Open Journal of Pediatrics
Vol. 3 No. 2 (2013) , Article ID: 33217 , 14 pages DOI:10.4236/ojped.2013.32027
Risk and benefit from clinical trials in minors: Making the case for transparent and consistent publications*
![]()
1Department of Pharmaceutical and Medical Chemistry—Clinical Pharmacy, University of Münster, Münster, Germany
2Department of Paediatric Haematology and Oncology, University Children’s Hospital Münster, Münster, Germany
3Institute of Ethics and History in Medicine, Center for Medicine, Society and Prevention, University of Tuebingen, Germany
4The Royal Marsden Hospital, Department of Paediatric Oncology, Surrey, UK
5Institute for Biostatistics and Clinical Research, University of Muenster, Germany
6Department of Pediatric Hematology and Oncology, University Children’s Hospital, Muenster Albert-Schweitzer-Campus, Muenster, Germany
Email: #boosj@uni-muenster.de
Copyright © 2013 Andreas Dirk Henschel et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 1 January 2013; revised 3 February 2013; accepted 10 February 2013
Keywords: Clinical Trials; Minors; SIGN; CONSORT; Ethics
ABSTRACT
Rationale, aim and objectives: The European (EU) regulation on medicinal products for pediatric use (EC 1901/2006), which became effective in 2007, aimed to stimulate the clinical testing of medications in minors in order to reduce off-label use. In consequence, the number of minors taking part in randomized controlled clinical trials (RCTs) is likely to increase. Clinical trials in minors require a complex methodological design, a careful consideration of risks and benefits and a high level of ethical reflection. Unfortunately, as to the quality of clinical trials and their publications in minors little is known. Therefore, we assessed published reports of randomized, controlled clinical trials in minors, focusing on a common disease (asthma) and a defined spectrum of lifethreatening diseases (malignant diseases). Method: In an exploratory design, we scanned the publications for methodological aspects as well as indicators of ethical soundness, e.g., statements that informed consent had been obtained before the start of the trial or that a Data and Safety Monitoring Board ensured the patients’ safety during the trial. We also looked for passages reflecting the debate on equipoise or other forms of weighing risks and benefits. Results: We found that many of these aspects, which according to the scientific literature and generally acknowledged guidelines are essential to ensure good-quality trials and trial reports, were not considered in the publications analyzed. Conclusion: Therefore, we call for a more transparent and consistent presentation of the trials, especially of safety aspects, relying on a more critical and transparent ethical reflection.
1. INTRODUCTION
The European (EU) regulation on medicinal products for pediatric use (EC 1901/2006) [1], which became effective in 2007, aimed to stimulate the clinical testing of medications in minors in order to reduce off-label use. In consequence, compared to the past decades, the number of minors taking part in randomized controlled clinical trials (RCTs) is very likely to increase, although increase has been moderate up to now [2]. Especially in pediatric oncology, attempts are made to enforce the inclusion of minor patients into clinical trials [3]. The introduction of conditional approval as a regulatory tool restricting the use of candidate drugs to clinical trials and considerations by public health services to push for the participation of children in clinical trials [4] raise a couple of ethical questions and have to be based on sound and transparent trial performance and publication as a precondition.
On the one hand, minors will thus be better able to participate in medical progress, as intended by the regulation. On the other hand, one has to keep in mind that participation in a clinical trial is associated with (more or less severe) risks [5] and special safeguards must be established for minors who are considered to be a highly vulnerable risk group. Finding the best risk-benefit ratio should be the guiding principle of any clinical trial. This demands a high level of ethical reflection in all stages of the trial. The process begins when the study design is planned and ends no sooner than the results are published, and it touches the journals’ cooperativeness to allow the necessary space for non-misleading reporting at least in the form of supplemental material on their websites.
Actually, there is a general debate on how to register and publish clinical trial data and how to open data to third parties to the best of the patients’ benefit today and in the future [6,7]. The EMA in 11/2012 organised a workshop on clinical trial data and transparency. Different advisory groups have been installed to propose policies for proactive publication of clinical trials data [8].
Taking this and the European (EU) regulation on medicinal products for pediatric use (EC 1901/2006) [1] into account it seems highly demanding to take a closer look at the ongoing practice of conducting and reporting clinical trials in children in the recent years.
1.1. The Criterion of Equipoise
A clinical trial concerns the interventions that will be compared as one of the first decisions in planning. In RCTs, trial participants are allocated to different groups (intervention versus control group) in random order. Randomization as a methodological instrument is used to avoid bias and thus get the best evidence possible; it is less concerned with therapeutic improvement or safety aspects concerning the individual trial participant [9]. The physician investigator, however, apart from running a clinical trial, has the moral obligation to give patients access to the best treatment available [10]. The equipoise criterion was introduced to reduce this dilemma in that it allows to sustain methodological integrity in a trial and to consider the individual benefit for the patient [11]. Equipoise in RCTs means that there has to be genuine uncertainty about the superior intervention. For the trial participant, and particularly for the vulnerable group of minors, this means that the risk-benefit-ratio is assumed to be equal in all test groups and that randomization is unlikely to lead to an ethically unacceptable disadvantage.
Although a broader discussion of the equipoise debate would reach beyond the scope of this article, it should be mentioned that there is still controversy about it within the scientific community. Critical authors reject the equipoise criterion and/or its application and interpretation [12-17]. Notwithstanding the controversy within the ethical debate, the Ethical Considerations For Clinical Trials On Medicinal Products Conducted With The Paediatric Population, an official paper published by the European Union in 2008, for the first time explicitly pointed out the importance of ensuring equipoise when conducting clinical trials in minors.
1.2. Publishing Clinical Trials—Benefit to Others
The physician investigator, whose first intention as a physician is to provide a direct benefit to the individual patient, is also obligated to generate scientific knowledge in a manner satisfying current research standards [5], i.e., the standards of Evidence-based Medicine (EbM). If these research standards are observed, medical research is also able to generate a benefit to others, i.e., science and society [18]. As a rule, the scientific knowledge gained is published in scientific journals. It is essential that the data from trials be reported soon after the final analysis and in a diligent manner, so that both the practicing physician and future scientists may be able to utilize the knowledge gained for optimizing treatments and in planning new, meaningful trials. This is the only way in which future patients and participants in clinical trials may benefit from clinical research. The previously generated benefit to others becomes potential benefit to future individual patients.
By the same reasoning, risk to others may become risk to self for future patients or trial participants if a clinical trial fails to meet the research standards and results are published inadequately or with great delay. In consequence, Murray [19] and the Declaration of Helsinki 2008 [5] conclude that properly conducted clinical trials must also to be properly reported and published.
The CONSORT statement [20], first published in 1996 [21], aims to guide researchers in writing good clinical trial reports and, meanwhile, is internationally accepted [22]. CONSORT lays down the most important quality criteria that biomedical scientific publishing of RCTs is supposed to meet for the sake of transparency, consistency and error prevention [23] and thus helps to improve publication quality. Suggestions for additional reporting requirements especially for trials with children in form of a CONSORT-C (children) checklist were already published in 2010 by Saint-Raymond et al. [24], but are not implemented up to know.
Other institutions like the Committee on Publication Ethics (COPE) (http://www.publicationethics.org) and the EQUATOR Network (http://www.equator-network.org) also offer guidance and help to promote good reporting of health research studies. However, it has also been found that these guidelines and/or quality criteria are often insufficiently observed; this was true for trial reports concerning a broad spectrum of diseases, mainly in adults, reviewed by Falagas et al. [25] and Levin and Palmer [26]. Other authors emphasized the problem of under-reporting [27,28].
