Food and Nutrition Sciences
Vol. 2 No. 4 (2011) , Article ID: 5446 , 10 pages DOI:10.4236/fns.2011.24051
Comparison of the Ability of Myricetin and Quercetin to Modulate the Oxidative DNA Damage Induced by Heterocyclic Amines
![]()
1Department of Food Nutrition and Technology, Veterinarian Faculty, Complutense University, Madrid, Spain; 2Department of Medicine, Medicine Faculty, Complutense University, Madrid, Spain.
Email: pmorales@vet.ucm.es
Received July 3rd, 2010; revised August 19th, 2010; accepted September 20th, 2010.
Keywords: Myricetin, Quercetin, Comet Assay, Oxidative DNA Damage, Heterocyclic Amines
ABSTRACT
The aim of the present study was to compare the ability of the myricetin and quercetin to modulate the oxidative DNA damage induced by 2-amino-3, 8-dimethylimidazo [4,5-f] quinoxaline (8-MeIQx), 2-amino-3, 4, 8-trimethylimidazo [4, 5-f]-quinoxaline (4,8-diMeIQx) and 2-amino-1-methyl-6-phenyl-imidazo [4,5-b] pyridine (PhIP), in human hepatoma cells. DNA damage (strand breaks and oxidized purines/pyrimidines) was evaluated by the alkaline single-cell gel electrophoresis or comet assay. None of the myricetin and quercetin concentrations tested protected against 8-MeIQx, 4, 8-diMeIQx and PhIP-induced DNA strand breaks. The oxidized pyrimidines induced by 4, 8-diMeIQx and PhIP were reduced by myricetin but not by quercetin. Quercetin reduced the oxidized purines induced by 8-MeIQx and PhIP, while myricetin also reduced the induced by 4, 8-diMeIQx. One feasible mechanism by which myricetin and quercetin exert their protective effect towards HCAs-induced oxidative DNA could be related in part to the reduction of human CYP1A1. Another mechanism claimed to be responsible for the protective effect of myricetin and quercetin is the induction of phase II metabolizing enzymes such as UDP-glucuronyltrasferase (UGT). The ethoxyresorufin O-deethylation (CYP1A1) activity was moderately inhibited by myricetin, while little effect was observed by quercetin. On the contrary, quercetin showed the greatest increase on UDP-glucuronyltransferase activity. However, these are not the only mechanisms by which myricetin and quercetin exert their protective effect, other mechanisms such as stimulation of the repair of carcinogen-induced DNA damage and or the free radical scavenging efficiency have been also implicated. In conclusion, our results clearly indicate that myricetin was more efficient than quercetin to prevent DNA damage (oxidized purines and pyrimidines) induced by the three HCAs evaluated. This protective effect depends on the chemical structure of flavonoid and the mutagen studied.
1. Introduction
Strong and consistent epidemiologic evidence suggests a diet that includes regular consumption of fruits and vegetables, significantly reduces the risk of many cancers [1]. Flavonoids are polyphenolic compounds, ubiquitous in plants, and found in significant quantities in vegetables, fruits, seeds, nuts and beverages such as tea and wine. Myricetin occurs in many plant-derived foodstuffs, notably tea, grapes and cranberries, while quercetin is found mainly in onions, apples and tea [2]. These flavonoids constitute an important group of phytochemicals that gained increased research attention since it was found that they could exert anticarcinogenic, antimutagenic, antiinflammatory, and antiviral actions [3]. The metabolism of flavonoids is also mediated by oxidation through cytochrome P450 (CYP) enzymes and conjugation through glucuronidation, sulfation and methylation [4]. Flavonoids are widely recognized as naturally occurring antioxidants, and they are suggested to act as chemopreventive compounds against tumors and cancer [5,6].
Evidence from epidemiologic and animal studies indicates that several cancers may be related to heterocyclic amines (HCAs) intake [7]. The evidence is stronger for colorectal, breast, pancreas, prostate, lung, larynx, liver, and renal cancers as well as lymphomas [8,9]. To date, more than 20 different HCAs have been identified in cooked foods [7] and they can be classified into twomain groups called carbolines and aminoimidazoazaarenes (AIAs) [10]. AIAs are formed at the normal cooking temperatures of 100˚C - 225˚C while heating foodstuffs in the presence of creatinine, aminoacids and sugar involving Maillard reaction [11]. Among those, 2-amino-3, 8-dimethylimidazo [4,5-f] quinoxaline(8-MeIx)2-amino-3, 4, 8-trimethylimidazo [4,5-f]-quinoxaline (4,8-diMeIQx) and 2-amino-1-methyl-6-phenyl-imidazo [4,5-b] pyridine (PhIP) are the most abundant [12]. They were categorized by the International Agency for Research on Cancer (IARC) in Group 2B: possibly carcinogenic to humans [13]. The AIAs require metabolic activation in order to exert its carcinogenicity [9]. HCAs are metabolized mainly by cytochrome P450 1A1 [14] to an N–OH intermediate and further metabolised by a series of conjugating enzymes to further activating or detoxifying, depending on the specific compound [15]. These reactive enzymatic products presumably act as a reactive free radicals that bind almost exclusively to the C-8 of guanine, causing DNA adducts, mutations, chromosomal abnormalities, and cancer [9,16]. Thus, the study of modulating species to control HCAs activity is an important area of interest [17].
The single cell gel electrophoresis (SCGE) or Comet assay has been established as a suitable method for assesing the ability of phytochemicals to protect cells against genotoxic effect of several xenobiotics [18]. In this study, we used the Comet assay with the lesion-specific enzymes endonuclease III (Endo III) and formamidopyrimidine DNA glycosylase (Fpg) that provide information on endogenous formation of oxidized pyrimidines and purines respectively. The human hepatoma cell line (HepG2) was used, since HepG2 cells are an excellent tool to detect genotoxic properties of environmental and dietary chemicals [19]. However, the levels of most phase I enzymes are low in HepG2 cells [20]. As a consequence, in this study microsomes from baculovirus infected cells expressing human CYP1A1 were used to evaluate one feasible mechanism by which flavonoids exerted its possible protective effects. In addition, microsomes from baculovirus-infected cells expressing human UDP-glucuronyltransferase (UGT1A4) have been also used.
The study of the reactions of flavonoids constituents of the diet with HCAs is an area of great promise in cancer preventive strategies. Thus, the aim of this study was to compare the ability of the myricetin and quercetin to modulate the oxidative DNA damage induced by HCAs in human hepatoma cells, using the single-cell gel electrophoresis (SCGE) assay.
