Open Journal of Veterinary Medicine
Vol. 2 No. 4 (2012) , Article ID: 25619 , 15 pages DOI:10.4236/ojvm.2012.24043
Anaphylatoxins: From Supposed Toxin Anaphylactics to Effective Mediators of the Early Events of Inflammation*
Laboratório de Imunoquímica, Instituto Butantan, São Paulo, Brazil
Email: wds@butantan.gov.br
Received September 5, 2012; revised November 2, 2012; accepted November 13, 2012
Keywords: Anaphylatoxins; Inflammation; Innate Immunity
ABSTRACT
This review updates original data describing the experiments showing the complement origin of anaphylatoxins unexplainable submerged under the surface of the articles related to the subject. Next, recalls subsequent data describing the anaphylatoxins peptide nature and sequences, the cell receptors with which they interact and activate and the outcome of the cell responses. The review continues by highlighting the anaphylatoxin biological properties focusing on the unequivocal participation of these mediators in inflammation. The review concludes bringing data reinforcing the promising use of these peptides as molecular primers to create specific and efficient pharmacological antagonists.
1. Introduction
1.1. Historic Facts
Guinea pigs intravenously injected with homologous serum samples previously treated with preformed immune complexes develop symptoms and tissue lesions indistinguishable from the anaphylactic shock, as persistent fall in the systemic blood pressure, constriction with obliteration of the bronchioles, and distention of alveoli [1]. The generated activity was coined anaphylatoxin to design a supposed “toxin anaphylactic”. Aliquots of serum recovered from guinea pigs intravenously injected with preformed immune complexes and suffering the anaphylactic-like symptoms when added onto isolated guinea pig ileum induced contractions, in a dose-dependent fashion, followed by tachyphylaxis after several additions of equal amounts of the treated serum. This in vitro smooth muscle contracting-activity induced by the immune complex treated serum resembles the typical anaphylactic Schultz-Dale reaction. Similar activity was also generated when serum was treated with complex polysaccharides as agar, inulin or dextran [2-4] . The main anaphylactic symptoms induced in guinea pigs by immune complexes treated serum could be reproduced by intravenous injecting guinea pigs with histamine [5]. Histamine release from isolated guinea lungs obtained from antigen-sensitized animals upon perfusion with the antigen reinforced the hypothesis that histamine could be one of the anaphylactic shock mediators [1,6-8] . The hypothesis that the complement system (CS) could be involved in anaphylatoxin formation was submitted to experimental approach by treating fresh serum with immune aggregates and assaying in parallel the emergence of a smooth muscle anaphylatoxin-like contraction and loss in hemolytic of the serum [9] . However, more rigorous definition of the involvement of the CS in anaphylatoxin formation awaited purification of the different CS components and how they interplay along the activation pathways.
1.2. Complement Activation
The CS comprises four sets of components: pro-enzymes/enzymes, stabilizers, aggregating proteins, and regulatory proteins. Pro-enzymes/enzymes: C1 complex (C1q-Ca++-C1r2-C1s2), C2, C4, MASPs, factor B (fB), factor D (fD), C3. Stabilizers: properdin (P). Aggregating factors: MBL, C5, C6, C7, C8, C9. Regulatory proteins: C1-INH, H, C4Bp, DAF (CD55), MCP (CD46), CR1 (CD35), CD59, S protein, clusterin and vitronectin. This system is activated by three routes, the alternative (AP), the classical (CP) and the lectin (LP) pathways, either of which leading to formation of a cell membrane attack complex and the generation of opsonins and anaphylatoxins. Anaphylatoxins derives by enzymatic cleavage of, C3, C4 or C5 components. The “Complement. Nomenclature” was discussed and approved by a WHO sponsored committee of specialist [10] .
C3. The native C3 molecule is a protein formed by a larger α-chain (115 kDa) and a smaller β-chain (75 kDa) held together by inter-chain disulfide bounds (Figure 1(a)). C3 derives from a pre-pro C3 native molecule after removal of the 22 N-terminus amino acid corresponding to the signal peptide and formation of the thioester bound by an intra-molecular transacylation involving the thiol group of Cys-998 and γ-amide of Gln-991 and removal
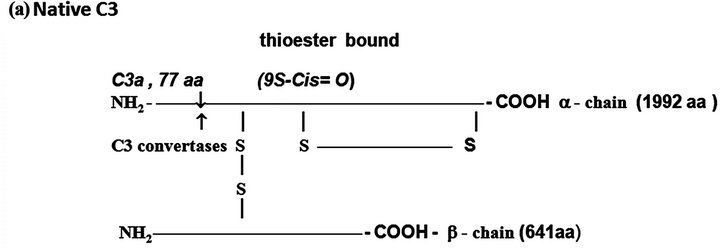
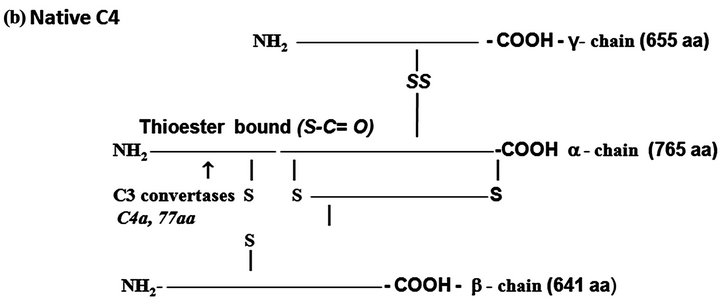
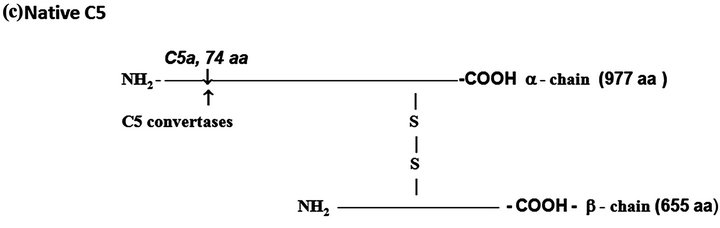
Figure 1. The protein complement molecules features directly involved in anaphylatoxin generation: Target sites for the activated C3 and C5 convertases. (a) C3 molecule. Native C3 is secreted into the circulation. The enzymes C3 convertases cleave the native C3 molecule at the peptide bond between the Arg 726 and Ser 727 residues releasing C3a, the peptide 77-residues from the α-chain, and C3b formed by the remaining α’-chain joined to intact ß-chain. C3a acquires inflammatory properties, and C3b expressed the abilities of covalently binding to the targets cell surface through the exposed thioester bound and binding sites for other CS components as C5, P, factors H, B and I, C4b-binding protein, C3b receptors and co-factor protein (MCP); (b) C4, molecule. Native C4 is secreted into the circulation. The enzymes C1 esterase and MBL-MASP-2 activated cleave the peptide Arg76 and Ala77 of the native C4 α-chain releasing the 77-residue peptide C4a. The resulting C4b formed by α’-chain, β-chain and γ-chain is prone of binding emerging activated C2a resulting also from cleavage of native C2 by the activated enzymes C1 esterase and MBL-MASP-2. As C4b is metastable, once in the absence of C2a is inactivated by the concerted action of factor I and C4-binding protein by cleavage of its α’-chain at two sites, with release of C4c into fluid phase and the remaining 380 residues fragment C4d retaining the thioester bound. If the C4 cleavage was performed sufficiently near of targets offering the reactive groups OHor NH2 groups to C4d to be covalently attached by ester or amide bond; and (c) C5 molecule. Native C5 is secreted into the circulation. Native C5 α-chain expresses a peptide bond susceptible to be cleaved by the C5 convertases either from CP, C4bC2aC3b, or from AP, C3bBbPC3b. C5 becomes susceptible to the cleavage only after be binding by the already adhered C3b on to C5 convertases complexes. Upon binding by C3b of these C5 convertases, C5 is cleaved in two fragments, C5a anaphylatoxin (74-residue peptide, 11 kDa) and C5b (180 kDa). After binding, the cleavage is then executed by the C2a or Bb proteinases domains. The C5b anchored onto cell surface targets initiates the assembling of MAC by successive attaching in the growing molecular complex one molecule each of C6, C7, C8, and at least four molecules of C9. As C9 is a perforin-like protein upon polymerization MAC acquires the membrane lytic properties.
of the tetrapeptide R-R-R-R) [11] .
The protected thiol group can be exposed by hydrolysis at the slow rates of 0.005% of the C3 molecules/ minute resulting in the inactive C3 (H2O) molecule, or by enzymatic cleavage of the peptide bound between the residues Arg-726 and Ser-727 of the C3 α-chain, resulting the free peptide C3a with 77 amino acid residues (9 kDa), and the C3b formed by the remaining α’ chain (106 kDa) attached by disulphide to intact β-chain (75 kDa). In both situations chemical energy is released after the thioester breakdown. If particles exhibiting OH− or NH2+ groups are sufficiently nearby ester or amide covalent bounds are constructed between C3b and the surface particles.
C4. The native C4 is a heterotrimer of 210 kDa formed by the polypeptides α, ß and γ chains held together by inter-chains disulfide bonds (Figure 1(b)). There are two isoforms of C4, C4A and C4B. This component shares with C3 the presence of the internal thioester bound also at the N-terminus of its α-chain.Activated C1 cleaves C4 molecule at the α-chain with removal of a 77-residue fragment (8.6 kDa) the C4a anaphylatoxin consisting of N-terminus of the α-chain, and C4b fragment consisting of the α-chain exposing the thioester bound and the entire ß and γ chains. C4A isoform whose thioester preferentially forms amide bonds links with surface proteins, while the isoform B whose thioester bound forms ester, attaches on carbohydrates.