It is evident that the scientific community needs transparent and clearly understandable published data in order to gauge a trial’s methodological quality and the adherence to principles of biomedical ethics [29]. Any trial report should supply answers to the following questions: Are the internal and external validity of the trial satisfactory? Was the risk-benefit-ratio well-balanced? Were stopping rules defined before the trial was started, so that the trial could be prematurely stopped when ethically required while keeping the loss of data as small as possible?
Against this background and in light of the European (EU) regulation on medicinal products for pediatric use (EC 1901/2006) [1] and the scarcity of findings about the quality of clinical trials in minors, especially concerning the risk-benefit ratio, we analyzed publications on clinical trials in minors with regard to the quality of the trial report and the presentation of the authors’ ethical reflections. In particular, we addressed the issue of internal validity and the more basic question if an overall conclusion on the ethical justification of a trial can actually be drawn on the basis of the published paper alone. Considering the complexity of these issues we refrained from phrasing a specific hypothesis and proceeded in an exploratory manner.
We would like to contribute our findings to the ongoing debate about the need for transparency where clinical research in minors and the resulting conclusions on the risk-benefit balance are concerned, and aim to reach first and foremost the clinical investigator. We present findings from an independent sub-project of the cooperative project Risks and benefits in clinical trials in humans: ethical, legal, and clinical studies, funded by the Federal Ministry of Research and Technology, Germany, from October 2006 to October 2009. The findings concerning publications dealing with malignant diseases have already been published in the journal Pediatric Blood and Cancer [30]. Here we present selected results from the earlier study for the purpose of comparison with so far unpublished findings on publications dealing with asthma in minors.
2. METHODS
We focused on publications of RCTs in minors, which we identified via MEDLINE, the most frequently used electronic database for medical science worldwide. All publications included in this review were analyzed using a previously developed evaluation form.
Results are given in absolute and relative numbers. Because of rounding effects, percentages based on the numbers will not necessarily sum to 100%.
2.1. Selecting a Database
As mentioned above, we decided to use MEDLINE as the only database. The European database EMBASE was not used as one may assume a high degree of overlap; the Cochrane Handbook (9/2009) [31] estimates about 26% more hits when a literature search is done in both of the databases rather than MEDLINE alone. Moreover, EMBASE is not free of charge.
2.2. Developing a Systematic, Algorithm-Guided Search
To develop the algorithm for our search, we selected appropriate terms from three search combinations for RCTs suggested by Cochrane [32] and adapted them to our specific research question. To keep the number of false negative results low, we decided to broaden our algorithm. In consequence, we obtained a number of false positive results that had to be excluded by hand in a second and, if necessary, third step, i.e., screening of the abstract or the full article.
Publications thus identified (Figure 1) and which ful-

Figure 1. MEDLINE algorithm used in our project.
filled the following inclusion criteria, were included in the assessment: Minors (at least one person <18 years of age included in the trial described in the publication) and RCT (at least one part of the trial randomized) and time frame (published 2001-2005; limits chosen for a reduction in number and focus on the years immediately before the EU regulation [1] became effective) and type of disease (asthma or malignant disease). Considering the language skills of our working team, only articles published in English or German were included to avoid misinterpretation.
2.3. Developing the Evaluation Form
We used a Microsoft Access database. The form included items on methodological aspects of EbM as well as indicators of ethical soundness including equipoise. The first part of the form, which referred to methodological aspects, consisted of items from the Methodology Checklist 2: Randomised Controlled Trials of the Scottish Intercollegiate Guidelines Network, published 2004 (hereafter called SIGN checklist) [33]. The second part with its focus on the reporting of the trial was derived from the methods part of the CONSORT statement (hereafter called CONSORT) published 2001 [32]. The third part was developed by our working group. Items in this section refer to indicators reflecting the authors’ consideration of the principles of bioethics as phrased by Beauchamp and Childress [29] and aspects concerning the equipoise criterion [10].
The rating categories used were adopted from SIGN (Table 1). SIGN offers six categories, which are explained in detail in the addendum to the checklist [34]. To maintain consistency, we used the same structure of six categories also in the section containing the CONSORT items and in the section with questions about ethical aspects.
Where a rating question was inappropriate, the categories yes, no and unclear were used. All in all, the evaluation form included n = 69 items.
For data collection and quantitative analysis we used Microsoft Access and Microsoft Excel 2003. Data are presented in absolute and relative numbers.
For further details on methods and the evaluation form, see also [30].
2.4. Inter-Rater-Analyses
Every tenth publication (on asthma as well as malignant diseases) was analyzed by two raters (A. H. and G. R.) and a third, external person (D. S.); all raters used the described evaluation form. Inter-rater-reliability was calculated as kappa coefficient according to Fleiss [35] for all three raters. Moreover, pair comparisons of two raters each were conducted using the kappa coefficient developed by Cohen [36]. The percentage of agreement was also determined. For a clearer distinction between the evaluations, the six categories given by SIGN were collapsed into three categories as shown in Table 2. Additionally, the inter-rater-agreement was calculated for all items of the yes, no, unclear answer type. For better distinction, the categories no and unclear were collapsed.
2.5. Comparison with External Study Data
At about the same time when the revised version of the CONSORT Statement was published in 2010, Hopewell et al. [37] published an analysis of all publications on RCTs indexed in PubMed, a MEDLINE search interface, published in December 2006. Hopewell et al. studied the degree to which these publications were in line with the CON-SORT Statement. Because of the proximity to our own analysis, we decided to include Hopewell’s findings in our comparison and discussion. As Hopewell et al. did not use the six SIGN-categories, we equated the first and second SIGN-category (well covered and adequately addressed) with Hopewell’s stated, which indicated a positive finding, and the last four SIGN-categories (poorly addressed, not addressed, not reported, not applicable) with Hopewell’s not stated in the sense of not described adequately. For lack of an international, binding excluded. Furthermore, Hopewell et al. used a much
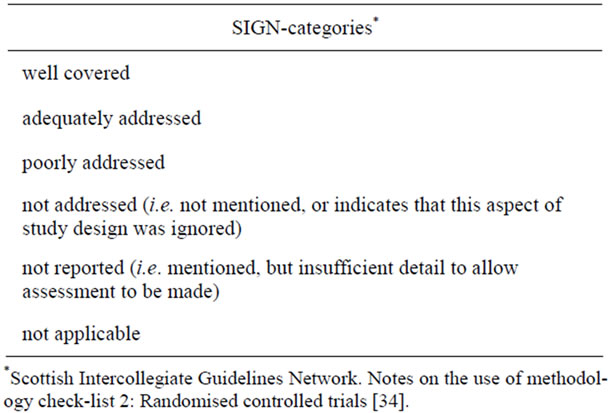
Table 1. Categories given by SIGN* and definitions used by the working group.
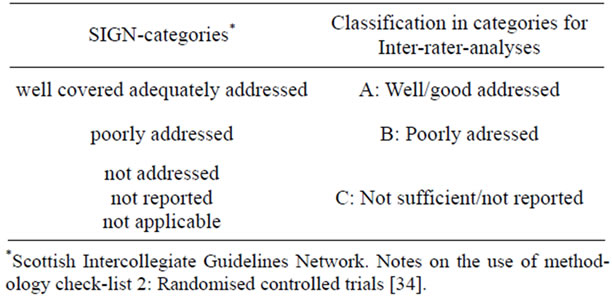
Table 2. Collapsing of SIGN-categories for better distinction in the inter-rater-analysis.
definition of stated and not stated, a mismatch cannot be narrower search algorithm than we did and did not differentiate between trials in minors and trials in adults. Nevertheless, in our view a comparison of the data may be useful.