2. Material and Methods
2.1. Chemicals
Quercetin, myricetin, dimethyl sulfoxide (DMSO) and low melting point agarose (LMP) were purchased from Sigma-Aldrich (St. Louis, MO). 2-amino-3, 8-dimethyl imidazo [4,5-f] quinoxaline (8-MeIQx), 2-amino-3,4,8- trimethyl -3H-imidazo [4,5-f] quinoxaline (4,8-diMeIQx) and 2-amino-1-methyl-6-phenylimidazo [4,5-b] pyridine (PhIP) (Figure 1) were purchased from Toronto Research Chemicals Inc. (North York, On. Canada). Formamidopyrimidine-DNA glycosylase (Fpg) and endonuclease III (Endo III) were obtained from Trevigen Inc. (Gaithersburg, MD). All other chemicals and solvents were of the highest grade commercially available. HCAs and flavonoids were dissolved in sterile DMSO. The stock solutions were stored deep frozen (–80˚C).
2.2. HepG2 cells
Human hepatocellular carcinoma (HepG2) cells were purchased from Biology Investigation Center Collection (BIC, Madrid, Spain). Only cells of passage 10 - 17 were used in the experiments. Cells were cultured as monolayer in Dulbecco’s Modified Eagle Medium supplemented with 10% v/v heat inactivated fetal calf serum, 50 U/ml penicillin and 50 g/ml streptomycin and 1% v/v L-glutamine. Culture medium and supplements required for the growth of the cells were purchased from Gibco Laboratories (Life Technologies, Inc., Gaithersburg, MD 20884-9980). Cell cultures were incubated at 37˚C and 100% humidity in a 5% CO2 atmosphere.
2.3. Human Microsomes
Microsomes from baculovirus-infected insect cells expressing human CYP1A1 + cytochrome b5 and UGT1A4 were obtained from Gentest (Woburn, MA).
2.4. Analysis of DNA Damage Induced by a Simultaneous Treatment of HCAs and Myricetin or Quercetin in the Alkaline Comet
Throughout the genotoxicity studies, viability was determined by the MTT (3-(4, 5-dimethylthiazo-2-yl)-2, 5-diphenyltetrazolium bromide) assay in order to select non-toxic concentrations of chemicals and only cultures with a cell viability of more than 80% were used for analysis [21]. The SCGE assay was carried out according to the protocol of Olive et al. [22] with minor modifications. Induction of DNA damage (strand breaks and oxidative DNA damage) by myricetin and quercetin [21] and by 2-amino-3, 8-dimethylimidazo [4,5-f] quinoxaline (8-MeIQx), 2-amino-3, 4, 8-trimethyl-3H-imidazo [4,5-f] quinoxaline (4, 8-diMeIQx) and 2-amino-ethyl-6-phenylimidazo [4,5-b] pyridine (PhIP) has been previously evaluated [23].
Briefly, HepG2 cells were plated on to multiwell systems at a density of 1.5 × 105 cells/ml culture medium.

Figure 1. Chemical structures of heterocyclic amines used in this study; (a) 2-amino-3, 8-dimethylimidazo [4,5-f] quinoxaline (8-MeIQx), (b) 2-amino-3, 4, 8- trimethyl-3Himidazo [4,5-f] quinoxaline (4, 8-di MeIQx) and (c) 2-amino-1- methyl-6-phenylimidazo [4,5-b] pyridine (PhIP).
24 h after seeding, cells were exposed to myricetin (0.1 – 5 M), or quercetin (0.1 - 5 M) or solvent, for another 24 h at 37˚C and 5% CO2. The solvent concentration in the incubation medium never exceeds 0.1%. After incubation, cells were simultaneously treated with: (1) 8-MeIQx (500 M) and myricetin (0.1 - 5 M) or quercetin (0.1 - 5 M) or (2) 4, 8-diMeIQx (200 M) and myricetin (0.1 - 5 M) or quercetin (0.1 - 5 M) or (3) PhIP (300 M) and myricetin (0.1 - 5 M) or quercetin (0.1 - 5 M). Then plates were incubated for another 24 hr at 37˚C and 5% CO2. After treatments, 10 l of a suspension 1 × 105 cells were mixed with 70 l of LMP agarose type VII (0.75% concentration in PBS), distributed on slides that had been pre-coated with LMP agarose type VII (0.30% concentration in PBS), and left to set on an ice tray. Three slides were prepared for each concentration of the compound tested, one slide for control and the others slides to be treated with Fpg or Endo III. After solidification, cells were lysed in darkness for 1 hour in a high salt alkaline buffer (2.5M NaCl, 0.1 M EDTA, 0.01 M Tris, 1% Triton X- 100, pH 10). The slides were then equilibrated 3 × 5 minutes in enzyme buffer (0.04 M HEPES, 0.1 M KCl, 0.5 mM EDTA, 0.2 mg/ml BSA, pH 8). After this time, slides were incubated with 30 l of Fpg or Endo III at 1 g/ml in enzyme buffer for 30 min at 37˚C in a humid dark chamber. Control slides were incubated with 30 l of enzyme buffer only. Following enzyme treatment, the slides were placed in electrophoresis buffer (0.3 M NaOH, 1 mM EDTA, pH 13, cooled in a refrigerator) in darkness for 40 min. Electrophoresis was performed in a cold-storage room, in darkness, in a Bio-Rad subcell GT unit containing the same buffer, for 30 min at 25 V. After electrophoresis, the slides were neutralized using 0.4 M Tris pH 7.5 and fixed in methanol. Subsequently, the DNA was stained with ethidium bromide (10 g/ml) in Tris acetate EDTA (TAE 1X) during 5 minutes and examined in a fluorescence microscope (OLYMPUS BH-2) connected to a computerized image analysis system (Comet ScoreTM 1.0). Olive tail moment (OTM), as defined by Olive et al. [22], was determined and expressed as arbitrary units (AU). OTM = I × L, where I is the fractional amount of DNA in the comet tail (%DNA in the tail) and L is the distance from the centre of the comet head to the centre of tail distribution.
