Perspective on Cancer Chemoprevention Research and Drug Development,” Advances in Cancer Research, Vol. 78, 1999, pp. 199-334. doi:10.1016/S0065-230X(08)61026-X
- M. El-Gaby, S. Abdel Gawad, M. Ghorab, et al., “Synthesis and Biological Activity of Some Novel Thieno[2,3-d]quinpline, Quinoline[3,2:4,5]thieno[3,2-d]pyrimidine and Pyrido[2,3:4,5]thieno[2,3-d]quinoline Derivatives,” Phosphorus, Sulphur, Silicon and Related Elements, Vol. 181, No. 2, 2006, pp. 279-297.
- V. V. Kouznetsov, C. O. Puentes, A. R. R. Bohorques, S. A. Zacchino, M. Sortino, M. Gupta, et al., “A Straightforward Synthetic Approach to Antitumoral Pyridinyl Substituted 7H-Indino [2,1-C] Quinoline Derivatives via Three-Component Imino Diels-Alder Reaction,” Letters in Organic Chemistry, Vol. 3, No. 4, 2006, pp. 300-304. doi:10.2174/157017806776114595
- G. Bouchain, S. Leit, S. Frechette, E. A. Khalil, R. Lavoie, O. Moradei, S. H. Woo, M. Fournel. P. T. Yan, A. Kalita, M. C. Trachy-Bourget, C. Beaulieu, M. F. Robert, A. R. MacLeod, J. M. Besterman and D. Delorme, “Development of Potential Antitumor Agents. Synthesis and Biological Evaluation of a New Set of Sulfonamide Derivatives as Histone Deacetylase Inhibitors,” Journal of Medicinal Chemistry, Vol. 46, No. 5, 2003, pp. 820-830. doi:10.1021/jm020377a
- P. W. Finn, M. Bandara, C. Butcher, A. Finn, R. Hollinshead, N. Khan, N. Law, S. Murthy, R. Romero and C. Watkins, “Novel Sulfonamide Derivatives as Inhibitors of Histone Deacetylase,” Helvetica Chimica Acta, Vol. 88, No. 7, 2005, pp. 1630-1657. doi:10.1002/hlca.200590129
- M. M. Ghorab, F. A. Ragab, E. Noaman, H. I. Heiba and E. M. El-Hossary, “Synthesis of Some Novel Quinolines and Pyrimido [4,5-B] Quinolines Bearing a Sulfonamide Moiety as Potential Anticancer and Radioprotective Agents,” Arzneimittelforschung, Vol. 57, No. 12, 2007, pp. 795-803.
- L. Hu, Z. R. Li, J. D. Jiang and D. W. Boykin, “Novel Diaryl or Heterocyclic Sulfonamides as Antimitotic Agents. Anti-Cancer Agents in Medicinal Chemistry,” Anti-Cancer Agents in Medicinal Chemistry, Vol. 8, No. 7, 2008, pp. 739-745.
- M. Fournel, M. C. Trachy-Bourget, P. T. Yan, A. Kalita, C. Bonfils, C. Beaulieu, S. Frechette, S. Leit, E. Abou-Khalil, S. H. Woo, D. Delorme, A. R. MacLeod, J. M. Besterman and Z. Li, “Sulfonamide Anilides, a Novel Class of Histone Deacetylase Inhibitors, Are Antiproliferative against Human Tumors,” Cancer Research, Vol. 62, No. 15, 2002, pp. 4325-4330.
- K. B. Glaser, M. J. Staver, J. F. Waring, J. Stender, R. G. Ulrich and S. K. Davidsen, “Gene Expression Profiling of Multiple Histone Deacetylase (HDAC) Inhibitors: Defining a Common Gene Set Produced by HDAC Inhibition in T24 and MDA Carcinoma Cell Lines,” Molecular Cancer Therapeutics, Vol. 2, No. 2, 2003, pp. 151-163.
- L. C. Hsi, X. Xi, R. Lotan, I. Shureiqi and S. M. Lippman, “The Histone Deacetylase Inhibitor Suberoylanilide Hydroxamic Acid Induces Apoptosis via Induction of 15-lipoxygenase-1 in Colorectal Cancer Cells,” Cancer Research, Vol. 64, No. 23, 2004, pp. 8778-8781. doi:10.1158/0008-5472.CAN-04-1867
- P. Peixoto and A. Lansiaux, “Histone-Deacetylases Inhibitors: From TSA to SAHA,” Bull Cancer, Vol. 93, No. 1, 2006, pp. 27-36.