3. RESULTS
We present details on selected indicators followed by a short discussion. The relevance of these findings within the overall context will be dealt with in the Discussion section. Figure 2 shows the flow diagram of publication inclusion; numbers are given separately for papers on asthma and malignant diseases. NAsthma = 800 publications on asthma and nOnko = 1000 publications on malignant diseases entered the screening process. Abstract screening of about half of all identified publications left n = 385 publications on asthma and n = 688 publications on malignant diseases for inclusion. The subsequent review of the full articles identified a number of publications that did not meet the inclusion criteria. Finally, n = 227 publications on asthma and n = 175 publications on malignant diseases were included in the analysis by evaluation form.
3.1. Indicators of Reporting Quality: Focus Trial Methodology
3.1.1. Randomization Procedure
The CONSORT statement and the SIGN checklist explicitly state that the procedure of randomization has to be clearly described in the publication. This is important to minimize bias and avoid (un)aware manipulation. The current state of the art is to use computer-generated random number tables for randomization. Allocation for example by day of the week or birthday of the participant are not considered valid randomization methods and are thus not acceptable: Such procedures are called pseudorandomization In our analysis, the SIGN-category poorly addressed was used when only a minimum of information was given on the randomization procedure; it was for example applied when the authors stated the fact of randomization as such, but did not describe how the procedure was done. We found that the procedure of randomization stayed unclear in about two thirds of all publications on asthma as well as malignant diseases (Table 3(A)). Hopewell et al. [37] reported similar results. In the worst case, randomization may not be adequate in the great majority of trials. Assessing risks and benefits is thus difficult or even impossible.
3.1.2. Allocation Concealment
Concealing the sequence of group allocation is essential for the process of randomization [38]. Concealment is ensured by calling on an external service for this task, e.g., the hospital’s pharmacy, an external call center or automatic systems operating by phone or fax, or, as suggested by SIGN [34], by using identically looking numbered containers. Allocation by sealed envelopes is also acceptable, but susceptible to manipulation [39]. Correct allocation concealment is of immense importance as trials with incorrect allocation concealment tend to overestimate treatment effects [40].
In our analysis, reporting of allocation concealment was adequate or better in only 10% of the papers on asthma and 16% of the papers on malignant diseases (Table 3(B)). Hopewell et al. [37] reported a higher percentage of about 25 %.
3.1.3. Trial Endpoints
Endpoints serve as the basis for sample size calculations. Hence, prospectively planned endpoints are essential for a valid RCT and for assessing the risk-benefit-ratio. Fur-
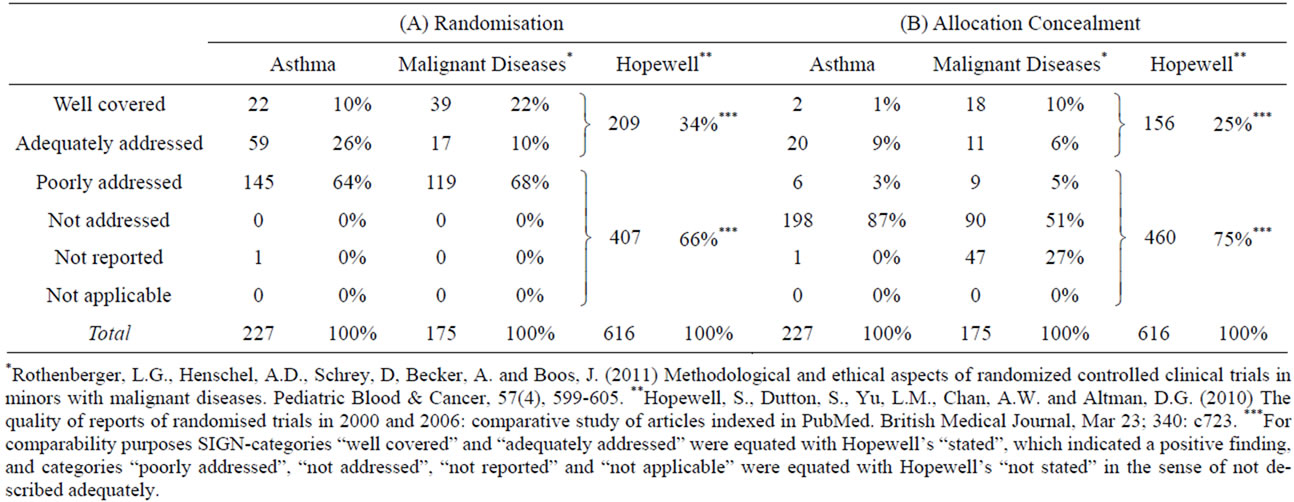
Table 3. Methodological indicators: Reporting of randomization and concealment.
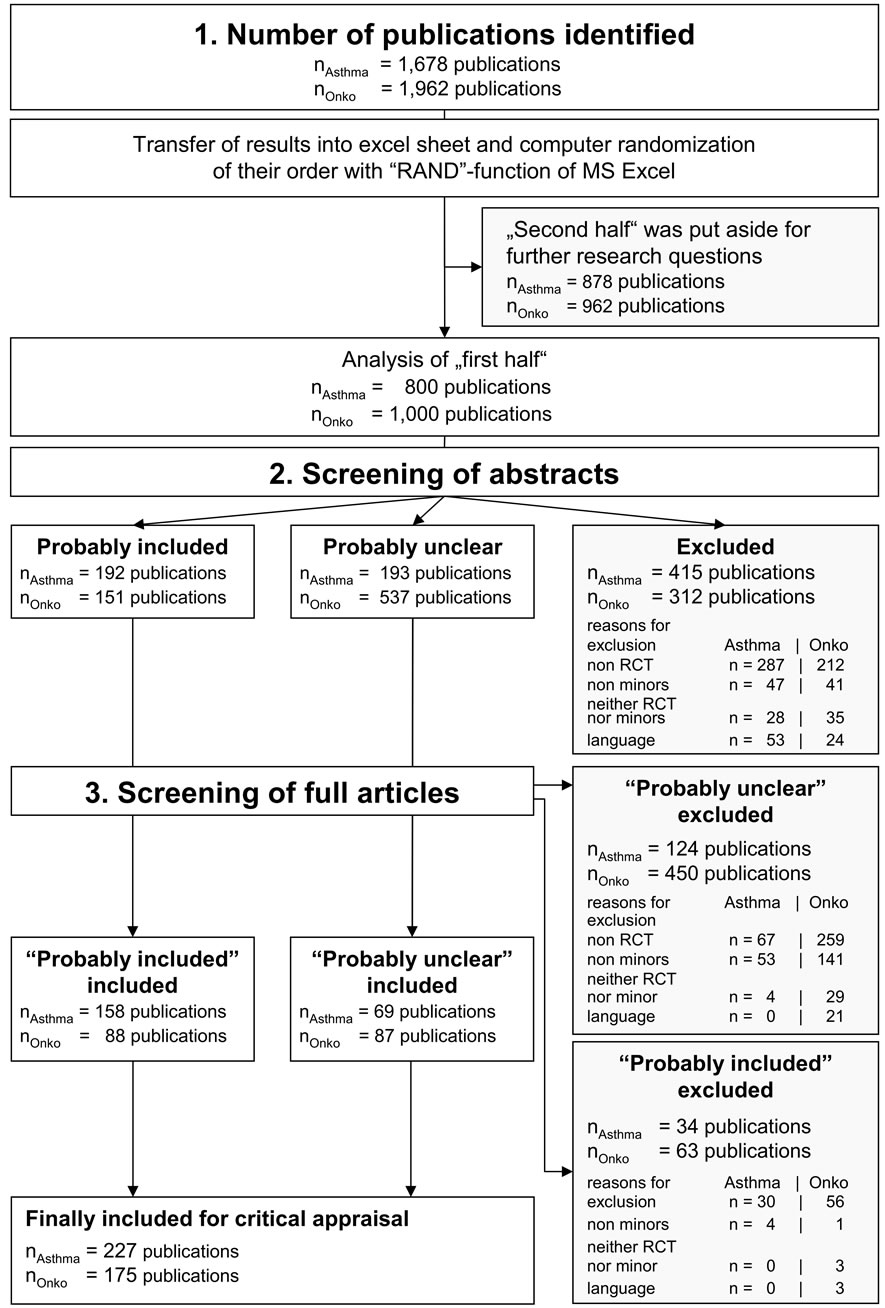
Figure 2. Flow diagram. Quantitative steps of the inclusion/exclusion procedure: 1) number of publications identified, 2) screening of abstracts, 3) screening of full articles.