2.5. Ethoxyresorufin O-Deethylation Assay
Deethylation of ethoxyresorufin to resorufin was detected with fluorescence spectroscopy according to the standard ethoxyresorufin O-deethylation (EROD) assay for CYP1A1 activity [24]. A 2 ml reaction mixture containing 2.5 pmol CYP1A1, 1.3 mM NADP+, 3.3 mM glucose-6-phosphate, 0.4 IU/ml glucose-6-phosphate dehydrogenase, 3.3 mM magnesium chloride and 1 g/ml ethoxy-resorufin in 100 mM potassium phosphate (pH 7.4) was incubated at 37˚C for 30 min. For inhibition study, these assays were evaluated with myricetin and quercetin concentrations of (0.1 - 5 M) used in the comet assay. After incubation, the fluorescence was determined with excitation at 550 nm and emission at 586 nm in a spectrofluorometer. The activity was quantified comparing to a standard curve for resorufin. The amount of resorufin formed was determined from a calibration curve containing known amounts of this compound. α-Naphthofla vone (10 M, 23% ethoxyresorufin O-deethylation activity) was shown to be a potent specific inhibitor of ethoxyresorufin O-deethy-lation [25].
2.6. 4-Methylumbellipherone Glucuronidation Assay
The equations UGT1A4 activity can be determined with different substrates and, among them 4-methyl umbellipherone is the most sensitive and can be used to quantify glucuronidation in vitro. The assay is based on the different fluorescence properties showed by 4-methylumbellipherone and its conjugate 4-methylumbellipheryl-β, D-glucuronide [26]. A sample 100 g of human UGT1A4 is incubated at 37˚C in 0.5 ml of 0.1 M Tris–HCl buffer (pH 7.4) containing 5 mol MgCl2 and 0.5 mol 4-methylumbellipherone. For glucuronidation studies, these assays were evaluated with the concen-trations of myricetin and quercetin (0.1 - 5 M) used by the comet assay. The reaction is started by the addition of 1.5 mol UDP-glucuronic acid and stopped 10 min after by addition of 0.5 ml of 0.5 M perchloric acid. Unreacted substrate is extracted with 2 ml of chloroform. After centrifugation at 1000 × g for 10 min, 0.5 ml of the upper water phase containing the glucoronide is transferred to another tube and 0.5 ml of 1.6 M glycine/NaOH (pH 10.3) is added. Fluorescence is measured at 368 nm excitation and 456 nm emissions. The amount of 4-methylumbellipheryl-β, D- -glucuronide formed was determined from a calibration curve containing known amounts of this compound.
Incubations without myricetin or quercetin were considered as negative controls (100% enzyme activity). Incubations with myricetin or quercetin relative to the negative control were calculated and expressed as percentage of enzyme activity (% residual activity) = A1/A0 × 100, where A1 is the absorbance of incubations with flavonoids and A0 is the absorbance of negative control.
2.7. Statistical Analysis
Images of 50 randomly selected cells per concentration were evaluated and the test was carried out three times. The reported OTM is the mean ± standard deviation (S.D.) of three independent experiments. Thus, we compare three mean of OTM from three independent experiments. Cultures without HCAs or flavonoids were considered as negative controls. In all experiments the following negative controls have been included: cells treated with solvents and treated without enzymes, cells incubated with Endo III and cells incubated with Fpg. Induction of DNA damage by HCAs was defined as 100% of genotoxicity. The Student’s t-test was used for statistical comparison between simultaneous treatments and controls, and differences were considered significant at p ≤ 0.05.
3. Results
3.1. DNA Damage (Strand Breaks and Oxidized Purines/Pyrimidines) Induced by Simultaneous Treatment of 8-MeIQx, 4,8-di MeIQx or PhIP and Myricetin or Quercetin in the Alkaline Comet Assay
Human Induction of DNA damage (strand breaks and oxidative DNA damage) by myricetin and quercetin [21] or by HCAs has been previously evaluated [23]. Results indicated that none of the myricetin (0.1 - 5 M) and quercetin (0.1 - 5 M) concentrations tested in presence or absence of Fpg or Endo III, caused DNA damage per se. The concentrations of 8-MeIQx, 4, 8-diMeIQx and PhIP required to cause a significant increase in DNA damage were 500 M, 200 M and 300 M respectively. For this reason these concentrations range was used in subsequent studies.
The effect of myricetin or quercetin on 8-MeIQx, -induced DNA damage in HepG2 cells is shown in Figure 2. Myricetin or quercetin did not protect HepG2 cells

Figure 2. Effect of myricetin and quercetin on 8-MeIQx-induced DNA damage in human HepG2 cells. OTM median values in control cells without enzymes and incubated with Endo III or Fpg were 1.71 ± 0.22 AU, 1.96 ± 0.32 AU, 2.09 ± 0.20 AU, respectively. (a) ( ) Cells treated with 8-MeIQx and incubated without enzymes. (
) Cells treated with 8-MeIQx and incubated without enzymes. ( ) Cells treated with 8-MeIQx and myricetin or quercetin incubated without enzymes. (b) (
) Cells treated with 8-MeIQx and myricetin or quercetin incubated without enzymes. (b) ( ) Cells treated with 8-MeIQx and incubated with Endo III. (
) Cells treated with 8-MeIQx and incubated with Endo III. ( ) Cells treated with 8-MeIQx and and myricetin or quercetin and incubated with Endo III. (c) (
) Cells treated with 8-MeIQx and and myricetin or quercetin and incubated with Endo III. (c) ( ) Cells treated with 8-MeIQx and incubated with Fpg. (
) Cells treated with 8-MeIQx and incubated with Fpg. ( ) Cells treated with 8-MeIQx and myricetin or quercetin and incubated with Fpg. Asterisks indicate significant difference from control *P ≤ 0.05.
) Cells treated with 8-MeIQx and myricetin or quercetin and incubated with Fpg. Asterisks indicate significant difference from control *P ≤ 0.05.
towards 8-MeIQx -induced DNA strand breaks and the formation of Endo sensitive sites (Figures 2(a) and 2(b)). However, myricetin (0.1 - 5 M) or quercetin (0.1 - 1 M) reduced the formation of Fpg sensitive sites (Figure 2(c)) induced by 8-MeIQx (26.2% - 24.2% and 32% - 26.7%, respectively).