- S. Sakajiri, T. Kumagai, N. Kawamata, T. Saitoh, J. W. Said and H. P. Koeffler, “Histone Deacetylase Inhibitors Profoundly Decrease Proliferation of Human Lymphoid Cancer Cell Lines,” Experimental Hematology, Vol. 33, No. 1, 2005, pp. 53-61. doi:10.1016/j.exphem.2004.09.008
- C. Zhang, V. Richon, X. Ni, R. Talpur and M. Duvic, “Selective Induction of Apoptosis by Histone Deacetylase Inhibitor SAHA in Cutaneous T-Cell Lymphoma Cells: Relevance to Mechanism of Therapeutic Action,” Journal of Investigative Dermatology, Vol. 125, No. 5, 2005, pp. 1045-1052. doi:10.1111/j.0022-202X.2005.23925.x
- H. Choy and L. Milas, “Enhancing Radiotherapy with Cyclooxygenase-2 Enzyme Inhibitors: A Rational Advance?” Journal of the National Cancer Institute, Vol. 95, No. 19, 2003, pp. 1440-1452.
- M. Entin-Meer, X. Yang, S. R. V. Berg, K. R. Lamborn, A. Nudelman, A. Rephaeli and D. A. Haas-Kogan, “In Vivo Efficacy of a novel Histone Deacetylase Inhibitor in Combination with Radiation for the Treatment of Gliomas,” Neuro-Oncology, Vol. 9, No. 2, 2007, pp. 82-88. doi:10.1215/15228517-2006-032
- M. Entin-Meer, A. Rephaeli, X. Yang, A. Nudelman, S. R. Vandenberg and D. A. Haas-Kogan, “Butyric Acid Prodrugs Are Histone Deacetylase Inhibitors That Show Antineoplastic Activity and Radiosensitizing Capacity in the Treatment of Malignant Gliomas,” Molecular Cancer Therapeutics, Vol. 4, No. 12, 2005, pp. 1952-1961. doi:10.1158/1535-7163.MCT-05-0087
- K. Camphausen, D. Cerna, T. Scott, M. Sproull, W. E. Burgan, M. A. Cerra, H. Fine and P. J. Tofilon, “Enhancement of in Vitro and in Vivo Tumor Cell Radiosensitivity by Valproic Acid,” International Journal of Cancer, Vol. 114, No. 3, 2005, pp. 380-386. doi:10.1002/ijc.20774
- A. Munshi, J. F. Kurland, T. Nishikawa, T. Tanaka, M. L. Hobbs, S. L. Tucker, S. Ismail, C. Stevens and R. E. Meyn, “Histone Deacetylase Inhibitors Radiosensitize Human Melanoma Cells by Suppressing DNA Repair Activity,” Clinical Cancer Research, Vol. 11, 2005, pp. 4912-4922. doi:10.1158/1078-0432.CCR-04-2088
- Y. Zhang, M. Jung, A. Dritschilo and M. Jung, “Enchancement of Radiation Sensitivity of Human Squamous Carcinoma Cells by Histone Deacetylase Inhibitors,” Radiation Research, Vol. 161, No. 916, 2004, pp. 667-674.
- M. Gupta, U. K. Mazumder, R. S. Kumar and T. S. Kumar, “Antitumor Activity and Antioxidant Role of Bauhinia Racemosa against Ehrlich Ascites Carcinoma in Swiss Albino Mice,” Acta Pharmacologica Sinica, Vol. 25, No. 8, 2004, pp. 1070-1076.
- F. M. Freimoser, C. A. Jakob, M. Aebi and U. Tuor, “The MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-Diphenyltetrazolium Bromide] Assay Is a Fast and Reliable Method for Colorimetric Determination of Fungal Cell Densities,” Applied and Environmental Microbiology, Vol. 65, No. 8, 1999, pp. 3727-3729.