C5. This component is synthesized as a single polypeptide chain (1658 amino acid residues) and posttranslationally processed resulting in two polypeptides, α (12.4 kDa) and ß (7.6 kDa) chains linked by disulfide bounds (Figure 1(c)). The resulting native protein (190 kDa) is both, the source of anaphylatoxin C5a and C5b the anchoring fragment for C6 during organization of the lytic complex MAC (Membrane Attach Complex).
The complement system is activated by alternative (AP), classical (CP) and lectin (LP) pathways (Figure 2).
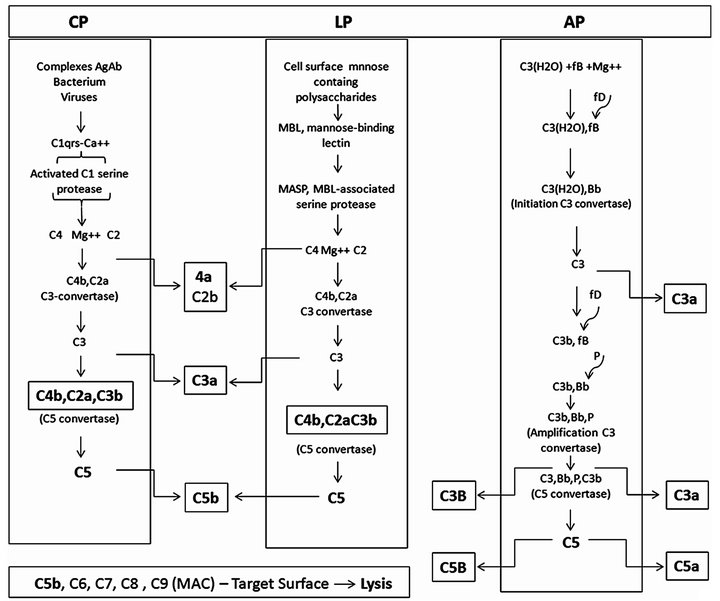
Figure 2. The complement activation pathways features directly involved in anaphylatoxin generation. Complement activation starts through three cascades: classical (CP), lectin (LP) and alternative (AP) pathways. Although initially distinct the pathways converge to product inflammatory mediating small peptides, larger opsonins fragments, and creation of robust polymolecular lytical complex. Subtle but important interplays among the pathways may occur along the activations aiming improving the utilization of some activated component. CP, initiates by interaction between the activators antibody antigen complexes, or some bacteria or viruses with C1 through the subcomponent C1q resulting in activation of the serine protease C1s domain. LP, initiates by interaction between the mannose-binding lectin (MBL) with the mannose-containing polysaccharides activators resulting in activation of MBL-associated serine proteases (MASPS). AP, initiates spontaneously, the “tickover” procedure involving the C3 thioester bound hydrolysis. The figure depicts the molecular interaction process.
AP. The AP involves the components C3, factor B (fB), factor D (fD) an active plasma protease and properdin (P) a stabilizer protein. Spontaneous reactive C3* is created at a rate of 0.005% - 1% of total C3 per hour generating hydrolyzed C3(H2O). Along the short hydrolysis process chemical energy is being released from the disrupted thioester bound. This C3* is capable of binding to both the cell targets and to fluid phase native fB. Once associated to C3* fB changes its conformation becoming susceptible to cleavage by fD into Ba an Bb. Ba is released into fluid phase and Bb remains associated to C3*, forming the complex C3*Bb. The exposed Bb intrinsic serine protease domain cleaves additional C3 molecules, generating C3b molecules. In a manner similar to C3* new generated C3b interacts with the cell targets surface and with fB, forming the molecular complex C3bBb. C3*Bb, the initiation C3 convertase, has a short life while C3bBb, upon stabilized by associating with P, resulting the trimolecular complex, C3bBbP, termed amplification C3 convertase. This enzyme is quite stable (over 10 min of life) and is responsible for saturate the target cell surface with covalently bound C3b and release C3a into the fluid phase (Figure 2).
CP. This pathway is triggered by preformed immune complexes, C-reactive protein (CRP) and by some bacteria and virus-capsular associated components. The activation involves the complement components, C1, C4 and C2. The trigger step initiates by interactions of the activator molecules with C1q. Binding distorts strategic C1q molecules domains resulting successively in C1r, next C1s activations into esterases. C1s esterase cleaves C4 and C2. C4 is cleaved into two fragments, C4b and C4a. C4b binds covalently to hydroxyl or amino groups on cellular membranes. C2, after binding into C4b is also cleaved into C2a and C2b.The fragment C2b remains associated with C4b, resulting the molecular complex, C4b2b, the C3 convertase of the CP that cleaves C3 also in C3b and C3a. Some CP generated C3b fragments can bindfB as in AP activation generating C3bBb. AP is therefore an important “amplification loop” for the CP (Figure 2).
LP. The LP activation is initiated upon recognition of pathogen-associated mannose and N-acetyl glucosamine oligosaccharides by a serum protein, the mannose-binding lectin (MBL) [12] . The next steps following MBL binding to pathogens associated carbohydrates is followed by activation of MBL-associated serine proteases (MASPs). Activated MASPs, similarly to C1s, act on C4 and C2, assembling the C4bC2b complex essentially as in CP activation [13,14] (Figure 2). The C3 convertases, C3bBbP and C4bC2a, upon aggregating an additional C3b fragment shifts their specificity from C3 to C5, transforming into the C5 convertases C3b, Bb, P, C3b and C4b, C2a, C3b.
Lyticpathway. This pathway comprises the components C5, C6, C7. C8 and C9.These proteins are constituted by TSR, LDLR, perforin, EGF, and 2-CCPR associated in different proportions. The C5 convertases cleave C5 into C5a and C5b. C5a anaphylatoxin is released into the fluid phase. C5b expresses transient binding sites for the target cell membrane and for C6. Upon binding C6 acquires the ability to binding C7. If the complex C5b, C6, C7 is formed C8 is attached, binds C9 and promotes its polymerization. The complex C5b, C6, C7, C8 and C9 (3 - 4 molecules), dimerizes forming a macromolecule complex (around 2000 kDa) which is inserted into the cell membrane resulting in permanent pore lesions of around 100Ǻ leading to the cell lysis.
1.3. Regulatory Mechanisms
The CS includes also regulatory components aiming amplify, coordinate and confine the main steps along its activation.
C1-inhibitor. C1-INH was described as a heat labile factor in human serum that inhibited the esterolytic activity of the first complement component [15]. This protein is a member of the serpin family of protease inhibittors. C1-INH differs from other members of the terpin family in that it contains a long N-terminal domain of approximately 100 amino acid residues that is heavily glycosilated with both Nand O-linked sugars.
Factor I. fI is a serum protein (35 μg/mL), formed by two chains with molecular mass of 50 and 38 kDa and contains one serine protease domain. fI cleaves: a) C4b into iC4b and in the presence of C4-binding protein (C4bBP) with the participation of complement receptor type 1 (CR1); b) C3b into iC3b with the participation of also of CR1, factor H and membrane-cofactor protein (MCP). The protein super-family formed by CR1, factor H, Cr2, C4BPA, C4bPB, MCP and DAF are modular molecules consisting of only one type of domain, the “complement control protein repeats” (CCPRs). CCPRs are present in varying numbers in the different factors as indicated in parentheses.
CR1 (30 CCPRS). Is a cell bound protein formed by two polypeptide chains, molecular mass of 190 and 280 kDa. Serves as co-factor for Fi, participates in the clearance of immune complexes and in the phagocytosis of C3b opsonized particles.
Factor H (20 CCPRS). fH is a 150 kDa circulating protein (±480 µg/mL). Inactivates the C3 and C5 convertases by dissociating the molecular subunits, and serves as co-factor for fI.
C4bBP (3 to 8 CCPRS).C4bBP is a circulating protein (±250µg/mL), molecular mass 570 kDa.
MCP or Membrane Co-factor Protein (5 CCPRS). MCP is a two chains cell molecular bound protein, 51 - 68 kDa. Participates in inactivation of C3b and C4b.
DAF or Decay Accelerating Factor (4 CCPRS). DAF is a cell-bound protein, molecular mass 70 kDa capable of inactivate the C3 convertases C4bC2a in CC4b e C2a and C3bBb in C3b and Bb by accelerating the normal decay of these enzymes.
1.4. Complement as Anaphylatoxins Source
Along the CS cascade activation the emerging enzymes, MBL-MASP-2, C1 esterase, C3(H2O)Bb, C3bBbP, C4bC2a, C3bBbPC3b and C4bC2aC3b, are specifically involved in anaphylatoxin generation. MBL-MASP-2 and C1 esterase, release C4a from C4; C3(H2O)Bb, C3bBbP and C4bC2a release C3a from C3; while C3bBbPC3b and C4bC2aC3b release C5a from C5. Anaphylatoxin-like peptides can also be generated as side-products of direct proteolysis of C3, C4 and C5 components by tissues or circulating generated proteolytic enzymes.