Figure 3 shows the effect of myricetin or quercetin on 4,8-diMeIQx-induced DNA damage in HepG2 cells. Myricetin did not protect HepG2 cells towards 4,8-diMeIQx-induced DNA strand breaks. On the contrary myricetin increased the genotoxic effect caused by 4,8-diMeIQx (Figure 3(a)). The formation of Endo III (17.3%) and Fpg (27.2%) sensitive sites induced by 4,8-diMeIQx (Figures 3(b) and 3(c)) was only reduced at the lowest concentration of myricetin (0.1 M). None of the quercetin concentrations tested reduced the DNA strand breaks and the formation of Endo III and Fpg sensitive sites induced by 4,8-diMeIQx (Figures 3(a), 3(b) and 3(c)).
The protective effect of myricetin or quercetin on PhIPinduced DNA damage is shown in Figure 4. Myricetin did not protect HepG2 cells towards PhIP-induced DNA strand breaks. The formation of Endo III (23.4%) and Fpg sensitive sites (34.6% - 18.4%) induced by PhIP was prevented by myricetin at all concentrations tested (0.1 – 5 M). However, quercetin (0.1 - 5 M) only reduced the formation of Fpg sensitive sites (29.6% - 41.9%).
3.2. Determination of Enzyme Activity
The effect of quercetin on ethoxyresorufin O-deethylation (CYP1A1) activity is shown in Figure 5. The results revealed that CYP1A1 activity decreases with increasing concentrations of myricetin and quercetin. The maximum inhibition of ethoxyresorufin activity was observed at 5 M myricetin and querctin (36.6 and 21.5%, respectively).
Finally, Figure 6 shows the effect of myricetin or quercetin on UDP-glucuronyltransferase (UGT1A4) activity. UGT1A4 activity increased with increasing concentrations of quercetin (1 - 5 M, 36 - 65%, Figure 6).
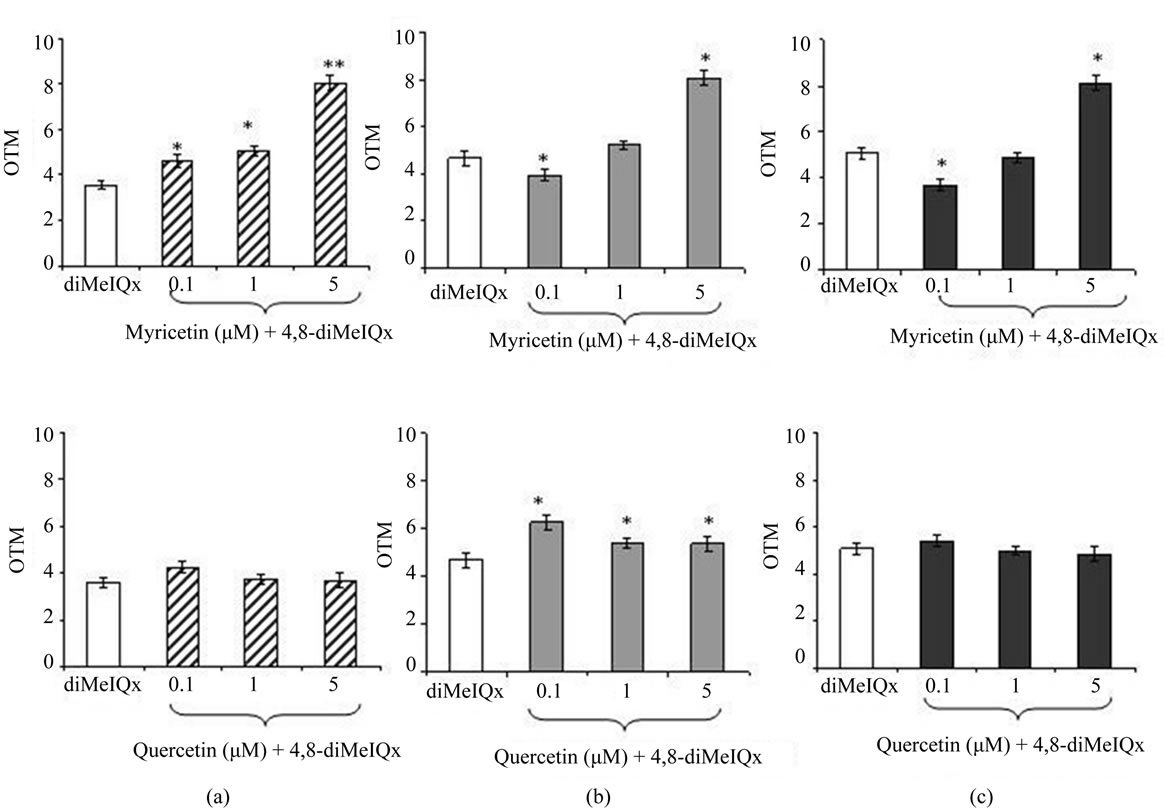
Figure 3. Effect of myricetin and quercetin on 4,8-diMeIQx-induced DNA damage in human HepG2 cells. OTM median values in control cells without enzymes and incubated with Endo III or Fpg were 1.71 ± 0.22 AU, 1.96 ± 0.32 AU, 2.09 ± 0.20 AU, respectively. (a) ( ) Cells treated with 4,8-diMeIQx and incubated without enzymes. (
) Cells treated with 4,8-diMeIQx and incubated without enzymes. ( ) Cells treated with 4,8-diMeIQx and myricetin or quercetin incubated without enzymes. (b) (
) Cells treated with 4,8-diMeIQx and myricetin or quercetin incubated without enzymes. (b) ( ) Cells treated with 4,8-diMeIQx and incubated with Endo III. (
) Cells treated with 4,8-diMeIQx and incubated with Endo III. ( ) Cells treated with 4,8-diMeIQx and and myricetin or quercetin and incubated with Endo III. (c) (
) Cells treated with 4,8-diMeIQx and and myricetin or quercetin and incubated with Endo III. (c) ( ) Cells treated with 4,8-diMeIQx and incubated with Fpg. (
) Cells treated with 4,8-diMeIQx and incubated with Fpg. ( ) Cells treated with 4,8-diMeIQx and myricetin or quercetin and incubated with Fpg. Asterisks indicate significant difference from control *P ≤ 0.05.
) Cells treated with 4,8-diMeIQx and myricetin or quercetin and incubated with Fpg. Asterisks indicate significant difference from control *P ≤ 0.05.