thermore, if necessary to evaluate additional effects of the tested intervention, secondary endpoints may be defined; these are, however, of minor relevance [41].
The commentary accompanying the SIGN Methodology Checklist 2: Randomised Controlled Trials [33] states clearly that publications without prospectively defined endpoints or whose main message is based on secondary endpoints are to be categorically rejected.
We found endpoints to be clearly described in about 40% of all analyzed publications on both, asthma and malignant diseases (Table 4(C)). A rating of adequately addressed was applied if the primary endpoint could be inferred, but was not explicitly named. For the purpose of comparison with Hopewell et al. [37], who claim that endpoints must be explicitly stated, we equated our SIGN rating category well covered with Hopewell’s category stated.
Concerning the papers on asthma, we found endpoints to be poorly addressed in about 35% of all publications analyzed, whereas a rating of poorly addressed was applied to only about 18% of publications on malignant diseases.
3.1.4. Blinding
Blinding is known to be the best methodological instrument to reduce performance bias and detection bias. Performance bias refers to a systematic difference in treatment management between two study arms based on group allocation alone. Detection bias means that the process of data ascertainment is influenced and thus not objectively done [42]. It is not always feasible to blind both the physician and the patient; if blinding involves giving placebo injections to children, the procedure might not be justified from an ethical point of view. At any rate, the person who analyzes the data can and should be blinded.
We found detailed descriptions of the blinding procedure in 39% (asthma) und 40% (malignant diseases) of publications analyzed, so that the reader could clearly identify who was blinded in the trial concerned (Table 4(D)). In more than half of all publications this remained unclear.
3.1.5. Sample Size Calculation
On the one hand, the sample size should be large enough to assure valid results. On the other hand, it should be small enough to keep the number of participants exposed to the potential risks of the trial as small as possible. A small sample size can be justified if the treatment effect is expected to be large. In a situation like that one might doubt that there was true equipoise before the trial was started. Therefore, when such a trial is approved as ethically justified, interim analyses are called for to control for equipoise.
We found that reporting of the sample size was sufficient in 43% (asthma) und 41% (malignant diseases) of the publications analyzed (Table 5(E)). Hopewell et al. [37] reported similar results (45%).
3.1.6. Flow Diagram
The CONSORT statement [32] recommends that a flow diagram be presented which gives the numbers of trial participants included respectively excluded at different stages of a trial and reasons for their inclusion or exclusion (similar to the flow diagram shown in Figure 2). Such information makes it possible to get a first impression on the comparability of the trial arms fast. The flow diagram is an essential element of the CONSORT statement’s recommendations [32].
We found that most of the publications reviewed did not provide a flow diagram.
3.2. Indicators of Ethical Soundness
3.2.1. Informed Consent
It is essential that clinical trial participants’ autonomy is respected. Therefore, informed consent has to be obtained in advance [5,29,43]. Where minors are concerned, obtaining informed consent from their legal representatives, e.g., their parents, should be complemented by giving age-appropriate information about the risks and benefits of the trial to the minor him/herself [5]. The Ethical Considerations for Clinical Trials on Medicinal Products Conducted with the Paediatric Population [44] of the EU suggest to try to obtain the minor’s assent from the age of three; from the age of six or seven, obtaining the minor’s written assent is recommended.
In our analysis, we applied a rating of well covered if informed consent and assent were clearly reported. This
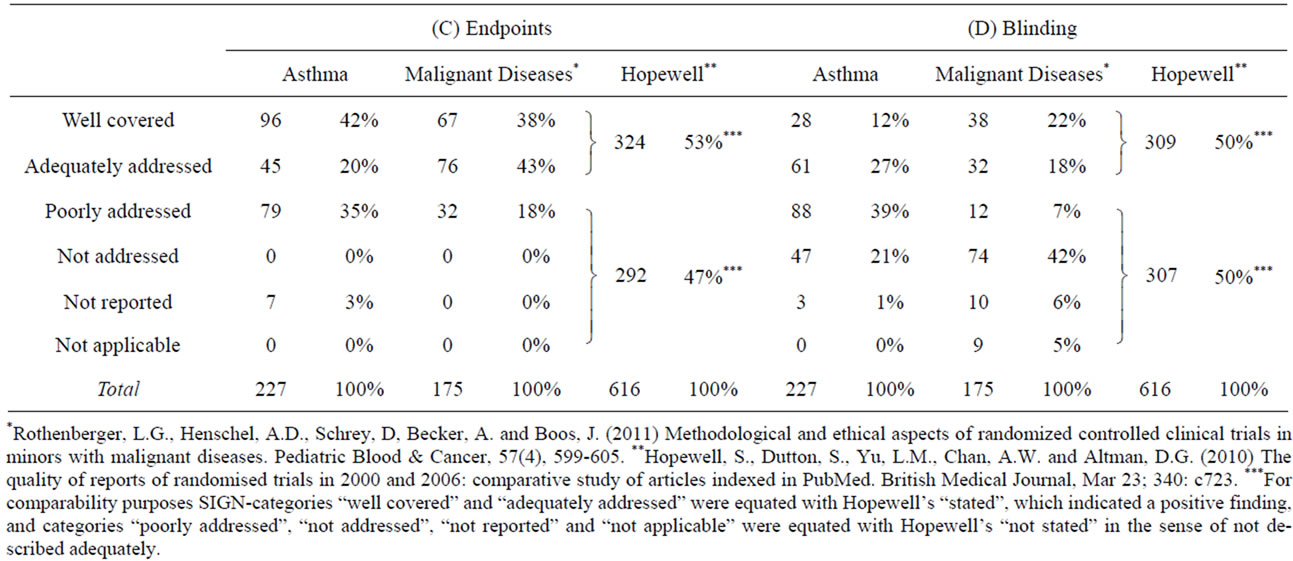
Table 4. Methodological indicators: Reporting of endpoints and blinding.
was the case in only about 10% of the analyzed publications in both samples (Table 6(G)). About another 10% of publications each did not report informed consent at all. While these indicators of ethical soundness do constitute an essential element of clinical trial reports, the checklist of the CONSORT statement contains no items on informed consent or ethical approval; pertinent recommendations are, however, given in the statement’s Explanation and Elaboration section [32].