C3a. Indication that CS could be anaphylatoxin source was the verification that purified human C1 esterase, the first complement enzyme purified to homogeneity, had the ability to increase vascular permeability in postcapillaryvenules in guinea pig skin [16]. This effect was a property of the active enzyme and was mediated by release of a histamine-like substance. In further experiments it was found that small concentrations of C1 esterase, sub-threshold for direct action in guinea pig skin, were capable of generating a permeability factor when mixed with normal guinea pig serum in vitro. The generation of this factor required heat-labile substances in serum and was correlated with consumption of hemolytic complement. The permeability factor itself was relatively heat-stable and its action in guinea pig skin was blocked by prior treatment of animals with triprolidine hydrochloride, an antihistamine drug. These observations were consistent with the hypothesis that interaction of purified C1 esterase and normal serum results in production of an anaphylatoxin like activity [17] . This hypothesis were further reinforced in in vitro experiments by using purified human C1 esterase as activator, normal guinea pig or rat serum as the permeability source, and guinea pig ileum or rat uterus preparations as the pharmacological assays. Human C1 esterase treated but not untreated serum exhibited the following properties: a) contraction of guinea pig ileum which was made unresponsive after a few additions of the same amount of treated serum; b) failure to contract guinea pig ileum previously desensitized to agarose-anaphylatoxin or treated with antihistaminic drug; c) degranulation of mast cells and release of histamine from guinea pig tissues; d) failure to contract rat uterus. These findings indicated that the activity liberated by C1 esterase was related to anaphylatoxin. Generation was prevented by prior inactivation of C1 esterase with DFP or partially purified inhibitor of C1 esterase, prior heating of the serum at 56˚C, the presence of salicylaldoxime or phlorizin, or by selective chelating of Mg++. A correlation between the formation of anaphylatoxin and complete consumption of hemolytic complement was also observed. The obtained dated data strongly suggested that C1 esterase generates anaphylatoxin by interacting with the subsequent complement components [18]. The Figures 3(a) and (b) show the pharmacological assay of the anaphylatoxin generated on rat serum by human purified C1 esterase.
At beginning of 1960 years the introduction of new techniques in protein chemistry allowed starting purification of some admitted complement proteins, as serum inhibitor of C1 esterase [19] , enzyme C1 esterase [20] , next C2 [21], C4 [22] , and C3 and C5 [23] .
Complete mixtures containing purified preparations of human C1 esterase, C4, C2 and C3, in VBS++ buffer, pH 7.5 plus 1.0 mM MgCl2 or incomplete mixtures lacking each onecomponent, were used as substitutes for whole serum. After incubation for 60 min, at 37˚C, aliquots were removed, and the presence of biologically active substances was assayed in vitro on isolated guinea pig ileum or rat uterus isolated preparations, and on guinea pig mesentery mast cells, or in vivo on guinea pig skin. The molecular integrity of C3 was evaluated by immunoelectrophoresis. The complete mixture, but not the incompletes exhibited: a) the ability to induce guinea pig ileum contraction followed by tachyphylaxis after two consecutive additions of the some dose (Figure 3(c)); b) it was fully blocked by the antihistaminic drug triprolidine performed in parallel assays using HA, histamine and bradikainin as agonists (Figure 3(c)); c) failed to contract rat uterus; d) induces mast cell degranulation and histamine release, and cleaves C3 (Figure 3(d)). It was concluded that anaphylatoxin is a product of the complement system [24-26] .
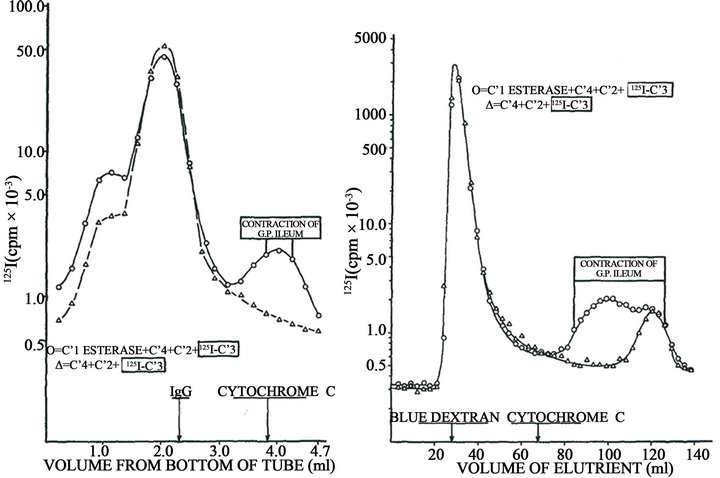
In order to demonstrate the molecular origin and nature of the biologically active product, purified preparations of human C1 esterase, C4, C2, C3, and C5 were labeled with 125I. Reaction mixtures were prepared containing a single labeled component and the other four components unlabeled. After incubation at 37˚C for 10 min at pH 7.4 in the presence of 5 × 10–4 Mg++, the pH was adjusted to 3.5 and subjected to sucrose density gradient ultracentrifugation and gel filtration at pH 3.5. In all cases the activity capable of contracting guinea pig ileum with tachyphylaxis was obtained in low molecular weight fractions. In addition, these fractions were labeled only when 125I-C3 was employed, indicating that the biological activity was associated with a cleavage product of C3 (Figure 4). This C3 fragment had a molecular weight of 6800 and exhibited all biological properties attributed to anaphylatoxin as the capacity to contract guinea pig ileum with tachyphylaxis, failure to contract rat uterus,
 (a)
(a) (b)
(b)
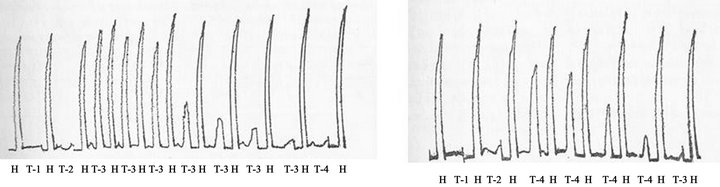
Figure 3. Cross-desensitization of isolated guinea pig ileum between anaphylatoxins generated in fresh rat serum (0.8 mL) upon incubation with 20 Units of purified human C1 esterase (T-3) or with agar (500 µg) (T-4) for 60 min at 37˚C. The mixtures were pharmacologically assayed by adding 0.1 mL of the mixtures on isolated guinea pig ileum suspended in Tyrode´s solution at 35˚C. H, 0.05 µg histamine base. Negative controls: Phosphate buffered saline plus C1 esterase (T-1) or plus fresh rat serum (T-2). (a) Desensitization of guinea pig ileum by anaphylatoxin generated by C1 esterase before contact with anaphylatoxin generated by agar. (b) Desensitization of guinea pig ileum by anaphylatoxin generated by agar before contact with anaphylatoxin generated by C1 esterase. (Dias d Silva and Lepow, 1966, Ref. # 18). (c) Generation of anaphylatoxin by incubating for 60 min at 37˚C aliquots of purified preparations of C1 esterase, C2, C3 and C4 diluted in PBS buffer containing 5 × 10–4M MgCl2 at expected normal serum concentrations, but not in the negative controls lacking C1 esterase (1C), C2 (2C), C3 (3C) or C4 (4C). The mixtures were also assayed on isolated guinea pig ileum suspended in Tyrode´s solution at 35˚C, using histamine base (H, 0.05 µg) as guinea pig ileum contraction positive control. (d) C3 cleavage in the complete mixture of purified human C1 esterase, C4, C2 and C3, but not in incomplete mixtures lacking one purified complement component, occurred in parallel with C3 cleavage from β1 original migrating protein to a α protein fragment as assayed by immunoelectrophoresis (Dias da Silva and Lepow, 1967).
enhancing the vascular permeability in guinea pig skin, degranulating mast cells in guinea pig mesentery, and release histamine from isolated rat mast cells. This C3 fragment did not cross-desensitize with guinea pig or rat agar anaphylatoxin and vice versa.
This result, coupled with immunoelectrophoretic conversion of C3 only in the complete reaction mixture, was highly suggestive of a mechanism involving cleavage of C3 to a biologically active fragment. Importantly, no active labeled fragment was detected in the mixtures in which 125I-C5, instead of 125I-C3 was used. Figure 4(a) shows these results. Generation of anaphylatoxin in HA mixture was accompanied by C3 cleavage into two fragmentsone moving faster band the other slowly (Figure 4(b)). The complete mixture HA induces skin inflammatory reaction in the injected tissue as indicated by the dilatation of small vessels, intercellular edema and afflux of neutrophils and eosinophils.
This C3 fragment was originally designed at that time F(a)C’3, latter C3a according to the WHO sponsored committee of specialist [27].
Similar conclusions were reached by [28] and by [29]. Similar results were also obtained when functional CS components were used as substitutes for the corresponding chemically isolated components. Using such preparations it was observed that the fifth component of complement that reacts hemolytically in the guinea pig or rat complement system, C’3b, is intimately associated with the generation of anaphylatoxin. Anaphylatoxin was formed 1) if this purified component reacted with a washed, intermediate complex consisting of antigen, antibody, and the first four components of complement; 2) if the component was treated with trypsin; or 3) if the component was incubated with a material isolated from
 (a) (b)
(a) (b)
Figure 4. Original data obtained by assays on isolated guinea pig ileum describing the complement origin of C3a. Left, sucrose density gradient ultracentrifugation of a reaction mixture on human purified C1 esterase, C4, C2 and 125I-C3, compared with mixture lacking C1 esterase. Right, Gel filtration on Biogel P-60 of the same mixtures. The area of detectable ileum-contracting activity refers only to the complete mixture of four components; no biological activity was present in the control mixture (Dias da Silva, Eisele and Lepow, 1967).
cobra venom together with a fraction from serum other than complement. The significance of this fifth component as anaphylatoxinogen is discussed and a common mechanism is proposed for the generation of anaphylatoxin in whole serum [30] . Amino acid sequence of the original human C3a peptide: CNYITELRRQHARASHLGLA.