- M. M. Jones, J. E. Schoenheit and A. D. Weaver, “Retreatment and Heavy Metal LD50 Values,” Toxicology and Applied Pharmacology, Vol. 49, No. 1, 1979, pp. 41-44. doi:10.1016/0041-008X(79)90274-6
- J. A. Nagle, Z. Ma, M. A. Byrne, M. F. White and L. M. Shaw, “Involvement of Insulin Receptor Substrate 2 in Mammary Tumor Metastasis,” Molecular and Cellular Biology, Vol. 24, No. 22, 2004, pp. 9726-9735. doi:10.1128/MCB.24.22.9726-9735.2004
- D. Baidyaroy, G. Brosch, S. Graessle, P. Trojer and J. D. Walton, “Characterization of Inhibitor-Resistant Histone Deacetylase Activity in Plant-Pathogenic Fungi,” Eukaryotic Cell, Vol. 1, No. 4, 2002, pp. 538- 547. doi:10.1128/EC.1.4.538-547.2002
- B. D. Strahl and C. D. Allis, “The Language of Covalent Histone Modifications,” Nature, Vol. 403, 2000, pp. 41- 45. doi:10.1038/47412
- P. Salgame, S. V. Arun, L. L. Primiano, J. E. Fincke, M. Sylviane and M. Monestier, “An ELISA for Detection of Apoptosis,” Oxford University Press, 1997, pp. 680-681.
- B. B. Mishell, S. M. Shiiqi, C. Henry, E. L. Chen, J. North, R. Gallily, M. Slomich, K. Miller, J. Marbrook, D. Parks and A. H. Good, “Selected Methods in Cellular Immunology,” W. H. Freeman, San Francisco, 1980, pp. 21-22.
- Y. H. Choi, L. Zhang, W. H. Lee and K. Y. Park, “Genistein-Induced G2/M Arrest Is Associated with the Inhibition of Cyclin B1 and the Induction of p21 in Human Breast Carcinoma Cells,” International Journal of Oncology, Vol. 13, No. 2, 1998, pp. 391-396.
- R. G. Steel, and T. H. Torrie, “Principles and Procedures of Statistics,” 2nd Edition, McCraw-Hill Book Co., New York, 1980.
- S. E. Touma, J. S. Goldberg, P. Moench, et al., “Retinoic Acid and the Histone Deacetylase Inhibitor Trichostatin a Inhibit the Proliferation of Human Renal Cell Carcinoma in a Xenograft Tumor Model,” Clinical Cancer Research, Vol. 11, No. 9, 2005, pp. 3558-3662.
- D. Z. Qian, X. Wang, S. K. Kachhap, et al., “The Histone Deacetylase Inhibitor NVP-LAQ824 Inhibits Angiogenesis and Has a Greater Antitumor Effect in Combination With the Vascular Endothelial Growth Factor Receptor Tyrosine Kinase Inhibitor,” Cancer Research, Vol. 64, No. 18, 2004, pp. 6626-6634. doi:10.1158/0008-5472.CAN-04-0540
- K. Camphausen, T. Scott, M. Sproull and P. J. Tofilon, “Enhancement of Xenograft Tumor Radiosensitivity by the Histone Deacetylase Inhibitor MS-275 and Correlation with Histone Hyperacetylation,” Clinical Cancer Research, Vol. 10, 2004, pp. 6066-6071. doi:10.1158/1078-0432.CCR-04-0537
- J. A. Plumb, P. W. Finn, R. J. Williams, et al., “Pharmacodynamic Response and Inhibition of Growth of Human Tumor Xenografts by the Novel Histone Deacetylase Inhibitor PXD101,” Molecular Cancer Therapeutics, Vol. 2, No. 8, 2003, pp. 721-728.
- Y. Komatsu, K. Y. Tomizaki, M. Tsukamoto, et al., “Cyclic Hydroxamicacid-Containing Peptide 31, a Potent Synthetic Histone Deacetylase Inhibitor with Antitumor Activity,” Cancer Research, Vol. 61, 2001, pp. 4459- 4466.
- N. Takai, J. C. Desmond, T. Kumagai, et al., “Histone Deacetylase Inhibitors Have a Profound Antigrowth Activity in Endometrial Cancer Cells,” Clinical Cancer Research, Vol. 10, 2004, pp. 1141-1149. doi:10.1158/1078-0432.CCR-03-0100
- J. J. Buggy, Z. A. Cao, K. E. Bass and E. Verner, “CRA- 024781: A Novel Synthetic Inhibitor of Histone Deacetylase Enzymes with Antitumor Activity in Vitro and in Vivo,” Molecular Cancer Therapeutics, Vol. 5, 2006, pp. 1309-1317. doi:10.1158/1535-7163.MCT-05-0442
- P. Rocchi, C. Camerin, S. Purgato, R. Fronza, F. Bianucci, F.Guerra, A. Pession and A. M. Ferreri, “p21Waf1/Cip1 Is a Common Target Induced by Short-Chain Fatty Acid HDAC Inhibitors (Valproic Acid, Tributyrin and Sodium Butyrate) in Neuroblastoma Cells,” Oncology Reports, Vol. 13, No. 6, 2005, pp. 1139-1144.