3.2.2. Ethical Approval
According to the Declaration of Helsinki [5] and the Uniform Requirements for Manuscripts Submitted to Biomedical Journals [45] it is required to obtain ethical approval before the start of a clinical trial.
We found that the majority of publications analyzed reported approval to have been obtained from an Ethics Committee or Institutional Review Board, which was rated as well covered (Table 6(H)).
3.2.3. Interim-Analyses and Stopping Rules
According to the CONSORT statement, interim-analyses should be reported and authors are asked to mention whether these were prospectively planned; prospectively planned interim-analyses help to control the trial participants’ risk and thus help to ensure the patients’ safety. Interim-analyses are not trivial at all [46] and require a complex biometrical data analysis. It is sometimes difficult to come to an ethically as well as scientifically sound decision. Consider this problem: When realised burden is more frequent than expected in the trial arm with the tested intervention and there is not yet enough
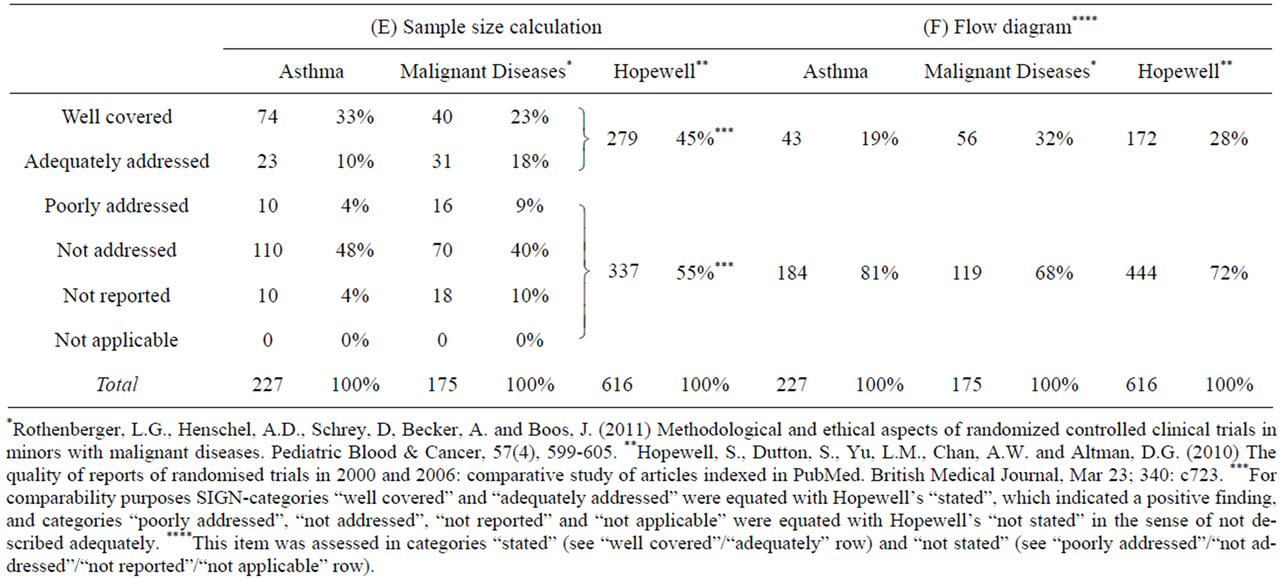
Table 5. Indicators of trial methodology: Reporting of sample size calculation and flow diagram.
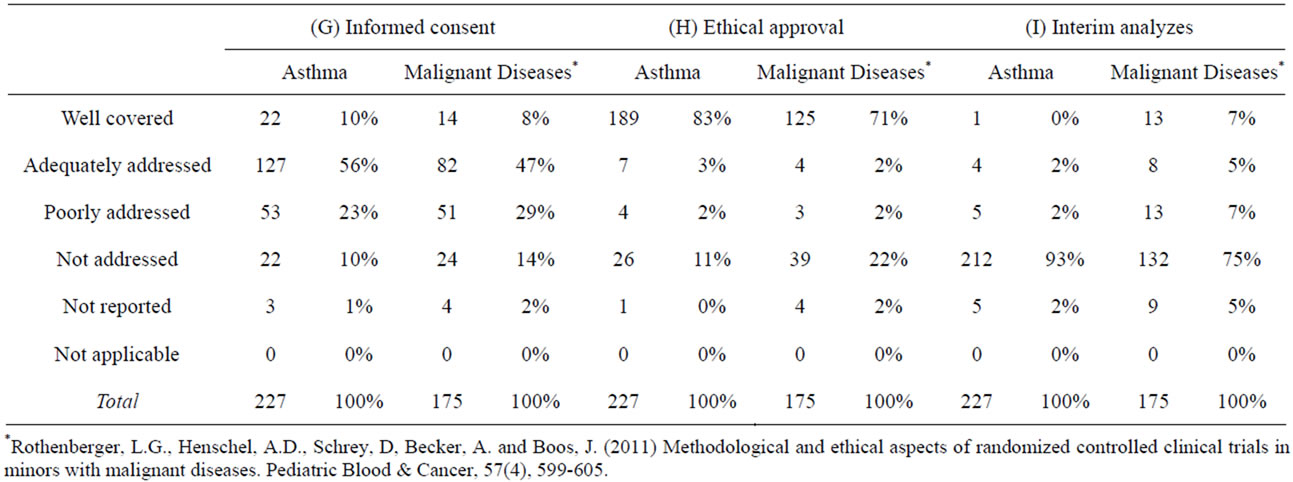
Table 6. Indicators of ethical soundness: Reporting of informed consent, ethical approval and interim analyzes.
scientific evidence to proof the substantial positive effects for several patients in the same arm, what would be a reliable basis for the investigator to decide? Without prospectively planned stopping rules, questions of this kind can hardly be answered adequately.
As to our findings, most of the analyzed publications mentioned neither interim-analyses nor stopping rules (Tables 6(I) and 7(J)).
3.2.4. Data and Safety Monitoring Board
It is recommended to establish an external Data and Safety Monitoring Board (DSMB) to ensure that strong imbalances between study arms and thus a violation of the equipoise criterion will be detected immediately [44].
Regarding our findings in the publications on asthma trials, only n = 2/227 publications (<1%) mentioned a DSMB. Concerning trials on malignant diseases, n = 9/175 publications (5%) reported that a DSMB had been implemented, an additional n = 6 publications (3%) reported a Data Monitoring Board and one publication reported a Safety Monitoring Board.
3.2.5. Reporting of Similar Studies
To avoid redundant research, it is necessary to conduct a thorough literature search before planning a clinical trial. The existing data should be summarized without prejudice and assessed for relevance concerning the research question. The results of this literature summary determine the research desideratum and, consequently, the reason why the clinical trial planned is in fact needed [5]. It is a task that falls within the responsibility of the investigator and cannot be delegated to the competent authorities or the ethics committee.