Figure 4. Effect of myricetin and quercetin on PhIP-induced DNA damage in human HepG2 cells. OTM median values in control cells without enzymes and incubated with Endo III or Fpg were 1.71 ± 0.22 AU, 1.96 ± 0.32 AU, 2.09 ± 0.20 AU, respectively. (a) ( ) Cells treated with PhIP and incubated without enzymes. (
) Cells treated with PhIP and incubated without enzymes. ( ) Cells treated with PhIP and myricetin or quercetin incubated without enzymes. (b) (
) Cells treated with PhIP and myricetin or quercetin incubated without enzymes. (b) ( ) Cells treated with PhIP and incubated with Endo III. (
) Cells treated with PhIP and incubated with Endo III. ( ) Cells treated with PhIP and myricetin or quercetin and incubated with Endo III. (c) (
) Cells treated with PhIP and myricetin or quercetin and incubated with Endo III. (c) ( ) Cells treated with PhIP and incubated with Fpg. (
) Cells treated with PhIP and incubated with Fpg. ( ) Cells treated with PhIP and myricetin or quercetin and incubated with Fpg. Asterisks indicate significant difference from control *P ≤ 0.05.
) Cells treated with PhIP and myricetin or quercetin and incubated with Fpg. Asterisks indicate significant difference from control *P ≤ 0.05.
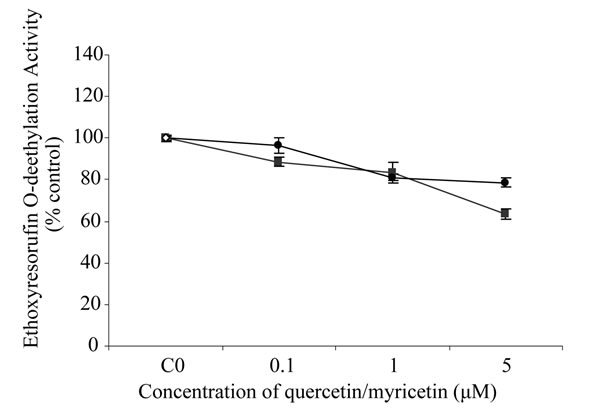
Figure 5. Effect of (●) quercetin and (■) myricetin, on ethoxyresorufin O-deethylation activity (CYP1A1). Values are mean of three samples ± SD and are expressed relative to the control (without inhibition). Asteriks indicate significant difference from control **P ≤ 0.01, *P ≤ 0.05.
However, myricetin at all concentrations tested, did not increased the UDP-glucuronyltransferase activity.

Figure 6. Effect of (●) quercetin and (■) myricetin, on UDPglucurony-ltransferase (UGT1A4) activity. Values are mean of three samples ± SD and are expressed relative to the control (without inhibition). Asteriks indicate significant difference from control **P ≤ 0.01, *P ≤ 0.05.
4. Discussion
The aim of the present study was to compare the ability of the myricetin and quercetin to modulate the oxidative DNA damage induced by HCAs in human hepatoma cells using the single-cell gel electrophoresis (SCGE) assay. This in vitro model is often used to study antioxidant defense mechanisms, as they express important protective enzymes against oxidative stress [27].
HCAs are potent genotoxins in bacterial and mamalian cell assays and exert strong carcinogenic effects in ex perimental animals [28]. Regarding the mechanisms of DNA damage by carcinogenic HCAs, DNA adduct formation has been considered to be a major causal factor. But the DNA adducts themselves may not be sufficient for the carcinogenic action, oxidative DNA damage also may play a role in carcinogenesis [29]. DNA oxidized bases are potentially mutagenic and so, are implicated in this process [18]. HCAs are believed to form DNA oxidized forms that stem from simultaneous formation of reactive oxygen species (ROS) during the hepatic metabolism by CYP450 enzymes [30]. Our previous results [23] also support this hypothesis indicating the presence of oxidized pyrimidines and purines bases respectively, induced by HCAs.
The mutagenic potency of HCAs is dependent upon the chemical structure and the ability of the HCAs to undergo N-oxidation to form the reactive nitrenium ion [31]. Nevertheless, a number of strong correlations have been found between the mutagenic potency and individual chemical properties. The number of fused rings, the number of heteroatoms in the non-imidazole ring, Nmethyl substitution on the imidazole ring, the methyl substitution on ring carbon atoms, and the extent of the pi electron system, with smaller contributions from dipole moment, the calculated stability of the nitrenium ion, and hydrophobicity were important determinants in the carcinogenic and mutagenic potencies of HCAs [31].
Genetic damage, which can lead to mutations, is a crucial event for the initiation of carcinogenesis, so that its inhibition can be the key step in cancer prevention [32]. Flavonoids have an important activity as antioxidants and also remarkable role on carcinogen activation in vivo and on carcinogenesis [33,34]. Myricetin and quercetin are two of the most frequently studied flavonoids in the class of flavonols [2].
In the present study, myricetin (3, 3’, 4’, 5, 5’, 7-hexahydroxyflavone) and quercetin (3, 3’, 4’, 5, 7-pentahydroxyflavone) did not protect against HCAsinduced DNA strand breaks. The oxidized reduced by myricetin but not by quercetin. Quercetin reduced the oxidized purines induced by 8-MeIQx and PhIP, while myricetin reduced also the induced by 4,8-diMeIQx. This variable effect of quercetin against oxidized pyrimidines and purines induced by HCAs is surprising given the similar chemical structure of quercetin and myricetin. Genetic damage, which can lead to mutations, is a crucial event for the initiation of carcinogenesis, so that its inhibition can be the key step in cancer prevention [32]. Flavonoids have an important activity as antioxidants and also a remarkable role oncarcinogen activation in vivo and on carcinogenesis [33,34]. Myricetin and quercetin are two of the most frequently studied flavonoids in the class of flavonols [2]. Quercetin and myricetin structures differ only by a hydroxyl group at the myricetin 5’ position. That gives rise to easily release an additional reducing agent from the B-ring hydroxyls and to form a more stable o-quinone [35]. Several groups established that the antioxidant activity of flavonols depends on the numbers of hydroxyl groups and their locations in the B ring [36, 37].