- N. K. Mukhopadhyay, E. Weisberg, D. Gilchrist, R. Bueno, D. J. Sugarbaker and M. T. Jaklitsch, “Effectiveness of Trichostatin A as a Potential Candidate for Anticancer Therapy in Non-Small-Cell Lung Cancer,” The Annals of Thoracic Surgery, Vol. 81, No. 3, 2006, pp. 1034-1042. doi:10.1016/j.athoracsur.2005.06.059
- R. S. Kumar, B. Jayakar and B. Rajkapoor, “Antitumor Activity of Indigofera Trita on Ehrlich Ascites Carcinoma Induced Mice,” International Journal of Cancer Research, Vol. 3, No. 4, 2007, pp. 180-185. doi:10.3923/ijcr.2007.180.185
- P. Gallinari, S. Di Marco, P. Jones, M. Pallaoro and C. Steinkühler, “HDACs, Histone Deacetylation and Gene Transcription: From Molecular Biology to Cancer Therapeutics,” Cell Research, Vol. 17, No. 3, 2007, pp. 195- 211.
- V. Baradari, A. Huether, M. Höpfner, D. Schuppan and H. Scherübl, “Antiproliferative and Proapoptotic Effects of Histone Deacetylase Inhibitors on Gastrointestinal Neuroendocrine Tumor Cells,” Endocrine-Related Cancer, Vol. 13, No. 4, 2006, pp. 1237-1250. doi:10.1677/erc.1.01249
- C. J. Phiel, F. Zhang, E. Y. Huang, M. G. Guenther, M. A. Lazar and P. S. Klein, “Histone Deacetylase Is a Direct Target of Valproic Acid, a Potent Anticonvulsant, Mood Stabilizer, and Teratogen,” Journal of Biological Chemistry, Vol. 276, No. 39, 2001, pp. 36734-36741. doi:10.1074/jbc.M101287200
- C. Monneret, “Histone Deacetylase Inhibitors,” European Journal of Medicinal Chemistry, Vol. 40, No. 1, 2005, pp. 1-13. doi:10.1016/j.ejmech.2004.10.001
- M. C. Myzak and R. H. Dashwood, “Histone Deacetylases as Targets for Dietary Cancer Preventive Agents: Lessons Learned with Butyrate, Diallyl Disulfide, and Sulforaphane,” Current Drug Targets, Vol. 7, No. 4, 2006, pp. 443-452. doi:10.2174/138945006776359467
- M. S. Finnin, J. R. Donigian, A. Cohen, V. M. Richon, R. A. Rifkind, P. A. Marks, R. Breslow and N. P. Pavletich, “Structures of a Histone Deacstylase Homologue Bound to the TSA and SAHA Inhibitors,” Nature, Vol. 401, 1999, pp. 188-193. doi:10.1038/43710
- A. H. Wyllie, J. F. R. Kerr and A. R. Currie, “Cell Death: The Significance of Apoptosis,” International Review of Cytology, Vol. 68, 1980, pp. 251-306. doi:10.1016/S0074-7696(08)62312-8
- X. Liu, P. Li, P. Widlak, H. Zou, X. Luo, W. T. Garrard and X. Wang, “The 40-kDa Subunit of DNA Fragmentation Factor Induces DNA Fragmentation and Chromatin Condensation during Apoptosis,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 95, No. 925, 1998, pp. 8461-8466.
- M. Enari, H. Sakahira, H. Yokoyama, K. Okawa, A. Iwamatsu and S. Nagata, “A Caspase-Activated DNase that Degrades DNA during Apoptosis, and Its Inhibitor ICAD,” Nature, Vol. 391, 1998, pp. 43-50. doi:10.1038/34112
- D. A. Hsia, S. K. Mitra, C. R. Hauck, D. N. Streblow, J. A. Nelson, D. Ilic, S. Huang, E. Li, G. R. Nemerow, J. Leng, K. S. Spencer, D. A. Cheresh and D. D. Schlaepfer, “Differential Regulation of Cell Motility and Invasion by FAK,” Journal of Cell Biology, Vol. 160, No. 5, 2003, pp. 753-767.