We found that similar studies were mentioned in many of the analyzed publications (Table 7(L)). Differences in the rating of publications on asthma and malignant diseases separately can probably be attributed to the fact that the distinction between the individual categories is not always clear-cut. When the two upper categories of well covered and adequately addressed, which indicate good reporting, were combined, most of the publications fulfilled this criterion (asthma: 91%; malignant diseases: 98%).
3.2.6. Statistical Testing—Oneor Two-Sided?
If the investigators assume equipoise, the prospective sample size calculation should allow testing for superiority and inferiority of the tested intervention relative to the standard treatment and/or placebo. For this purpose, a two-sided statistical test is needed (as opposed to a one-sided test which checks for superiority or inferiority). If a one-sided test on superiority was used and the results were negative for the tested intervention, one must not conclude inferiority to the control treatment as this was not investigated.
Our findings showed that the reporting of statistical tests was not always sufficient (Table 7(M)). Concerning the papers on asthma, one-sided tests were reported in about 10% (n = 23) of all publications analyzed, twosided tests, in about 30% (n = 69). In publications focusing on malignant diseases, the result was 8% (n = 14) for one-sided tests and 39% (n = 38) for two-sided tests. Any conclusions on equipoise must thus remain vague.
3.2.7. Documentation of Adverse Events
Adverse events can be very rare and may thus be difficult to determine accurately.
Nevertheless, it is of great importance to detect adverse events early and collect pertinent information as systematically as possible, because only then will it be possible to provide a valid estimate of the risk-benefitratio of the tested intervention. This is why trial proto-

Table 7. Indicators of ethical soundness: Reporting of stopping rules, similar studies and two-sided tests.
cols call for good documentation of adverse events [5] and the CONSORT statement stipulates reporting of adverse events in publications on clinical trials. Tabular lists on adverse events will provide a quick overview and help to assess the risk of occurrence.
Although our study results showed that adverse events were mainly reported adequately in the analyzed publications (Malignant Diseases: n = 140/175; 80%), the Common Toxicity Criteria (CTC) [47] or Common Terminology Criteria for Adverse Events (CTCAE) [47] were only marginally used. In the publications about asthma, 62% (n = 140) reported adverse events. No information on adverse events was found in n = 35/175 publications (malignant diseases, 20%) and n = 87/227 publications (asthma, 38%).
3.2.8. Inter-Rater-Analyses
Following a randomization sequence established at the beginning of our review process, we performed inter-rater-analyses on the review of n = 17 papers on malignant diseases and n = 22 papers on asthma, i.e. a total of n = 39 publications. Inter-rater-reliability for three raters was calculated according to Fleiss (kappa coefficient) [35]. The kappa coefficient was κFleiss = 0.57 for n = 3 × 1443 = 4329 items rated according to the SIGNcategories and κFleiss = 0.66 for n = 3 × 658 = 1974 items rated according to the yes/no/unclear answer categories. According to Landis and Koch [48], kappa coefficients from 0.40 to 0.59 can be assessed as satisfactory, kappa coefficients from 0.60 to 0.79 as substantially good. The items with SIGN answer categories were compared in pairs (two raters each) using the kappa coefficient according to Cohen (Cohen, 1960). The results from this analysis concerning the items presented in this article were assessed as substantially good (raters A. H. and G. R.: κCohen = 0.65, agreement: 80%). Inter-rater-reliability between one internal and one external rater was satisfactory (G. R. and D. S. (external rater): κCohen = 0.56, agreement: 74%; D. S. (external) and A, H.: κCohen = 0.51, agreement: 70%). The majority of differences in rating (n = 583/1103 difference: about 53%) concerned categories A and B (s. Table 2). G. R. tended to prefer category A, whereas A. H. tended to prefer category B. This is not entirely surprising as the distinction between the SIGNcategories well covered (inter-rater-category A), adequately addressed (inter-rater-category A) and poorly addressed (inter-rater-category B) is more or less vague. The rating by D. S. showed no tendency in preference.
4. DISCUSSION
Authors find themselves in a dilemma between the limited length of an article and the necessity to explain all relevant issues in as much detail as possible. The CONSORT statement [20,32] suggests how to cope with this problem concerning the methodological aspects of an RCT. Our findings show that these suggestions were often not followed in the analyzed publications on RCTs in asthma and malignant diseases in minors. Data published by Hopewell et al. [37], even though they did not focus on minors, support our results.
Regarding the assessment of ethical aspects of an RCT, publications need to present additional information, e.g., if ethical approval and informed consent/assent were obtained. For an assessment on whether or not the equipoise criterion was fulfilled at the start of the trial, a (short) reflection on the risk-benefit-ratio, which is already required for obtaining approval of the study protocol, should also be part of the publication [5]. We found that this kind of reflection was rarely presented. For the reader, a reflection on the interplay of ethical and methodological aspects is of particular interest for a valid assessment of equipoise. Such an assessment is only possible if the relevant information is clearly described in the article. For example, lack of information on the following aspects may result in serious problems concerning the ethical assessment of an RCT: 1) Scientific data already published on the research topic: without a detailed discussion a valid estimate of the risk-benefitratio can be given neither for the intervention nor for the control group of the trial; 2) Primary endpoint and sample size calculation: without a clear description, the reader cannot conclude the size of the treatment effect expected by the investigators conducting the trial; 3) Prospectively planned stopping rules: without pertinent information, risk management and the control of equipoise during the trial cannot be assessed; 4) Type of statistical tests, oneor two-sided: without appropriate information, prospective assumptions regarding the direction of the effect (e.g., testing for superiority or inferiority) stay unclear; 5) Data Safety and Monitoring Board: without a DSMB, a continuous, independent assessment of adverse events seems problematic and a control for equipoise during the trial seems impossible.
Another aspect, which was not in the focus of our study, but has to be kept in mind when discussing the assessment of ethical trial justification based on publications alone, is the potential discrepancy between the published report and the trial protocol. With regard to our findings, it is thus important to be aware that the analyzed publications may not represent exactly what was done or written in the study protocol. It goes without saying that any publication should truly and unmistakably report how the trial was conducted. Our findings, however, indicate that the authors of the analyzed publications often failed to report some essential information because, in the worst case, they did not know about these aspects, because they did not consider them essential for reporting, or because journal guidelines—e.g., by insisting on a certain structure and/or not insisting on the inclusion of missing, relevant aspects during the review process—did not allow for an article covering all of these aspects. Concerning the last point, data from Meerpohl et al. [49] indicate that endorsement of established reporting recommendations is only moderate in pediatric open access journals. Further exploration of these issues is warranted.
More transparent publications that provide a link to where the study protocol can be found, as demanded by the 2010 version of the CONSORT Statement [20], would help to reduce potential misinterpretation. In pointing out these aspects, the clinical research team could also signal that it is aware of their ethical responsibility not only for the present, but also for future research possibly conducted by another research team.
It stays unclear why clinical trial reporting is incomplete and often insufficient despite guidance and help offered by so many institutions like CONSORT, COPE or the EQUATOR Network. Probably, these offers are still not well-known enough among clinical investigators to be routinely used. It is essential that senior scientists advise young research fellows about the available guidelines and checklists and help them to understand their methodological and ethical background. Obviously, ethical research and good reporting is a question of scientific education. Here, we have identified potential for improvement, which seems to be a condition sine qua non if the impact of economic interests on clinical trials in minors continues to increase as a consequence of the regulatory demands. Therefore, full transparency to the public, e.g., publicly available research protocols on the journals’ websites, and extensive ethical debate within the scientific community and in journals, but also including trial participants/parents and the public as well as non-profit organizations funding medical research and the pharmaceutical industry are required. Especially when participation in clinical trials becomes a precondition for access to new health care options or new drugs, voluntariness becomes critical. It is an unanswered ethical question if the principles of bioethics respect for autonomy and justice are still sufficiently fulfilled under this condition.