C4a. The C4 cleavage product C4a exhibits similar function compared to C3a regarding smooth muscle contraction and enhancement of vascular permeability [31]. C4a release was demonstrated by treating purified human C4 with purified C1 esterase. The released peptide appeared to be homogeneous by electrophoresis on cellulose acetate and by polyacrylamide gel electrophoresis. Generated C4a is an 8.65 kDa peptide with an electrophoretic mobility at pH 8.6 of +2.1. One mol of Arg/mol of the generated C4a can be released by carboxypeptidase B. The partial COOH-terminal sequence was determined to be Leu-Gln-Arg-COOH. The isolated C4a was spasmogenic for guinea pig ileum at a concentration of 1 μM and it induced tachyphylaxis with respect C3a anaphylatoxin (at 0.33 μM) but not with respect to human C5a anaphylatoxin. These data suggested similar cell receptors for C3a and C4a, but distinct for C5a. The injection of 1 nmol of C4a induced immediate local erythema and edema formation. The spasmogenic, tachyphylactic, and vascular activities of C4a were abrogated by removal of the COOH-terminal Arg, a property that is characteristic also of the C3a and C5a anaphylatoxins [31].
C5a. Human C5a was next isolated from complementactivated serum and was characterized for protein and carbohydrate content, and compared with C3a. The purified C5a was judged to be homogeneous by both polyacrylamide gel electrophoresis and immunologic techniques. The polypeptide moiety of C5a contains 73 amino acid residues, which represent a molecular mass of 8.2 kDa. Analysis of the carbohydrate moiety in C5a indicated 4 moles of glucosamine, 3 to 4 moles of sialic acid, 4 moles of mannose and 2 moles of galactose. The total carbohydrate content in C5a corresponds approximately 25% of the apparent molecular mass of the C5a anaphylatoxin molecule. The protein and carbohydrate portions of C5a together equal a molecular mass of approximately 11 kDa which is considerably less than the indicated by physical measurements. Human C5a contains a COOH-terminal-Arg, which is essential for anaphylatoxin activity and a sequence of Gln-Leu-Gly-ArgCOOH at the COOH-terminus comparable with that of human C3a (Gly-Leu-Ala-Arg-COOH). Additional similarities between the C3a and C5a molecules include length of the polypeptide chain, number of disulfide bonds and an absence of Try residues. The differences amongst the two human anaphylatoxins include, carbohydrate is associated with C5a but is absent in the C3a molecule and little homology of the partial NH2-terminal sequence of C5a NH2-Thr-Leu-Glx-Lys-Ile-Glx-Glx-IleAla-with the corresponding NH2-terminal sequence of C3a [32] . C5a is cleaved from the N-terminal end of the C5 α-chain by a specific C5 convertase [33] during C activation [34] . Rat C5a is unusual in that the desArg derivative maintains a higher level of potency than does C5adesArg from other species [35] . IntactC5a exhibits a number of potent biological activities primarily associated with the inflammatory response. As a spasmogensub-nanomolar concentrations of C5a can induce contraction of smooth muscle tissue from a variety of animal sources [34].
C5a promotes histamine release from mast cells and basophils and can enhance vascular permeability [36,37] . Nanomolar concentrations of C5a also induce directed migration (i.e., chemotaxis) and degranulation of both leukocytes and monocytes [38-40] . The C-terminal Arg that is exposed when C5a is released from C5 is rapidly removed in vivo by a serum carboxypeptidase (serum carboxypeptidase N) [41] to produce C5adesArg. Removal of Arg and concomitant reduction in C5a potency is presumed to be a control mechanism to inactivate C5a in vivo. However, it has been shown that Arg removal reduces C5a potency to a variable extent depending both on the animal source of C5a and the biological response or assay used for measurement of C5a potency [42,43] .
Elucidation of the primary structures of human, porcine, bovine, and mouse C5a [32,44,45] indicate that these 74 residues peptide share a high degree of similarity with respect to size residues for human, porcine, and bovine and mouse as well as the primary structure (overall = 70% identity). In addition, the tertiary structures of human and porcineC5a are highly similar, as determined by NMR [46]. C5a consists of 4 helical bundles arranged in an anti-parallel orientation. The N-terminal and Cterminal regions are largely helical and are stabilized by hydrophobic side chain interactions with neighboring residues [47]. The internal “core” is stabilized by 3 disulfide linkages [46,48] .
In summary, C3a, C4a and C5a are the terminal part of the corresponding C3, C4 and C5 α-chains the first two released by the action of C3 convertases and the third by the C5 convertases. C3a and C4a are the 77 amino acid peptides removed from the C3 and C4 N-terminus of C3 and C4 molecules, while C5a is 74 amino acid peptide removed from the N-terminus of C5.
1.5. Anaphylatoxins Sequencing and Three-Dimensional Structure
The three anaphylatoxins, C3a, C4a and C5a represent the N-terminal portions of the α-chains of their respective components. They are similar in structure and functions. Anaphylatoxin C3a consists of a 74-residue polypeptide. Its N-terminal domain is intra-connected by three native disulfide bonds (Cys22-Cys49, Cys23-Cys56, and Cys36- Cys57) and a helical C-terminal domain. Its three-dimensional structure is depicted in the. Anaphylatoxin C4a consists of 74 amino acids residues. Anaphylatoxin C5a consists of a 74-residue peptide. The eight C-terminal residues, 74 to 66 aa, are disordered when the molecule is in solution. Three disulfide bounds formed by the Cys residues 21 and 47, 22 and 54, 34 and 54) are disposed around great part of the α-helix. A single carbohydrate moiety is attached to Asn 64 residue.
Circular dichroism (CD) spectra for C3a anaphylatoxins from human and porcine sources were compared in the region of 190 to 250 nm. The spectra were indistinguishable in this region, although an estimated difference of approximately 20% exists between the primary structures of human and porcine C3a. Calculations indicated a total of 40% to 45% α-helical content based on either the 208 or 222 nm extremum of the CD spectra. Biological activity was not affected upon mercaptoethanol or 6 M guanidinium chloride treatments even with the resulting decreasing in the molecule ellipicity [49]. CD spectrum distortion and biological inactivation however occur when C3a was simultaneously treated with mercaptoethanol and guanidinium chloride, both events being reversed after removal of the denaturant and reducing agents. Heat treatment or reduction and alkylation of human C3a produced biologically inactive and conformational modified anaphylatoxin. In contrast, C3a inactivated by enzymatic removal of the COOH-terminal Arg residue was structurally unchanged as judged by CD measurements. Consequently, it is proposed that in addition to the essential COOH-terminal Argresidue, a highly ordered conformation is required for biological functionality of the C3a molecule [49] .
The circular dichroism spectra of C5a in the 200 - 250 nm region at pH 7.2 and 3.7 are nearly identical of C3a anaphylatoxin. Treatment of C5a with 2-mercaptoethanol progressively diminishes the elliptic curvature at 222 nm and its anaphylatoxic activity to limiting values. Removal of the reducing agent by dialysis completely restored both the elliptic form at 222 nm and the activity. This finding indicates that the integrity of the secondary conformation of the C5a molecule is largely dependent on disulfide bonds and is essential for its full activity ADDIN EN.CITE ADDIN EN.CITE.DATA [49] .
1.6. Anaphylatoxins Inactivation
The noticeable damage caused by anaphylatoxinsare strictly in vivo controlled by naturally plasma circulating carboxypeptidase N. Once formed the anaphylatoxins C-terminal residues are removed by these enzymes yielding the C3ades-Arg derivative [50]. Carboxypeptidase N is a tetramericmetalloprotease, belonging to a family of a protein-cleaving, metal atom-containing enzymes [51]. Although C3a-desArg is completely free of inflammatory properties, C5-desArg retains around 1% - 10% of this activity [52] .
1.7. Anaphylatoxin Cell Targets
Anaphylatoxin receptors are expressed on mast cells, granulocytes, smooth muscle, endothelial cells, and monocytes-macrophages-dendriticcells.Upon interacting with the corresponding anaphylatoxins they are activated responding with release of inflammatory mediators and/or accumulation in inflammation sites.
1.8. Anaphylatoxins Receptors
To exert their pharmacological effects anaphylatoxins first bind to specific receptors, collectively designed ATRs. The existence of these anaphylatoxin-type recaptors wasanticipated when C3a was described by experiments showing clearly that guinea pig ileum preparations first desensitized to C3a, continues responding by the usual contraction to C5a, and vice-versa. ATRs, C3aR, C5aR and C5a-like receptor belong to a super-family of G-proteins coupled receptors GPCRs.
GPCRs exist as a superfamily of integral membrane protein receptors that contain seven transmembrane alpha-helical regions, which bind to a wide range of ligands. Upon activation by the specific ligand, the GPCR undergoes a conformational change and then activate the G proteins by promoting the exchange of GDP/GTP associated with the Galpha subunit. This leads to the dissociation of Gbeta/Ggamma dimmer from Galpha. Both these moieties then become free to act upon their downstream effectors and thereby initiate unique intracellular signaling responses. After the signal propagation, the GTP of Galpha-GTP is hydrolyzed to GDP and Galpha becomes inactive (Galpha-GDP), which leads to its reassociation with the Gbeta/Ggamma dimmer to form the inactive heterotrimeric complex. The GPCR can also transduce the signal through G protein independent pathway. GPCRs also regulate cell cycle progression [53] . Activation of GPCRs leads to the dissociation of heterotrimeric G-proteins into Gα and Gβγ subunits, which go on to regulate various effectors involved in a panoply of cellular responses. During chemotaxis, Gβγ subunits regulate actin assembly and migration, but the protein(s) linking Gβγ to the actin cytoskeleton remains unknown. The Gβγ effector, ElmoE in Dictyostelium, had demonstrated that it is required for GPCR-mediated chemotaxis [54] . As ElmoE associates with Gβγ and Dock-like proteins to activate the small GTPaseRac, in a GPCR-dependent manner, and also associates with Arp2/3 complex and F-actin, ElmoE serves as a link between chemoattractant GPCRs, G-proteins and the actin cytoskeleton. The pathway, consisting of GPCR, Gβγ, Elmo/Dock, Rac, and Arp2/3, spatially guides the growth of dendritic actin networks in pseudopods of eukaryotic cells during chemotaxis [54] .