This feature in chemical structure, may give a better protective effect of myricetin than quercetin against HCAs-induced oxidative DNA damage. This indicates that only an additional hydroxyl group at position 5′ in the chemical structure of myricetin would significantly affect the biological activity against HCAs. Thus, the protective effect of flavonols against the mutagens tested is close related to the number of hydroxyl groups in their structure. These results are supported by previous works of our research group [21] where myricetin and quercetin were evaluated against N-nitrosamines and benzo (a) pyrene (BaP)-induced DNA damage. Myricetin protected against oxidized pyrimidines induced by BaP and N.nitrosodimethylamine and towards oxidized pyrimidines and purines induced by N-nitrosopyrrolidine. However, quercetin did not protect against oxidized purines induced by N-nitrosamines and BaP. In addition, the protective effect of myricetin and quercetin was higher towards BaP than against N-nitrosamines. In the present study, myricetin was more efficient than quercetin to prevent DNA damage (oxidized purines and pyrimidines) induced by the three HCAs evaluated. Thus, the presence and number of polyhydroxyl groups in the chemical structure and/or the kind of mutagen used to induce DNA damage has an important role in the protective effect against N-nitrosamines and benzo (a) pyrene (BaP)-induced DNA damage. Myricetin protected against oxidized pyrimidines induced by BaP and N.nitrosodimethylamine and towards oxidized pyrimidines and purines induced by N-nitrosopyrrolidine. However, quercetin did not protect against oxidized purines induced by N-nitrosamines and BaP. In addition, the protective effect of myricetin and quercetin was higher towards BaP than against N-nitrosamines. In the present study, myricetin was more efficient than quercetin to prevent DNA damage (oxidized purines and pyrimidines) induced by the three HCAs evaluated. Thus, the presence and number of polyhydroxyl groups in the chemical structure and/or the kind of mutagen used to induce DNA damage has an important role in the protective effect.
The cancer protective effects of flavonoids have been attributed to a wide variety of mechanisms, including the free radical scavenging ability [32]. Other mechanism proposed to explain this protective effect might also include inhibition of phase I metabolizing enzymes, such as CYP450, which metabolically activates procarcinogens to reactive intermediates that trigger carcinogenesis [5]. The HCAs require a two-step metabolic activation to DNA-reactive genotoxins, and also lead to an oxidative modification of DNA [38]. Several authors reported that flavonoids could inhibit cancer at the initiation phase through down-regulation of CYP450, a phase I metabolizing enzyme [39,40]. As a general rule, flavonoids possessing hydroxyl groups inhibit CYP-dependent monooxygenase activity, whereas those lacking hydroxyl groups can stimulate the enzyme activity [41]. In the present work, we showed that the protective effect of myricetin and quercetin towards DNA damage could be related to the reduction of CYP1A1 activity. CYP450 enzymes belonging to the CYP1A family are highly inducible by HCAs [42] and our results showed that human CYP1A1 activity was moderately reduced by myricetin while little effect was observed by quercetin. Accordingly, the protective effect of myricetin, and quercetin towards HCAs-induced oxidative DNA damage could be related in part to the reduction of human CYP1A1. However this is not the only mechanism by which myricetin and quercetin exert their protective effect, other mechanisms such as stimulation of the repair of carcinogen-induced DNA damage and or the free radical scavenging efficiency [43] have been also implicated. In addition, not well correlation between human CYPs inhibition and inhibition of DNA damage-induced by HCAs could be attributed to the incubation of polyphenols with P-450 enzymes under cell-free conditions, such as microsomes from baculovirus-infected insect cells expressing human CYP1A1, instead living cells (HepG2 cells) as in the comet assay.
Finally, another mechanism claimed to be responsible for the protective effect of myricetin and quercetin is the induction of phase II metabolizing enzymes such as UDP-glucuronyltrasferase (UGT) by which carcinogens are detoxified and therefore more readily eliminated from the body. Thus, activation of phase II detoxifying enzymes represents one mechanism of their anticarcinogenic effects [44]. Our results showed quercetin as the most potent inducer of UGT1A4 activity. However, myricetin at all concentrations tested, did not increase the UDP-glucuronyltransferase activity. In conclusion, our results clearly indicate that myricetin was more efficient than quercetin to prevent oxidative DNA damage (oxidized purines and pyrimidines) induced by HCAs evaluated. This protective effect depends on the chemical structure of flavonoid and the mutagen studied.
5. Acknowledgements
This work has been supported by Grant ALI2005-01517 from the Ministerio de Educación y Ciencia (Spain) and by Grant 910177 from the Comunidad de Madrid and the Universidad Complutense (UCM).
REFERENCES
- World Cancer Research Fund, “Food, Nutrition and the Prevention of Cancer: A Global Perspective,” Nutrtion, Vol. 15, No. 6, 1997, pp. 523-526.
- S. J. Duthie, A. R. Collins, G. C. Duthie and V. L. Dobson, “Quercetin and Myricetin Protect against Hydrogen Peroxide-Induced DNA Damage Strand Breaks and Oxidized Pyrimidines, in Human Lymphocytes,” Mutation Research, Vol. 393, No. 3, 1997, pp. 223-231.
- M. Kampa, A. Nifli, G. Notas and E. Castanas, “Polyphenols and Cancer Cell Growth,” Reviews of Physiology, Biochemistry & Pharmacology, Vol. 159, No. 2, 2007, pp. 79-113. doi:10.1007/112_2006_0702
- K. Min and S. E. Ebeler, “Flavonoid Effects on DNA Oxidation at Low Concentrations Relevant to Physiological Levels,” Food and Chemical Toxicology, Vol. 46, No. 1, 2008, pp. 96-104. doi:10.1016/j.fct.2007.07.002
- M. Maggiolini, A. G. Recchia, D. Bonofiglio, S. Catalano, A. Vivacqua, A. Carpino, V. Rago, R. Rossi and S. Ando, “The Red Wine Phenolics Piceatannol and Myricetin Act as Agonists for Estrogen Receptor {Alpha} in Human Breast Cancer Cells,” Journal of Molecular Endocrinology, Vol. 35, No. 2, 2005, pp. 269-281. doi:10.1677/jme.1.01783
- M. L. Neuhouser, “Dietary Flavonoids and Cancer Risk: Evidence From Human Population Studies,” Nutrition and Cancer, Vol. 50, No. 1, 2004, pp. 1-7. doi:10.1207/s15327914nc5001_1
- T. Sugimura, “Overview of Carcinogenic Heterocyclic Amines,” Mutation Research, Vol. 376, No. 1, 1997, pp. 211-219. doi:10.1016/S0027-5107(97)00045-6
- R. Sinha, “An Epidemiologic Approach to Studying Heterocyclic Amines,” Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, Vol. 506-507, 2002, pp. 197-204. doi:10.1016/S0027-5107(02)00166-5
- J. Felton and M. Malfatti, “What Do Diet-Induced Changes in Phase I and II Enzymes Tell Us about Prevention from Exposure to Heterocyclic Amines?” Journal of Nutrition, Vol. 136, No. 10, 2006, pp. 2683-2684.