- F. Abel, R. M. Sjoberg, C. Krona, S. Nilsson and T. Martinsson, “Mutations in the N-Terminal Domain of DFF45 in a Primary Germ Cell Tumor and in Neuroblastoma Tumors,” International Journal of Oncology, Vol. 25, No. 5, 2004, pp. 1297-1302.
- G. Driessens, L. Harsan, P. Browaeys, X. Giannakopoulos, T. Velu and C. Bruyns, “Assessment of in Vivo Chemotherapy-Induced DNA Damage in a p53-Mutated Rat Tumor by Micronuclei Assay,” Annals of the New York Academy of Sciences, Vol. 1010, 2003, pp. 775-779. doi:10.1196/annals.1299.139
- P. Widlak, J. Lanuszewska, R. B. Cary and W. T. Garrard, “Subunit Structures and Stoichiometries of Human DFF Proteins before and after Induction of Apoptosis,” Journal of Biological Chemistry, Vol. 278, No. 29, 2003, pp. 26915-26922. doi:10.1074/jbc.M303807200
- H. Kamitani, S. Taniura, K. Watanabe, M. Sakamoto, T. Watanabe and T. Eling, “Histone Acetylation May Suppress Human Glioma Cell Proliferation When p21WAF/ Cip1 and Gelsolin Are Induced,” Neuro Oncology, Vol. 4, 2002, pp. 95-101.
- K. Suzuki, M. Ojima, S. Kodama and M. Watanabe, “Radiation Induced DNA Damage and Delayed Induced Genomic Instability,” Oncogene, Vol. 2, 2003, pp. 6988- 6993. doi:10.1038/sj.onc.1206881
- Y. Higuchi, “Chromosomal DNA Fragmentation in Apoptosis and Necrosis Induced by Oxidative Stress,” Biochemical Pharmacology, Vol. 66, No. 8, 2003, pp. 1527-1535. doi:10.1016/S0006-2952(03)00508-2
- K. Bhalla and A. List, “Histone Deacetylase Inhibitors in Myelodysplastic Syndrome,” Best Practice & Research Clinical Haematology, Vol. 17, 2004, pp. 595-611.
- J. L. Aron, M. R. Parthun, G. Marcucci, S. Kitada, A. P. Mone, M. E. Davis, T. Shen, T. Murphy, J. Wickham, C. Kanakry, D. M. Lucas, J. C. Reed, M. R. Grever and J. C. Byrd, “Depsipeptide (FR901228) Induces Histone Acetylation and Inhibition of Histone Deacetylase in Chronic Lymphocytic Leukaemia Cells Concurrent with Activation of Caspase 8-Mediated Apoptosis and Down-Regulation of C-FLIP Protein,” Blood, Vol. 102, No. 2, 2003, pp. 652-658. doi:10.1182/blood-2002-12-3794
- F. Guo, C. Sigua, J. Tao, P. Bali, P. George, Y. Li, S. Wittmann, L. Moscinski, P. Atadja and K. Bhalla, “Cotreatment with Histone Deacetylase Inhibitor LAQ824 Enhances Apo-2l/Tumor Necrosis Factor-Related Apoptosis Inducing Ligand-Induced Death Inducing Signaling Complex Activity and Apoptosis of Human Acute Leukemia Cells,” Cancer Research, Vol. 64, 2004, pp. 2580- 2589. doi:10.1158/0008-5472.CAN-03-2629
- R. R. Rosato, J. A. Almenara and S. Grant, “The Histone Deacetylase Inhibitor MS-275 Promotes Differentiation or Apoptosis in Human Leukemia Cells through a Process Regulated by Generation of Reactive Oxygen Species and Induction of p21CIP1/WAF1,” Cancer Research, Vol. 63, 2003, pp. 3637-3645 .
- E. S. Arner and A. Holmgren, “Physiological Functions of Thioredoxin and Thioredoxin Reductase,” European Journal of Biochemistry, Vol. 267, No. 20, 2000, pp. 6102-6109. doi:10.1046/j.1432-1327.2000.01701.x
- S. Y. Archer, J. Johnson, H. J. Kim, Q. Ma, H. Mou, V. Daesety, S. Meng and R. A. Hodin, “The Histone Deacetylase Inhibitor Butyrate Down Regulates Cyclin B1 Gene Expression via a p21/WAF-1-Dependent Mechanism in Human Colon Cancer Cells,” American Journal of Physiology—Gastrointestinal and Liver Physiology, Vol. 289, No. 4, 2005, pp. 696-703.