With regard to the work of clinical ethics committees, our study results may encourage them to randomly check forms on clinical trials submitted for ethical approval (ex ante perspective) with the clinical trial report and finally the publication (ex post perspective) for consistency. Blümle et al. [50] conducted a cohort study comparing the reporting of eligibility criteria in RCTs in trial protocols submitted to the ethics committee of a German medical faculty and the subsequent publication. They found differences for all of the submitted n = 52 trials. However, it is left to discussion whether the clinical ethics committee is the best authority for this ex ante/ex post comparison as ethics committees should not turn into an ethics police. In everyone’s interest, new policies leading to more bureaucracy should be avoided. To increase transparency, it seems more efficient e.g. for ethics committees, journals and editors first to enforce policies that already exist. If necessary, these policies could be gradually extended in a second step. A more in-depth analysis should be left to further discussion as this article is primarily addressed not to ethics committees, journals and editors, but to the clinical investigator himor herself. Giving an impetus for these scientists to reflect more critically on the ethical aspects of their work—that was the intended purpose of our study. No more and no less.
5. CONCLUSION
The ethical responsibility of the investigator does not end with the last patient leaving the trial. Instead, it extends much further and includes the responsibility 1) to publish the data obtained in a timely fashion (even if the study results are negative or in any other way fail to support the research hypothesis, and although the publication process may be time consuming) as a basis for further research and 2) to report all relevant issues truly, clearly and in a consistent manner (concerning scientific background, methods, results, adverse events and safety monitoring, and including a reflection on ethical aspects). Sound publications that meet these quality criteria will increase the likelihood of a fruitful research process in that they realize what most investigators and physicians probably wish for today, i.e., to maximize the benefit from clinical trials with minimal risks, for the sake of all participants today and in the future—and especially for minors.
6. ACKNOWLEDGEMENTS
This study was part of the cooperative project Risks and benefits in clinical trials in humans: ethical, legal, and clinical studies granted by the federal Ministry of Research and Technology, Germany, and supported by the Cancer Kids and Families Support Group (Verein zur Förderung krebskranker Kinder Münster e.V.), Rishon-Le-Zion-Ring 26, 48149 Muenster, Germany; grant number: 01GP0614.
We would like to thank Gabriele Braun-Munzinger for editing the manuscript.
REFERENCES
- Commission of the European Communities (2006) Regulation (EC) No 1901/2006 on medicinal products for paediatric use and amending Regulation (EC) No 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC) No 726/2004. Official Journal of the European Communities, L378, 1-19.
- European Medicines Agency (2011) Report to the European Commission on companies and products that have benefited from any of the rewards and incentives in the Paediatric Regulation and on the companies that have failed to comply with any of the obligations in this Regulation, covering the year 2010. EMA/163613/2011, 3 May 2011, Human Medicines Development and Evalution.
- European Medicines Agency (2010) Reflection paper on the strategy for paediatric onco-logy early authorization and availability. Review, proposal and stakeholders’ views. Draft. EMA/410778/2010, 3 October 2010, Human Medicines Development and Evalution.
- Gemeinsamer Bundesausschuss (GBA) (2009) Agreement of the Federal Joint Committee on quality assurance measures in the inpatient care of children and adolescents with hemato-oncological diseases according to §137 Part 1 Sentence 3 No. 2 SGB V for hospitals qualified according to § 108 SGB V, dated 16 May 2006, taking effect on 1 Jan 2007, last modified on 18 Dec 2008, taking effect on 1 Jan 2009. http://www.g-ba.de/informationen/richtlinien/47/
- World Medical Association (2008) Declaration of Helsinki. http://www.wma.net/en/30publications/10policies/b3/index.html
- Drazen, J.M. (2012) Transparency for clinical trials—The TEST act. The New England Journal of Medicine, 367, 863-864. doi:10.1056/NEJMe1209433
- Eichler, H.-G., Abadie, E., Breckenridge, A., Leufkens, H. and Rasi, G. (2012) Open clinical trial data for all? A view from regulators. PLOS Medicine, 9, e1001202. doi:10.1371/journal.pmed.1001202
- Pignatti F. European Medicines Agency (2013) Proactive publication of clinical trial data—Feedback from the EMA workshop. http://www.ema.europa.eu/docs/en_GB/document_library/Presentation/2013/05/WC500143654.pdf
- Schulz, K.F. (1998) Randomized controlled trials. Clinical Obstetrics and Gynecology, 41, 245-56.
- Hoffmann, M. (2009) Definitions and clarifications: An Introduction. In: Boos, J., Merkel, R., Raspe, H. and Schöne Seifert, B. Eds., Risks and Benefits in Clinical Trials in Humans: Ethical, Legal and Clinical Studies, Deutscher Ärzte-Verlag, Köln, 1-12.
- Fried, C. (1974) Medical experimentation: Personal integrity and social policy. North Holland Publishing, Amsterdam.
- Emanuel, E.J., Wendler, D. and Grady, C. (2000) What makes clinical research ethical? JAMA, 283, 2701-2711. doi:10.1001/jama.283.20.2701
- Miller, F.G. and Brody, H. (2002) What makes placebo-controlled trials unethical? The American Journal of Bioethics, 2, 3-9. doi:10.1162/152651602317533523
- Miller, F.G. and Brody, H. (2003) A critique of clinical equipoise. Therapeutic miscon-ception in the ethics of clinical trials. The Hastings Center Report, 33, 19-28. doi:10.2307/3528434
- Miller, F.G. and Brody, H. (2007) Clinical equipoise and the incoherence of research ethics. The Journal of Medicine and Philosophy, 32, 151-165. doi:10.1080/03605310701255750
- Veatch, R.M. (2002) Indifference of subjects: An alternative to equipoise in randomized clinical trials. Social Philosophy and Policy, 19, 295-323. doi:10.1017/S0265052502192120
- Veatch, R.M. (2007) The irrelevance of equipoise. The Journal of Medicine and Philosophy, 32, 167-183. doi:10.1080/03605310701255776
- Raspe, H. and Hüppe, A. (2009) Analysis and considerations of risks and benefits in clinical research. In: Boos, J., Merkel, R., Raspe, H. and Schöne-Seifert, B. Eds., Risks and Benefits in Clinical Trials in Humans: Ethical, Legal and Clinical Studies, Deutscher Ärzte-Verlag, Köln, 13- 52.
- Murray, G.D. (2000) Promoting good research practice. Statistical Methods in Medical Research, 9, 17-24. doi:10.1191/096228000670253839
- Schulz, K.F., Altman, D.G. and Moher, D. (CONSORT Group) (2010) CONSORT 2010 statement: Updated guidelines for reporting parallel group randomised trials. British Medical Journal, 340: c332.