- F. Toribio, R. Busquets, L. Puignou and M. T. Galceran, “Heterocyclic Amines in Griddled Beef Steak Analysed Using a Single Extract Clean-Up Procedure,” Food and Chemical Toxicology, Vol. 45, No. 4, 2007, pp. 667-675. doi:10.1016/j.fct.2006.10.016
- A. Ristic, M. Cichna and G. Sontag, “Determination of Less Polar Heterocyclic Aromatic Amines in Standardised Beef Extracts and Cooked Meat Consumed in Austria by Liquid Chromatography and Fluorescence Detection,” Journal of Chromatography B, Vol. 802, No. 1, 2004, pp. 87-94. doi:10.1016/j.jchromb.2003.09.028
- K. I. Skog, M. A. E. Johansson and M. I. Jägerstad, “Carcinogenic Heterocyclic Amines in Model Systems and Cooked Foods: A Review on Formation, Occurrence and Intake,” Food and Chemical Toxicology, Vol. 36, No. 9-10, 1998, pp. 879-896. doi:10.1016/S0278-6915(98)00061-1
- International Agency for Research on Cancer, “Some Natural Occurring Substances: Food Items and Constituents Heterocyclic Amines and Mycotoxins: IARC Monographs on the Evaluation of Carcinogenic Risks to Humans by International Agency for Research on Cancer,” World Health Organization, Lyon, 1993, pp. 165-231.
- S. Knasmuller, C. E. Schwab, S. J. Land, C. Y. Wang, R. Sanyal, M. Kundi, W. Parzefall and F. Darroudi, “Genotoxic Effects of Heterocyclic Aromatic Amines in Human Derived Hepatoma (HepG2) Cells,” Mutagenesis, Vol. 14, No. 6, 1999, pp. 533-540. doi:10.1093/mutage/14.6.533
- J. Felton, M. Knize, L. M. Bennett, M. Malfatti, M. Colvin and K. Kulp, “Impact of Environmental Exposures on the Mutagenicity/Carcinoge Nicity of Heterocyclic Amines,” Toxicology, Vol. 198, No. 1-3, 2004, pp. 135-145. doi:10.1016/j.tox.2004.01.024
- Y. He, M. D. Friesen, R. J. Ruch and H. A. J. Schut, “Indole-3-Carbinol as a Chemopreventive Agent in 2-Amino-1-Methyl- 6-Phenyl Imdazo [4,5-b] Pyridine (PhIP) Carcinogenesis: Inhibition of PhIP–DNA Adduct Formation, Acceleration of PhIP Metabolism, and Induction of Cytochrome P450 in Female F344 Rats,” Food and Chemical Toxicology, Vol. 38, No. 1, 2000, pp. 15-23. doi:10.1016/S0278-6915(99)00117-9
- R. H. Dashwood, “Modulation of Heterocyclic Amine-Induced Mutagenicity and Carcinogenicity: An ‘A-to-Z’ Guide to Chemopreventive Agents, Promoters, and Transgenic Models,” Mutation Research, Vol. 511, No. 2, 2002, pp. 89-112. doi:10.1016/S1383-5742(02)00005-4
- A. R. Collins, “Assays for Oxidative Stress and Antioxidant Status: Applications to Research into the Biological Effectiveness of Polyphenols,” The American Journal of Clinical Nutrition, Vol. 81, No. 1, 2005, pp. 261-267.
- V. Mersch-Sundermann, S. Knasmuller, X. Wu, F. Darroudi and F. Kassie, “Use of a Human-Derived Liver Cell Line for the Detection of Cytoprotective, Antigenotoxic and Cogenotoxic Agents,” Toxicology, Vol. 198, No. 1-3, 2004, pp. 329-340. doi:10.1016/j.tox.2004.02.009
- M. A. Westerink and G. E. J. Schoonen, “Cytochrome P450 Enzyme Levels in HepG2 Cells and Cryopreserved Primary Human Hepatocytes and Their Induction in HepG2 Cells,” Toxicology in Vitro, Vol. 21, No. 8, 2007, pp. 1581-1591. doi:10.1016/j.tiv.2007.05.014
- M. E. Delgado, A. I. Haza, N. Arranz, A. García and P. Morales, “Dietary Polyphenols Protect against N-Nitrosamines and Benzo(a) Pyrene-Induced DNA Damage (Strand Breaks and Oxidized Purines/Pyrimidines) in HepG2 Human Hepatoma Cells,” European Journal of Nutrition, Vol. 47, No. 11, 2008, pp. 479-490. doi:10.1007/s00394-008-0751-6
- P. L. Olive, D. Wlodek, R. E. Durand and J. P. Banáth, “Factors Influencing DNA Migration from Individual Cells Subjected to Gel Electrophoresis,” Experimental Cell Research, Vol. 198, No. 2, 1992, pp. 259-267. doi:10.1016/0014-4827(92)90378-L
- A. I. Haza and P. Morales, “Effects of (+) Catechin and (-) Epicatechin on Heterocyclic Amines-Induced Oxidative DNA Damage,” Journal of Applied Toxicology, Vol. 31, No. 1, 2011, pp. 53-62. doi:10.1002/jat.1559
- M. D. Burke and R. T. Mayer, “Differential Effects of Phenobarbitone and 3-Methyl Holanthrene Induction on the Hepatic Microsomal Metabolism and Cytochrome P-450-Binding of Phenoxazone and a Homologous Series of Its n-Alkyl Ethers (Alkoxyresorufins),” Chemico-Biological Interactions, Vol. 45, No. 2, No. 2, 1983, pp. 243-258. doi:10.1016/0009-2797(83)90072-8
- M. D. Burke, S. Thompson, C. R. Elcombe, J. Halpert, T. Haaparanta and R. T. Mayer, “Ethoxy-Penthoxy and Benzyloxyphexazones and Homologues: A Series of Substrates to Different Induced Cytochromes P-450,” Biochemical Pharmacology, Vol. 34, No. 18, 1985, pp. 3337- 3345. doi:10.1016/0006-2952(85)90355-7