- Y. X. Chen, J. Y. Fang, J. Lu and D. K. Qiu, “Regulation of Histone Acetylation on the Expression of Cell Cycle-Associated Genes in Human Colon Cancer Cell Lines,” Chinese Medical Journal, Vol. 84, No. 4, 2004, pp. 312-317.
- A. Adamczyk and A. Gasinska, “The Influence of Fractionated Radiation on Proliferation, Cell Cycle and Apoptosis of Normal Human Dermal Fibroblasts,” Nukleonika, Vol. 50, No. 2, 2005, pp. 9-12.
- L. Geng, K. C. Cuneo, A. Fu, T. Tu, P. W. Atadja and D. E. Hallahan, “Histone Deacetylase (HDAC) Inhibitor LBH589 Increases Duration of γ-H2AX Foci and Confines HDAC4 to the Cytoplasm in Irradiated Non–Small Cell Lung Cancer,” Cancer Research, Vol. 66, No. 23, 2006, p. 11298. doi:10.1158/0008-5472.CAN-06-0049
- I. A. Kim, J. H. Shin, Il H. Kim, J. H. Kim, J. S. Kim, H. G. Wu, E. K. Chi, S. W. Ha, C. I. Park and G. D. Kao, “Histone Deacetylase Hong Gyun Wu, Inhibitor Mediated Radio Sensitization of Human Cancer Cells: Class Differences and the Potential Influence of p53,” Clinical Cancer Research, Vol. 12, No. 3, 2006, p. 940. doi:10.1158/1078-0432.CCR-05-1230
- K. Camphausen, W. Burgan, M. Cerra, K. A. Oswald, J. B. Trepel, M. J. Lee and P. J. Tofilon. “Enhanced Radiationinduced Cell Killing and Prolongation of γH2AX Foci Expression by the Histone Deacetylase Inhibitor MS- 275,” Cancer Research, Vol. 64, 2004, pp. 316-321. doi:10.1158/0008-5472.CAN-03-2630
- J. Luo, F. Su, D. Chen, A. Shiloh and W. Gu, “Deacetylation of p53 Modulates Its Effect on Cell Growth and Apoptosis,” Nature, Vol. 408, 2000, pp. 377-381. doi:10.1038/35042612
- J. Joseph, N. Wajapeyee and K. Somasundaram, “Role of p53 Status in Chemosensitivity Determination of Cancer Cells against Histone Deacetylase Inhibitor Sodium Butyrate,” International Journal of Cancer, Vol. 115, No. 1, 2005, pp. 11-18. doi:10.1002/ijc.20842
- A. Eastman, “Cell Cycle Checkpoints and Their Impact on Anticancer Therapeutic Strategies,” Journal of Cellular Biochemistry, Vol. 91, 2004, pp. 223-231. doi:10.1002/jcb.10699
- S. J. Collis, M. J. Swartz, W. G. Nelson and T. L. DeWeese, “Enhanced Radiation and Chemotherapy-Mediated Cell Killing of Human Cancer Cells by Small Inhibitory RNA Silencing of DNA Repair Factors,” Cancer Research, Vol. 63, 2003, pp. 1550-1554.
- S. P. Jackson, “Sensing and Repairing DNA DoubleStrand Breaks,” Carcinogenesis, Vol. 23, No. 5, 2002, pp. 687-696. doi:10.1093/carcin/23.5.687
- S. Omori, Y. Takiguchi, A. Suda, T. Sugimoto, H. Miyazawa, Y. Takiguchi, N. Tanabe, K. Tatsumi, H. Kimura, P. E. Pardington, F. Chen, D. J. Chen and T. Kuriyama, “Suppression of a DNA Doublestrand Break Repair Gene, Ku70, Increases Radioand Chemosensitivity in a Human Lung Carcinoma Cell Line,” DNA Repair, Vol. 1, No. 4, 2002, pp. 299-310. doi:10.1016/S1568-7864(02)00006-X
- E. Marangoni, N. Foray, M. O’Driscoll, S. Douc-Rasy, J. Bernier, J. Bourhis, P. Jeggo, “A Ku80 Fragment with Dominant Negative Activity Imparts a Radiosensitive Phenotype to CHO-K1 Cells,” Nucleic Acids Research, Vol. 28, 2000, pp. 4778-4782.