- Begg, C., Cho, M., Eastwood, S., et al. (1996) Improving the quality of reporting of randomized controlled trials. The CONSORT statement. JAMA, 276, 637-639. doi:10.1001/jama.1996.03540080059030
- CONSORT (2011) CONSORT endorsers—Journals. http://www.consort-statement.org/about-consort/consort-endorsement/consort-endorsers---journals/
- Windeler, J. and Koch, A. (2000) What a good scientific publication has to cover. Medizinische Klinik (Munich), 95, 171-177. doi:10.1007/PL00002102
- Saint-Raymond, A., Hill, S., Martines, J., Bahl, R., Fonaine, O. and Bero, L. (2010) CONSORT 2010. The Lancet, 376, 229-230. doi:10.1016/S0140-6736(10)61134-8
- Falagas, M.E., Grigori, T. and Ioannidou, E. (2009) A systematic review of trends in the methodological quality of randomized controlled trials in various research fields. Journal of Clinical Epidemiology, 62, 227-231.
- Levin, L.A. and Palmer, J. G. (2007) Institutional review boards should require clinical trial registration. Archives of Internal Medicine, 167, 1576-1580. doi:10.1001/archinte.167.15.1576
- Antes, G. and Chalmers, I. (2003) Under-reporting of clinical trials is unethical. Lancet, 361, 978-979. doi:10.1016/S0140-6736(03)12838-3
- McHenry, L. (2006) Ethical issues in psychopharmacolgy. Journal of Medical Ethics, 32, 405-410. doi:10.1136/jme.2005.013185
- Beauchamp, T.L. and Childress, J.F. (1994) Principles of biomedical ethics. 4th Edition, Oxford University Press, New York.
- Rothenberger, L.G., Henschel, A.D., Schrey D., Becker, A. and Boos, J. (2011) Metho-dological and ethical aspects of randomized controlled clinical trials in minors with malignant diseases. Pediatric Blood & Cancer, 57, 599-605. doi:10.1002/pbc.23171
- Higgins, J.P.T. & Green, S. (2009) Cochrane handbook for systematic reviews of interventions version 5.0.2 [updated September 2009]. The Cochrane Collabortion. www.cochrane-handbook.org
- Altman, D.G., Schulz, K.F., Moher, D., Egger, M., Davidoff, F., Elbourne, D., Gøtzsche, P. C. and Lang, T. (CONSORT Group) (Consolidated Standards of Reporting Trials) (2001) The revised CONSORT statement for reporting randomized trials: Explanation and elaboration. Annals of Internal Medicine, 134, 663-694. doi:10.7326/0003-4819-134-8-200104170-00012
- Scottish Intercollegiate Guidelines Network (SIGN) (2004a) Methodology Checklist 2: Randomised controlled trials. http://www.sign.ac.uk/guidelines/fulltext/50/checklist2.html
- Scottish Intercollegiate Guidelines Network (SIGN) (2004b) Notes on the use of Methodology Checklist 2: Randomised controlled trials. http://www.sign.ac.uk/guidelines/fulltext/50/notes2.html
- Fleiss, J.L. (1981) The measurement of interrater agreement. In: Statistical Methods for Rates and Proportions, 2nd Edition, John Wiley, NewYork, 212-236.
- Cohen, J. (1960) A coefficient of agreement for nominal scales. Educational and Psychological Measurement, 20, 37-46. doi:10.1177/001316446002000104
- Hopewell, S., Dutton, S., Yu, L.M., Chan, A.W. and Altman, D.G. (2010) The quality of reports of randomised trials in 2000 and 2006: Comparative study of articles indexed in PubMed. British Medical Journal, 340, c723.
- Schulz, K.F., Chalmers, I., Grimes, D.A. and Altman, D.G. (1994) Assessing the quality of randomization from reports of controlled trials published in obstetrics and gynecology journals. The Journal of the American Medical Association, 272, 125-128. doi:10.1001/jama.1994.03520020051014
- Schulz, K.F. (1995) Subverting randomization in controlled trials. The Journal of the American Medical Association, 274, 1456-1458. doi:10.1001/jama.1995.03530180050029
- Wood, L., Egger, M., Gluud, L.L., Schulz, K.F., Jüni. P., Altman. D.G., Gluud. C., Martin, R.M., Wood, A.J. and Sterne, J.A. (2008) Empirical evidence of bias in treatment effect estimates in controlled trials with different interventions and outcomes: Meta-epidemiological study. British Medical Journal, 336, 601-605. doi:10.1136/bmj.39465.451748.AD
- Green, S. and Higgins, J. (2005) Glossary. Cochrane handbook for systematic reviews of interventions version 4.2.5 [updated May 2005]. http://www.cochrane.org/resources/handbook/
- Noseworthy, J.H., Ebers, G.C., Vandervoort, M.K., Farquhar, R.E., Yetisir, E. and Roberts, R. (1994) The impact of blinding on the results of a randomized, placebo-controlled multiple sclerosis clinical trial. Neurology, 44, 16-20. doi:10.1212/WNL.44.1.16
- US Department of Health & Human Services (1979) The Belmont Report. http://www.hhs.gov/ohrp/humansubjects/guidance/belmont.html
- European Union (2008) Ethical considerations for clinical trials on medicinal products conducted with the paediatric population. European Journal of Health Law, 15, 223- 250.
- International Committee of Medical Journal Editors (ICMJE) (2010) Uniform requirements for manuscripts submitted to biomedical journals: Writing and editing for biomedical publication. http://www.icmje.org/urm_full.pdf
- Tukey, J.W. (1977) Some thoughts on clinical trials, especially problems of multiplicity. Science, 198, 679-684. doi:10.1126/science.333584
- NIH (2011) National Institute of Health (NIH)-Cancer Therapy Evaluation Program. Common Terminology Criteria for Adverse Events (CTCAE) and Common Toxicity Criteria (CTC). http://ctep.cancer.gov/protocolDevelopment/electronic_applications/ctc.htm
- Landis, J.R. and Koch, G.G. (1977) The measurement of observer agreement for cate-gorical data. Biometrics, 33, 159-174. doi:10.2307/2529310
- Meerpohl, J.J., Wolff, R.F., Antes, G. and von Elm, E. (2011) Are pediatric open access journals promoting good publication practice? An analysis of author instructtions. BioMed Central Pediatrics, 11, 27. http://www.biomedcentral.com/1471-2431/11/27
- Blümle, A., Meerpohl, J.J., Rücker, G., Antes, G., Schumacher, M. and von Elm, E. (2011) Reporting of eligibility criteria of randomised trials: Cohort study comparing trial protocols with subsequent articles. British Medical Journal, 342, d1828.
ABBREVIATIONS (ALPHABETICAL ORDER)
EbM Evidence-based Medicine EU European Union CONSORT Consolidated Standards of Reporting Trials COPE Committee on Publication Ethics DSMB Data and Safety Monitoring Board EMBASE Excerpta Medica Database EQUATOR Enhancing the Quality and Transparency of Health Research ICMJE International Committee of Medical Journal Editors MEDLINE Online Database of the US National Library of Medicine MeSH Medical Subject Headings NIH US National Institute of Health PDAT Publication Date RCT Randomized controlled clinical trial SIGN Scottish Intercollegiate Guidelines Network
NOTES
*Grant sponsor: Federal Ministry of Research and Technology; Grant Cancer Kids and Families Support Group Verein zur Förderung krebskranker Kinder Münster e.V.; grant number: 01GP0614.
Conflict of interest: Nothing to declare.
#Equal authorship of A.D. Henschel and L.G. Rothenberger.
†Formerly Department of Pediatric Hematology and Oncology, University Children’s Hospital, Muenster, Germany.
‡Formerly Department of Pediatric Hematology and Oncology, University Children’s Hospital, Muenster, Germany.
§Corresponding author.