- W. Lilienblum, A. K. Walli and K. W. Bock, “Differential Induction of Rat Liver Microsomal UDP-Glucuronosyl-Transferase Activites by Various Inducing Agents,” Biochemical Pharmacology, Vol. 31, No. 6, 1982, pp. 907-913. doi:10.1016/0006-2952(82)90319-7
- J. Plazar, B. Žegura, T. T. Lah and M. Filipič, “Protective Effects of Xanthohumol against the Genotoxicity of Benzo(a)pyrene (BaP), 2-Amino-3-Methylimidazo [4,5-f] Quinoline (IQ) and Tert-Butyl Hydroperoxide (t-BOOH) in HepG2 Human Hepatoma Cells,” Mutation Research/ Genetic Toxicology and Environmental Mutagenesis, Vol. 632, No. 1-2, 2007, pp. 1-8. doi:10.1016/j.mrgentox.2007.03.013
- T. Sugimura, “Food and Cancer,” Toxicology, Vol. 181- 182, 2002, pp. 17-21. doi:10.1016/S0300-483X(02)00250-0
- M. Murata, M. Kobayashi and S. Kawanishi, “Mechanism of Oxidative DNA Damage Induced by a Heterocyclic Amine, 2-Amino-3,8-Dimethylimidazo[4,5-F]Quinoxaline,” Cancer Science, Vol. 90, No. 3, 1999, pp. 268- 275. doi:10.1111/j.1349-7006.1999.tb00743.x
- J. H. Weisburger, “Comments on the History and Importance of Aromatic and Heterocyclic Amines in Public Health,” Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, Vol. 506-507, No. 9, 2002, pp. 9-20. doi:10.1016/S0027-5107(02)00147-1
- F. T. Hatch, M. G. Knize and M. E. Colvin, “Extended Quantitative Structure-Activity Relationships for 80 Aromatic and Heterocyclic Amines: Structural, Electronic, and Hydropathic Factors Affecting Mutagenic Potency,” Environmental and Molecular Mutagenesis, Vol. 38, No. 4, 2001, pp. 268-291. doi:10.1002/em.10028
- S. De Flora and L. R. Ferguson, “Overview of Mechanisms of Cancer Chemopreventive Agents,” Mutation Research/Fundamental and Molecular Mechanisms of Mutagenesis, Vol. 591, No. 1-2, 2005, pp. 8-15. doi:10.1016/j.mrfmmm.2005.02.029
- P. Dolara, C. Luceri, C. D. Filippo, A. P. Femia, L. Giovannelli, G. Caderni, C. Cecchini, S. Silvi, C. Orpianesi and A. Cresci, “Red Wine Polyphenols Influence Carcinogenesis, Intestinal Microflora, Oxidative Damage and Gene Expression Profiles of Colonic Mucosa in F344 Rats,” Mutation Research, Vol. 591, No. 1-2, 2005, pp. 237-246. doi:10.1016/j.mrfmmm.2005.04.022
- J. D. Lambert, J. Hong, G. Yang, J. Liao and C. S. Yang, “Inhibition of Carcinogenesis by Polyphenols: Evidence from Laboratory Investigations,” The American Journal of Clinical Nutrition, Vol. 81, No. 1, 2005, pp. S284- S291.
- K. Kanazawa, M. Uehara, H. Yanagitani and T. Hashimoto, “Bioavailable Flavonoids to Suppress the Formation of 8-OHdG in HepG2 Cells,” Archives of Biochemistry and Biophysics, Vol. 455, No. 2, 2006, pp. 197- 203. doi:10.1016/j.abb.2006.09.003
- C. Rice-Evans, N. Miller and G. Paganga, “StructureAntioxidant Activity Relationships of Flavonoids and Phenolic Acids,” Free Radical Biology & Medicine, Vol. 20, No. 7, 1996, pp. 933-956. doi:10.1016/0891-5849(95)02227-9
- P. Makena and K. Chung, “Effects of Various Plant Polyphenols on Bladder Carcinogen Benzidine-Induced Mutagenicity,” Food and Chemical Toxicology, Vol. 45, No. 10, 2007, pp. 1899-1909. doi:10.1016/j.fct.2007.04.007
- J. H. Weisburger and F. Chung, “Mechanisms of Chronic Disease Causation by Nutritional Factors and Tobacco Products and Their Prevention by Tea Polyphenols,” Food and Chemical Toxicology, Vol. 40, No. 8, 2002, pp. 1145-1154. doi:10.1016/S0278-6915(02)00044-3
- F. T. Hatch, F. C. Lightstone and M. E. Colvin, “Quantitative Structure-Activity Relationship of Flavonoids for Inhibition of Heterocyclic Amine Mutagenicity,” Environmental and Molecular Mutagenesis, Vol. 35, No. 4, 2000, pp. 279-299. doi:10.1002/1098-2280(2000)35:4<279::AID-EM3>3.0.CO;2-9
- Z. Apostolides, D. A. Balentine, M. E. Harbowy, Y. Hara and J. H. Weisburger, “Inhibition of PhIP Mutagenicity by Catechins, and by Theaflavins and Gallate Esters,” Mutation Research/Genetic Toxicology and Environmental Mutagenesis, Vol. 389, No. 2-3, 1997, pp. 167-172. doi:10.1016/S1383-5718(96)00143-X
- P. Hodek, P. Trefil and M. Stiborová, “Flavonoids-Potent and Versatile Biologically Active Compounds Interacting with Cytochromes P450,” Chemico-Biological Interactions, Vol. 139, No. 1, 2002, pp. 1-21. doi:10.1016/S0009-2797(01)00285-X
- J. Hümmerich, C. Zohm and W. Pfau, “Modulation of Cytochrome P450 1A1 by Food-Derived Heterocyclic Aromatic Amines,” Toxicology, Vol. 199, No. 2-3, 2004, pp. 231-240. doi:10.1016/j.tox.2004.02.028
- R. J. Williams, J. P. E. Spencer and C. Rice-Evans, “Flavonoids: Antioxidants or Signalling Molecules?” Free Radical Biology and Medicine, Vol. 36, No. 7, 2004, pp. 838-849. doi:10.1016/j.freeradbiomed.2004.01.001
- Y. J. Moon, X. Wang and M. E. Morris, “Dietary Flavonoids: Effects on Xenobiotic and Carcinogen Metabolism,” Toxicology in Vitro, Vol. 20, No. 2, 2006, pp. 187- 210. doi:10.1016/j.tiv.2005.06.048