C3aR. This receptor is expressed on a large variety of cells as of myeloid origin like mast cells, basophils, neutrophils, eosinophils, monocytes-macrophages, dendritic cells and microglia [55-60] , as well as in non-myeloid cells like astrocytes [61,62] , endothelial cells [63] , and epithelioid cells, smooth muscle cells and parenchymal cells [64] . The presence of C3aR on activated but not non-activated T lymphocytes has also been reported [65] . C3aR is a 482 amino acids molecule (54 kDa) protein glycosylated at the Asp-9 and Asp-195 residues and sulfated at the Tyr-174 [66,67] . The protein second extracellular loop is it the larger part accommodating the sulfated Tir-174 important molecular region involved on C3a-C3aR interactions [68]. Upon interaction the resulting signal is transduced via heterotrimeric G-proteins mobilizing Ca++ fluxes from extra-cellular stores. G protein coupled receptors (GPCRs) comprise a superfamily of proteins involved in transducing signals emitted by the partner receptors upon it combining with the specific agonists. Over eight hundred GPCRs from various cell types have been cloned [69] . The next signaling steps diverge with the cell type. In endothelial cells C3aR is connected with pertussis-toxin insensitive Gα16 [70] , following activation of protein-kinase C by phospholipase C. In astrocytes the protein (MAP) kinases Erk1 and Erk2 are activated [52,71] . In mast cells C3aR binding by C3a activates cytokine production through a complex intracellular signals involving PI3K, Akt-phosohorylation and MAP kinase Erk1 and ERK2 [72] .
C5aR. C5a exerts its effects through the C5a receptor (C5aR or CD88), also a member of the rhodopsin family of seven-transmembrane(seven-TM) G protein-coupled receptors [73]. The membrane bound protein C5aR (CD88) is also expressed in many cell types. Higher expression is found in neutrophils, eosinophils, basophils, monocytes-macrophages, dendritic cells and mast cells [38,43,74-76] . C5aR expression in lymphocytes subtype cells is controversial. Although some reports confirms its presence [77,78] ; other indicates its absence [79-81] , or scarcely near the negative controls in some population of lymphoid cells [82] or even not detected by anti-C5aE antibodies [83]. GPCRs perceive many extracellular signals and transduce them to heterotrimeric G proteins, which further transduce these intracellular signals to appropriate downstream effectors allowing play important role in various signaling pathways.
1.9. Anaphylatoxin Antagonists
The anaphylatoxins activities can be blocked by specific antibodies directed to their relevant immunogenic epitopes and by competitive antagonists with their cellular receptors accessing molecular domains.
1.10. Specific Antibodies
Development of blocking monoclonal antibodies (mAb) to prevent C3, C4 or C5 cleavage and the subsequent C3a, C4a and C5a formation is a potential therapeutic approach to treat complement-induced inflammation.
C3amAb. Monoclonal Ab anti-C3a blocks in vitro and in vivo the activities of C3a.
C5amAb. Rat anti-mouse C5 mAb (BB5.1) was utilized to investigate the effects of inhibition of C5 cleavage on disease progression and severity in experimental autoimmune uveoretinitis (EAU), a model of organ-specific autoimmunity in the eye characterized by structural retinal damage mediated by infiltrating macrophages. In vivo, systemic treatment with BB5.1 results in significantly reduced disease scores compared with control groups, while local administration results in an earlier resolution of disease. Systemic treatment with BB5.1 results in significantly reduced disease scores compared with control groups, while local administration results in an earlier resolution of disease [84] . Structural and functional studies of the interaction between the mAb F20 and C5a was used to investigate the functional epitope in C5a. Association of molecular modeling, docking methods and biological mutant experiment, were used to identify the key epitope. Based on theoretically and predicted discreet sites in antigen and antigen the key epitope in the antigen was identified. The predicted results allowed determine Lys68 in C5a as the key C5a epitope. This epitope can be used in C5a antagonist screening and design [85] .
1.11. Competitive Antagonists
The central strategy used is the competitive blockage the anaphylatoxins effective interaction with their cellular receptors. Homologous peptides mimicking the anaphylatoxin peptides sequences recognized by their cell target receptors are the useful focus.
In a pioneering approach the compound N2-[(2,2-diphenylethoxy) acetyl]-L-arginine designated SB 290157 was delineated having as model a molecule selected from 240,000 compounds synthesized by SmithKline. The molecule N2-[(2,2-diphenylethoxy)acetic acid]-L-arginine (SK&F 63649) serving as immediate template to synthesize SB 290157 was used as control in agonist versus antagonists assays.
The ability of the modeled molecules to competitively binding with C3a for C3aR was analyzed in vitro and in vivo assays. First, membranes of RBL-C3aR cells were prebound to SPA beads mixed previously with SB 290157 or SK&F 63649 compounds, next with the 125-C3a radioligand in appropriated buffer, and bound radiolabeled ligand measured by counting. 125IC3a binds to RBL-C3aR cells with high affinity (Kd = 8pM) and is saturated. The detected affinities of SB 290157 and SK&F 63649 were 200 and 3000 nM, respectively. To determine whether the competitive binding reflected on functional activity of C3a, a FLIPR-based C3a-induced Ca++ mobilization assay in RBL-C3aR measured in terms of IC50S units were more efficient with SB 290157 (30 nM) than with SK&F 63649 (308 nN). As C3a also induces mast cell chemotaxis, the ability of the SB 290157 compound to attract inflammatory cells was tested on HMC-1, a human mast cell line that naturally expresses the C3aR and for which C3a is chemotactic. At the concentration of 5μM SB 2901157 markedly inhibition of C3a-mediated chemotaxis of HMC-cells was detected. This effect was not detected on C5a-mediated chemotaxis. SB 290157 compound exhibited also potent ability to inhibit C3a-induced internalization of the C3aR [86] .
In in vivo animals model the anti-C3a antagonist SB 290157 was also capable of inhibiting some C3a manifestation as neutrophil recruitment in a guinea pig, the LPS-induced neutrophilia in airway model and the paw edema in a rat adjuvantinduced arthritis.
The key strategy in the construction of N2-[(2, 2- diphenylethoxy) acetyl]-L-arginine (SB290157) was the selection of Arg, the essential amino acid residue involved in the interaction of the C3a with C3aR. The compound [(2,2-Diphenylethoxyl)acetyl]-L-arginine expresses higher C3a-C3aR competitive activity with the Diphenyl radicals separately attached to the acetic-Larginine moiety.
1.12. Anaphylatoxins and Inflammation
Complement activation at local tissue sites forms a gradient of anaphylatoxins C3a, C4a and C5a which due to their properties to contract smooth muscles, to attract neutrophils, eosinophils, monocytes-macrophages and to activate these cells as well as resident mast cells and endothelial cells, unequivocally participate in the early events of the inflammatory process. Other biological anaphylatoxins attributes including regulation of cationic protein synthesis by eosinophils, adhesion of these cells to endothelial cells [87,88] , stimulation of serotonin release from platelets and modulation of the synthesis of IL-6 and TNF-α by B lymphocytes and monocytes by C3a [89-91] , expands further the anaphylatoxins roles in inflammation. In situ complement activation strongly supports the involvement of these active peptides in the pathogenesis of important lesions. Activation of the CS is also a common event along tissue homing of microbes or in non-infective lesions. The participation of anaphylatoxins have been demonstrated in the pathogenesis of several pathological conditions as in asthma [92] , in the diarrhea-associated hemolytic syndrome and acute renal failure induced by E. coli-producing Shiga toxin [93-95] , in the liver regenerative process [96] , and in arthritis [97] .
2. Concluding Remarks
Since the unequivocal demonstration that anaphylatoxins are pieces of three complement components, C3, C4 and C5, emerging during the biologically activation of this system, a continued prolific scientific dedication was invested to clarify their nature, biological functions, and involvement in the pathogenesis of important human disease.
3. Acknowledgements
We wish to recall particularly to three scientists for their very idealistic vision of complement system either as a basic source of fundamental information as well as of promising applications in pathology:
Dr. Irving H. Lepow (In memoriam). The complement system, a monotone secondarily reagent used in the past years to identify some preformed immune complexes, has become along the recent fifty years a fascinating source of first class of important biologically mediator agents. Amongst the distinguished scientists deeply involved in investigating the nature, properties, origin and destiny of the now over thirteen complement proteins, Dr. Irving H. Lepow certainly is included. Particular complement subjects received his undeniable creativity signature: the complement alternative pathway with the demonstration that properdin is not a “natural anti-polysaccharide antibody”, that factor B is a pro-enzyme, and that C3 is part and target of the C3 convertase, and the direct source of anaphylatoxin C3a.
Dra. Thereza Liberman Kipnis (In memoriam). The first clear interplay between complement system and Trypanosoma cruzi, the causal agent of Chaga’s disesase, was clarified by her description of DAF-like components present in infective and complement resistant trypomastigotes surface but absent in complement susceptible non-infective epimastigote forms of this parasites. Her particular ability to identify young students potentially capable of pursuing good scientific research was also undeniable.
Dra. Denise V. Tambourgi. For her creativity and dignifying persistence in pursuing successfully fundamental data involved on interactions between Trypanosoma cruziand animal toxinswith the complement.
We thank Estevam José Baldon and Angela Alice Amadeu for the kind assistance in preparing and editing the manuscript.
Finally, we thank our many past and present students, for their patience in addressing to our talks on anaphylatoxins details.
This work was conducted along contract with the “Fundação Butantan—Instituto Butantan” and supported also by National Counsel of Technological and Scientific Development—CNPq (Bolsa de Produtividade, (WDS) Nível 1A, Proc. No: 301836/2005-1).
REFERENCES
- I. Dally, S. Peat and H. Schild, “The Release of a Histamine-Like Substance from the Lungs of Guinea Pigs during Anaphylactic Shock,” Experimental Physiology, Vol. 25, No. 1, 1935, pp. 33-59.
- J. Bordet, “Gélose et Anaphylatoxine,” Comptes rendus des Seances de la Societe de Biologie, Vol. 74, 1913, p. 877.
- E. Nathan, “Über Anaphylatoxinbildund durch Inulin Zuglich ein Beitrag über die Bedeutung des Physiukalischen Zustandes für Anaphylatoxinbildung,” ZeitschriftfürImmunitätsforschung, Vol. 23, 1915, p. 204.
- F. G. Novy and P. D. Kruif, “Anaphylatoxin and Anaphylaxis. III. Ager Anaphylatoxin: Rabbit Serum. IV. Agar Anaphylatoxin: Rat Serum,” The Journal of Infectious Diseases, Vol. 20, No. 5, 1913, pp. 499-535. doi:10.1093/infdis/20.5.499
- H. H. Dale and C. H. Kelleway, “Anaphylaxis and Anaphylatoxins,” Philosophical Transactions of the Royal Society, Vol. 211, No. 382-390, 1922, pp. 273-315. doi:10.1098/rstb.1923.0006
- R. Bartosch, W. Feldberg and E. Nagel, “Das Freiwerden eines Histaminähnlichen Stoffes bey der Anaphylxie des Meerschweinchens,” Pflüger’s Archiv für die Gesamte Physiologie des Menschen und der Tiere, Vol. 230, No. 1, 1932, pp. 129-153. doi:10.1007/BF01751974
- H. O. Schild, “Histamine Release in Anaphylactic Shock from Various Tissues of the Guinea-Pig,” The Journal of Physiology, Vol. 95, No. 3, 1939, pp. 393-403.
- M. R. E. Silva, O. G. Bier and M. Aronson, “Histamine Release by Anaphylaxis,” Nature, Vol. 168, 1951, pp. 465- 466. doi:10.1038/168465a0
- A. G. Osler, H. G. Randall, B. M. Hill and Z. Ovary, “Studies on the Mechanism of Hypersensitivity Phenomena. III. The Participation of Complement in the Formation of Anaphylatoxin,” The Journal of Experimental Medicine, Vol. 110, No. 2, 1959, pp. 311-339.
- M. W. Turner and J. B. Nativig, “Nomenclature of Complement,” In: M. W. Turner and J. B. Nativig, Eds., Immunology Nomenclature, Hogrefe & Huber Publishers, Seatle, 1999, pp. 2-93.
- B. F. Tack, R. A. Harrison, J. Janatova, M. L. Thomas and J. W. Prahl, “Evidence for Presence of an Internal Thiolester Bond in Third Component of Human Complement,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 77, No. 10, 1980, pp. 5764-8576.
- J. C. Jensenius, “The Mannan-Binding Lectin (MBL) Pathway of Complement Activation: Biochemistry, Biology and Clinical Implications,” Advances in Experimental Medicine and Biology, Vol. 564, 2005, pp. 21-22. doi:10.1007/0-387-25515-X_6
- M. W. Turner and R. M. Hamvas, “Mannose-Binding Lectin: Structure, Function, Genetics and Disease Associations,” Reviews in Immunogenetics, Vol. 2, No. 3, 2000, pp. 305-322.
- J. K. van de Wetering, L. M. van Golde and J. J. Batenburg, “Collectins: Players of the Innate Immune System,” European Journal of Biochemistry, Vol. 271, No. 7, 2004, pp. 1229-1249. doi:10.1111/j.1432-1033.2004.04040.x
- O. Ratinoff and I. H. Lepow, “Some Properties of an Esterase Derived from Preparations of the First Component of Complement,” The Journal of Experimental Medicine, Vol. 106, No. 2, 1957, pp. 327-343. doi:10.1084/jem.106.2.327
- O. D. Ratinoff and I. H. Lepow, “Complement as Mediator Inflammation: Enhancement of Vascular Permeability by Purified Human C1 Esterase,” The Journal of Experimental Medicine, Vol. 118, No. 5, 1963, pp. 681-698. doi:10.1084/jem.118.5.681
- R. D. Smink Jr., R. W. Abernethy, O. D. Ratnoff and I. H. Lepow, “Enhancement of Vascular Permeability by Mixtures of C’1 Esterase and Normal Serum,” Proceedings of the Society for Experimental Biology and Medicine, Vol. 116, No. 2, 1964, pp. 280-283.
- W. D. Silva and H. I. Lepow, “Anaphylatoxin Formation by Purified C1 Esterase,” The Journal of Immunology, Vol. 95, 1965, pp. 1080-1089.
- J. Pensky, L. R. Levy and I. H. Lepow, “Partial Purification of a Serum Inhibitor of C’1-Esterase,” The Journal of Biological Chemistry, Vol. 236, 1961, pp. 1674-1679.
- A. L. Haines and I. H. Lepow, “Studies on Human C’1-Esterase. I. Purification and Enzymatic Properties,” The Journal of Immunology, Vol. 92, 1964, pp. 456-467.
- I. H. Lepow, E. W. Todd, R. D. Smink and J. Pensky, “Purification of the Second Component (C2) of Human Complement,” Federation Proceedings, Vol. 24, 1965, p. 446.
- H. J. Muller-Eberhard and C. E. Biro, “Isolation and Description of the Fourth Component of Human Complement,” The Journal of Experimental Medicine, Vol. 118, No. 3, 1963, pp. 447-466. doi:10.1084/jem.118.3.447
- H. J. Muller-Eberhard, U. Nilsson and T. Aronsson, “Isolation and Characterization of Two Beta1-Glycoproteins of Human Serum,” The Journal of Experimental Medicine, Vol. 111, No. 2, 1960, pp. 201-215. doi:10.1084/jem.111.2.201
- W. D. Silva, J. W. Eisele and I. H. Lepow, “Complement as a Mediator of Inflammation. III. Purification of the Activity with Anaphylatoxin Properties Generated by Interaction of the First Four Components of Complement and Identification as a Cleavage Product of C3,” The Journal of Experimental Medicine, Vol. 126, No. 6, 1967, p. 1027. doi:10.1084/jem.126.6.1027
- W. D. Silva and H. I. Lepow, “Properties of Anaphylatoxin Prepared from Purified Components of Human Complement,” Biochemistry, Vol. 3, 1966, p. 497.
- W. D. Silva and I. H. Lepow, “Complement as a Mediator of Inflammation. II. Biological Properties of Anaphylatoxin Prepared with Purified Components of Human Complement,” The Journal of Experimental Medicine, Vol. 125, No. 5, 1967, pp. 921-946.
- WHO, “Nomenclature of Complement,” Bulletin of the World Health Organization, 1968, pp. 935-938.
- H. J. Muller-Eberhard, M. J. Polley and M. A. Calcott, “Formation and Functional Significance of a Molecular Complex Derived from the Second and the Fourth Component of Human Complement,” The Journal of Experimental Medicine, Vol. 125, No. 2, 1967, pp. 359-380.
- C. G. Cochrane and H. J. Müller-Eberhard, “Biological Effects of C3 Fragmentation,” Federation Proceedings, Vol. 26, 1967, p. 362.
- J. Jensen, “Anaphylatoxin in Its Relation to the Complement System,” Science, Vol. 155, No. 3766, 1967, pp. 1122-1123.
- J. P. Gorski, T. E. Hugli and H. J. Muller-Eberhard, “C4a: The Third Anaphylatoxin of the Human Complement System,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 76, No. 10, 1979, pp. 5299-5302.
- H. N. Fernandez and T. E. Hugli, “Partial Characterization of Human C5a Anaphylatoxin. I. Chemical Description of the Carbohydrate and Polypeptide Prtions of Human C5a,” The Journal of Immunology, Vol. 117, No. 5, 1976, pp. 1688-1694.
- T. E. Hugli, “Biochemistry and Biology of Anaphylatoxins,” Complement, Vol. 3, No. 3, 1986, pp. 111-127.
- T. E. Hugli, “Chemical Aspects of the Serum Anaphylatoxins,” Contemporary Topics in Molecular Immunology, Vol. 7, 1978, pp. 181-214.
- T. E. Hugli, F. Marceau and C. Lundberg, “Effects of Complement Fragments on Pulmonary and Vascular Smooth Muscle,” The American Review of Respiratory Disease, Vol. 135, No. 6, 1987, pp. S9-S13.
- A. R. Johnson, T. E. Hugli and H. J. Muller-Eberhard, “Release of Histamine from Rat Mast Cells by the Complement Peptides C3a and C5a,” Immunology, Vol. 28, No. 6, 1975, pp. 1067-1080.
- P. J. Jose, M. J. Forrest and T. J. Williams, “Human C5a des Arg Increases Vascular Permeability,” The Journal of Immunology, Vol. 127, No. 6, 1981, pp. 2376-2380.
- D. E. Chenoweth and M. G. Goodman, “The C5a Receptor of Neutrophils and Macrophages,” Agents and Actions Supplement, Vol. 12, 1983, pp. 252-273.
- D. E. Chenoweth and T. E. Hugli, “Human C5a and C5a Analogs as Probes of the Neutrophil C5a Receptor,” Molecular Immunology Journal, Vol. 17, No. 2, 1980, pp. 151-161.
- M. S. Hausman, R. Snyderman and S. E. Mergenhagen, “Humoral Mediators of Chemotaxis of Mononuclear Leukocytes,” The Journal of Infectious Diseases, Vol. 125, No. 6, 1972, pp. 595-602.
- C. Gerard and T. E. Hugli, “Identification of Classical Anaphylatoxin as the Des-Arg form of the C5a Molecule: Evidence of a Modulator Role for the Oligosaccharide Unit in Human Des-Arg74-C5a,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 78, No. 3, 1981, pp. 1833-1837.
- N. P. Gerard and C. Gerard, “The Chemotactic Receptor for Human C5a Anaphylatoxin,” Nature, Vol. 349, No. 6310, 1991, pp. 614-617.
- N. P. Gerard, M. K. Hodges, J. M. Drazen, P. F. Weller and C. Gerard, “Characterization of a Receptor for C5a Anaphylatoxin on Human Eosinophils,” The Journal of Biological Chemistry, Vol. 264, No. 3, 1989, pp. 1760- 1766.
- H. N. Fernandez, P. M. Henson, A. Otani and T. E. Hugli, “Chemotactic Response to Human C3a and C5a Anaphylatoxins. I. Evaluation of C3a and C5a Leukotaxis in Vitro and Under Stimulated in Vivo Conditions,” The Journal of Immunology, Vol. 120, No. 1, 1978, pp. 109-115.
- R. A. Wetsel, “Expression of the Complement C5a Anaphylatoxin Receptor (C5aR) on Non-Myeloid Cells,” Immunology Letters, Vol. 44, No. 2-3, 1995, pp. 183-187.
- M. P. Williamson and V. S. Madison, “Three-Dimensional Structure of Porcine C5adesArg from 1H Nuclear Magnetic Resonance Data,” Biochemistry, Vol. 29, No. 12, 1990, pp. 2895-2905.
- D. F. Carney and T. E. Hugli, “Interaction of the N-Terminal Region of Human C5a with the Neutrophil C5a Receptor,” Protein Science, Vol. 2, 1993, pp. 1391-1399. doi:10.1002/pro.5560020904
- E. R. Zuiderweg, D. G. Nettesheim, K. W. Mollison and G. W. Carter, “Tertiary Structure of Human Complement Component C5a in Solution From Nuclear Magnetic Resonance Data,” Biochemistry, Vol. 28, No. 1, 1989, 172-185.
- T. E. Hugli, W. T. Morgan and H. J. Muller-Eberhard, “Circular Dichroism of C3a Anaphylatoxin. Effects of pH, Heat, Guanidinium Chloride, and Mercaptoethanol on Conformation and Function,” The Journal of Biological Chemistry, Vol. 250, No. 4, 1975, pp. 1479-1483.
- V. A. Bokisch and H. J. Muller-Eberhard, “Anaphylatoxin Inactivator of Human Plasma: Its Isolation and Characterization as a Carboxypeptidase,” Journal of Clinical Investigation, Vol. 49, No. 12, 1970, pp. 2427-2436.
- E. W. Mathews, S. L. Mueller-Ortiz and R. A. Wetsel, “Carboxypeptidases Pleiotropic Regulator of Inflammation,” Molecular Immunology, Vol. 40, No. 11, 2004, pp. 785-793.
- S. Sayah, A. C. Jauneau, C. Patte, M. C. Tonon, H. Vaudry and M. Fontaine, “Two Different Transduction Pathways Are Activated by C3a and C5a Anaphylatoxins on Astrocytes,” Brain Research. Molecular Brain Research, Vol. 112, No. 1-2, 2003, pp. 53-60.
- N. Tuteja, “Signaling through G Protein Coupled Receptors,” Plant Signaling and Behavior, Vol. 4, No. 10, 2009, pp. 942-947.
- J. Yan, V. Mihaylov, X. Xu, J. A. Brzostowski, H. Li, L. Liu, T. D. Veenstra, C. A. Parent and T. Jin, “A Gbetagamma Effector, ElmoE, transduces GPCR Signaling to the Actin Network During Chemotaxis,” Developmental Cell, Vol. 22, No. 1, 2012, pp. 92-103.
- P. J. Daffern, P. H. Pfeifer, J. A. Ember and T. E. Hugli, “C3a Is a Chemotaxin for Human Eosinophils but Not for Neutrophils. I. C3a Stimulation of Neutrophils Is Secondary to Eosinophil Activation,” The Journal of Experimental Medicine, Vol. 181, No. 6, 1995, pp. 2119-2127.
- M. M. Glovsky, T. E. Hugli, T. Ishizaka, L. M. Lichtenstein and B. W. Erickson, “Anaphylatoxin-Induced Histamine Release with Human Leukocytes: Studies of C3a Leukocyte Binding and Histamine Release,” Journal of Clinical Investigation, Vol. 64, No. 3, 1979, pp. 804-811.
- R. Gutzmer, M. Lisewski, J. Zwirner, S. Mommert, C. Diesel, M. Wittmann, A. Kapp and T. Werfel, “Human Monocyte-Derived Dendritic Cells Are Chemoattracted to C3a after Up-Regulation of the C3a Receptor with Interferons,” Immunology, Vol. 111, No. 4, 2004, pp. 435-443.
- A. Klos, S. Bank, C. Gietz, W. Bautsch, J. Kohl, M. Burg and T. Kretzschmar, “C3a Receptor on dibutyryl-cAMPDifferentiated U937 Cells and Human Neutrophils: The Human C3a Receptor Characterized by Functional Responses and 125I-C3a Binding,” Biochemistry, Vol. 31, No. 46, 1992, pp. 11274-11282.
- J. Zwirner, O. Gotze, A. Sieber, A. Kapp, G. Begemann, T. Zuberbier and T. Werfel, “The Human Mast Cell Line HMC-1 Binds and Responds to C3a but Not C3a(desArg),” Scandinavian Journal of Immunology, Vol. 47, No. 1, 1998, pp. 19-24.
- [61] J. Zwirner, T. Werfel, H. C. Wilken, E. Theile and O. Gotze, “Anaphylatoxin C3a but Not C3a(desArg) Is a Chemotaxin for the Mouse Macrophage Cell Line J774,” European Journal of Immunology, Vol. 28, No. 5, 1998, pp. 1570-1577.
- [62] P. Gasque, S. K. Singhrao, J. W. Neal, P. Wang, S. Sayah, M. Fontaine and B. P. Morgan, “The Receptor for Complement Anaphylatoxin C3a Is Expressed by Myeloid Cells and Nonmyeloid Cells in Inflamed Human Central Nervous System: Analysis in Multiple Sclerosis and Bacterial Meningitis,” The Journal of Immunology, Vol. 160, No. 7, 1998, pp. 3543-3554.
- [63] A. Ischenko, S. Sayah, C. Patte, S. Andreev, P. Gasque, M. T. Schouft, H. Vaudry and M. Fontaine, “Expression of a Functional Anaphylatoxin C3a Receptor by Astrocytes,” Journal of Neurochemistry, Vol. 71, No. 6, 1998, pp. 2487-2496.
- [64] T. Monsinjon, P. Gasque, P. Chan, A. Ischenko, J. J. Brady and M. C. Fontaine, “Regulation by Complement C3a and C5a Anaphylatoxins of Cytokine Production in Human Umbilical Vein Endothelial Cells,” The FASEB Journal, Vol. 17, No. 9, 2003, pp. 1003-1014.
- [65] L. Fregonese, F. J. Swan, A. van Schadewijk, M. Dolhnikoff, M. A. Santos, M. R. Daha, J. Stolk, T. Tschernig, P. J. Sterk, P. S. Hiemstra, K. F. Rabe and T. Mauad, “Expression of the Anaphylatoxin Receptors C3aR and C5aR Is Increased in Fatal Asthma,” Journal of Allergy and Clinical Immunology, Vol. 115, No. 6, 2005, pp. 1148-1154.
- [66] T. Werfel, K. Kirchhoff, M. Wittmann, G. Begemann, A. Kapp, F. Heidenreich, O. Gotze and J. Zwirner, “Activated Human T Lymphocytes Express a Functional C3a Receptor,” The Journal of Immunology, Vol. 165, No. 11, 2000, pp. 6599-6605.
- [67] R. S. Ames, Y. Li, H. M. Sarau, P. Nuthulaganti, J. J. Foley, C. Ellis, Z. Zeng, K. Su, A. J. Jurewicz, R. P. Hertzberg, D. J. Bergsma and C. Kumar, “Molecular Cloning and Characterization of the Human Anaphylatoxin C3a Receptor,” The Journal of Biological Chemistry, Vol. 271, No. 34, 1996, pp. 20231-20234.
- [68] T. Crass, R. S. Ames, H. M. Sarau, M. A. Tornetta, J. J. Foley, J. Kohl, A. Klos and W. Bautsch, “Chimeric Receptors of the Human C3a Receptor and C5a Receptor (CD88),” The Journal of Biological Chemistry, Vol. 274, No. 13, 1999, pp. 8367-8370.
- [69] J. Gao, H. Choe, D. Bota, P. L. Wright, C. Gerard and N. P. Gerard, “Sulfation of Tyrosine 174 in the Human C3a Receptor Is Essential for Binding of C3a Anaphylatoxin,” The Journal of Biological Chemistry, Vol. 278, No. 39, 2003, pp. 37902-37908.
- [70] R. B. Penn, A. N. Pronin and J. L. Benovic, “Regulation of G Protein-Coupled Receptor Kinases,” Trends in Cardiovascular Medicine Journal, Vol. 10, No. 2, 2000, pp. 81-89.
- [71] I. U. Schraufstatter, K. Trieu, L. Sikora, P. Sriramarao and R. DiScipio, “Complement C3a and C5a Induce Different Signal Transduction Cascades in Endothelial Cells,” The Journal of Immunology, Vol. 169, No. 4, 2002, pp. 2102-2110.
- [72] P. Langkabel, J. Zwirner and M. Oppermann, “LigandInduced Phosphorylation of Anaphylatoxin Receptors C3aR and C5aR Is Mediated by G Protein-Coupled Receptor Kinases,” European Journal of Immunology, Vol. 29, No. 9, 1999, pp. 3035-3046.
- [73] R. T. Venkatesha, E. B. Thangam, A. K. Zaidi and H. Ali, “Distinct Regulation of C3a-Induced MCP-1/CCL2 and Rantes/CCL5 Production in Human Mast Cells by Extracellular Signal Regulated Kinase and PI3 Kinase,” Molecular Immunology Journal, Vol. 42, No. 5, 2005, pp. 581-587.
- [74] J. A. Ember and T. E. Hugli, “Complement Factors and Their Receptors,” Immunopharmacology, Vol. 38, No. 1- 2, 1997, p. 3. doi:10.1016/S0162-3109(97)00088-X
- [75] D. E. Chenoweth and T. E. Hugli, “Demonstration of Specific C5a Receptor on Intact Human Polymorphonuclear Leukocytes,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 75, No. 8, 1978, pp. 3943-3947.
- [76] A. Morelli, A. Larregina, I. Chuluyan, E. Kolkowski and L. Fainboim, “Expression and Modulation of C5a Receptor (CD88) on Skin Dendritic Cells. Chemotactic Effect of C5a on Skin Migratory Dendritic Cells,” Immunology, Vol. 89, No. 1, 1996, pp. 126-134.
- [77] T. Werfel, M. Oppermann, G. Begemann, O. Gotze and J. Zwirner, “C5a Receptors Are Detectable on Mast Cells in Normal Human Skin and in Psoriatic Plaques but Not in Weal and Flare Reactions or in Uticaria Pigmentosa by Immunohistochemistry,” Archives of Dermatological Research, Vol. 289, No. 2, 1997, pp. 83-86.
- [78] S. Nataf, N. Davoust, R. S. Ames and S. R. Barnum, “Human T Cells Express the C5a Receptor and Are Chemoattracted to C5a,” The Journal of Immunology, Vol. 162, No. 7, 1999, pp. 4018-4023.
- [79] S. Nataf, P. F. Stahel, N. Davoust and S. R. Barnum, “Complement Anaphylatoxin Receptors on Neurons: New Tricks for Old Receptors?” Trends in Neurosciences, Vol. 22, No. 9, 1999, pp. 397-402.
- [80] M. A. Connelly, R. A. Moulton, A. K. Smith, D. R. Lindsey, M. Sinha, R. A. Wetsel and C. Jagannath, “Mycobacteria-Primed Macrophages and Dendritic Cells Induce an Up-Regulation of Complement C5a Anaphylatoxin Receptor (CD88) in CD3+ Murine T Cells,” Journal of Leukocyte Biology, Vol. 81, No. 1, 2007, pp. 212- 220.
- [81] M. G. Strainic, J. Liu, D. Huang, F. An, P. N. Lalli, N. Muqim, V. S. Shapiro, G. R. Dubyak, P. S. Heeger and M. E. Medof, “Locally Produced Complement Fragments C5a and C3a Provide Both Costimulatory and Survival Signals to Naive CD4+ T Cells,” Immunity, Vol. 28, No. 3, 2008, pp. 425-435.
- [82] P. N. Lalli, M. G. Strainic, M. Yang, F. Lin, M. E. Medof and P. S. Heeger, “Locally Produced C5a Binds to T CellExpressed C5aR to Enhance Effector T-Cell Expansion by Limiting Antigen-Induced Apoptosis,” Blood, Vol. 112, No. 5, 2008, pp. 1759-1766.
- [83] D. E. van Epps and D. E. Chenoweth, “Analysis of the Binding of Fluorescent C5a and C3a to Human Peripheral Blood Leukocytes,” The Journal of Immunology, Vol. 132, No. 6, 1984, pp. 2862-287.
- [84] A. Soruri, S. Kim, Z. Kiafard and J. Zwirner, “Characterization of C5aR Expression on Murine Myeloid and Lymphoid Cells by the Use of a Novel Monoclonal Antibody,” Immunology Letters, Vol. 88, No. 1, 2003, pp. 47-52.
- [85] D. A. Copland, K. Hussain, S. Baalasubramanian, T. R. Hughes, B. P. Morgan, H. Xu, A. D. Dick and L. B. Nicholson, “Systemic and Local Anti-C5 Therapy Reduces the Disease Severity in Experimental Autoimmune Uveoretinitis,” Clinical & Experimental Immunology, Vol. 159, No. 3, 2010, pp. 303-314.
- [86] H. Wei, Z. Lin, J. Feng, H. Peng, R. Guo, G. Han, S. Geng, X. Lang, Y. Sun, B. Shen and Y. Li, “Identification of Conformational Core Epitope Lys(6)(8) in C5a Based on the 3-D Modeling Complex C5a and Its Functional Antibody F20,” Molecular Immunology Journal, Vol. 48, No. 12-13, 2011, pp. 1377-1383.
- [87] R. S. Ames, D. Lee, J. J. Foley, A. J. Jurewicz, M. A. Tornetta, W. Bautsch, B. Settmacher, A. Klos, K. F. Erhard, R. D. Cousins, A. C. Sulpizio, J. P. Hieble, G. McCafferty, K. W. Ward, J. L. Adams, W. E. Bondinell, D. C. Underwood, R. R. Osborn, A. M. Badger and H. M. Sarau, “Identification of a Selective Nonpeptide Antagonist of the Anaphylatoxin C3a Receptor That Demonstrates Antiinflammatory Activity in Animal Models,” The Journal of Immunology, Vol. 166, No. 10, 2001, pp. 6341-6348.
- [88] R. G. DiScipio, P. J. Daffern, M. A. Jagels, D. H. Broide and P. Sriramarao, “A Comparison of C3a and C5a-Mediated Stable Adhesion of Rolling Eosinophils in Postcapillary Venules and Transendothelial Migration in Vitro and in Vivo,” The Journal of Immunology, Vol. 162, No. 2, 1999, pp. 1127-1136.
- [89] S. Takafuji, K. Tadokoro, K. Ito and C. A. Dahinden, “Degranulation from Human Eosinophils Stimulated with C3a and C5a,” International Archives of Allergy and Immunology, Vol. 104, Suppl. 1, 1994, pp. 27-29.
- [90] W. H. Fischer and T. E. Hugli, “Regulation of B Cell Functions by C3a and C3a(desArg): Suppression of TNFAlpha, IL-6 and the Polyclonal Immune Response,” The Journal of Immunology, Vol. 159, No. 9, 1997, pp. 4279- 4286.
- [91] W. H. Fischer, M. A. Jagels and T. E. Hugli, “Regulation of IL-6 Synthesis in Human Peripheral Blood Mononuclear Cells by C3a and C3a(desArg),” The Journal of Immunology, Vol. 162, No. 1, 1999, pp. 453-459.
- [92] Y. Fukuoka and T. E. Hugli, “Demonstration of a Specific C3a Receptor on Guinea Pig Platelets,” The Journal of Immunology, Vol. 140, No. 10, 1988, pp. 3496-3501.
- [93] A. A. Humbles, B. Lu, C. A. Nilsson, C. Lilly, E. Israel, Y. Fujiwara, N. P. Gerard and C. Gerard, “A Role for the C3a Anaphylatoxin Receptor in the Effector Phase of Asthma,” Nature, Vol. 406, No. 6799, 2000, pp. 998- 1001.
- [94] M. Noris and G. Remuzzi, “Hemolytic Uremic Syndrome,” Journal of the American Society of Nephrology, Vol. 16, 2005, pp. 1035-1050. doi:10.1681/ASN.2004100861
- [95] J. Scheiring, S. P. Andreoli and L. B. Zimmerhackl, “Treatment and Outcome of Shiga-Toxin-Associated Hemolytic Uremic Syndrome (HUS),” Pediatric Nephrology, Vol. 23, No. 10, 2008, pp. 1749-1760.
- [96] P. I. Tarr, C. A. Gordon and W. L. Chandler, “ShigaToxin-Producing Escherichia Coli and Haemolytic Uraemic Syndrome,” Lancet, Vol. 365, No. 9464, 2005, pp. 1073-1086.
- [97] C. W. Strey, M. Markiewski, D. Mastellos, R. Tudoran, L. A. Spruce, L. E. Greenbaum and J. D. Lambris, “The Proinflammatory Mediators C3a and C5a Are Essential for Liver Regeneration,” The Journal of Experimental Medicine, Vol. 198, No. 6, 2003, pp. 913-923.
- [98] G. Moxley and S. Ruddy, “Elevated Plasma C3 Anaphylatoxin Levels in Rheumatoid Arthritis Patients,” Arthritis & Rheumatism, Vol. 30, No. 10, 1987, pp. 1097-1104.
NOTES
*Conflict of interest: the author has no financial conflicts of interest.