Paper Menu >>
Journal Menu >>
 J. Biomedical Science and Engineering, 2009, 2, 63-69 Published Online February 2009 in SciRes. http://www.scirp.org/journal/jbise JBiSE 1 The pattern of co-existed posttranslational modifications-A case study Zheng-Rong Yang School of Biosciences, University of Exeter, Correspondence should be addressed to Zheng- Rong Yang (Z.R.Yang@exeter.ac.uk) Received August 20th, 2008; revised December 1st, 2008; accepted December 5th, 2008 ABSTRACT Posttranslational modifications are a class of important cellular activities in various bio- chemical processes including signalling trans- duction, gene/metabolite networks, and disease development. It has been found that multiple posttranslational modifications with the same or different modification residues can co-exist in the same protein and this co-occurrence is critical to signalling networks in cells. Although some biological studies have spotted this phe- nomenon, little bioinformatics study has been carried out for understanding its mechanism. Four data sets were downloaded from NCBI for the study. The joint probabilities of any two neighbouring posttranslational modification sites of different modification residues were analyzed. The Bayesian probabilistic network was derived for visualizing the relationship be- tween a target modification and the contributing modifications as the predictive factors. Keywords: posttranslational modification, bio- informatics, pattern analysis, posttranslational modification, pattern analysis, probability model 1. INTRODUCTION Posttranslational modifications (PTMs) are a chemical process of modifying a protein’s chemical or structural properties after the translation of the protein has been completed. Posttranslational modifications are closely related with signalling networks of molecules in cells. The modifications include attaching a chemical, chemi- cal structural change of amino acids, or protein structural change. The modification will also alter the functions of a protein in both ways, adding functions or removing functions, for instance, phosphorylation and dephos- phorylation, carboxylation and decarboxylation. Because of these changes, proteins will carry different signals for functioning in cells. Posttranslational modifications are then the focus of many signalling transduction network studies. For instance, properly folded and posttransla- tional modified endoplasmic reticulum is related with stress and will lead to different pathological states [1]. Poly (ADP-ribosyl)ation is related with DNA repair and cell cycle checkpoint pathways as the unique signal for protein function modulations [2]. Posttranslational modi- fications are the mediators for the transporters for multi- ple functions of human copper-transporting ATPases [3]. In the study of various chronic diseases, it is found that protein 3-nitrotyrosine (nitration) plays an important role in pathological conditions [4]. Together mutations and aberrant mRNA splicing, hyperphosphorylation will lead to a number of neurodegenerative disorders [5]. S-Nitrosation has been recently found to have similar function as phosphorylation and acetylation because of its association with various pathological cell reactions in signalling networks [6]. In cell-cycle control, differen- tiation, metabolism, stress response and programmed cell-death, the FOXO subgroup of forkhead transcription factors have been found being tightly controlled by phosphorylation, acetylation and ubiquitination [7]. In biological experiments, it has been found that the co-occurrence of posttranslational modifications is criti- cal for many cellular functions in recent a few years. For instance, it has been found that the necessary condition for stable transcriptional activity of p53 is the coopera- tion of multiple posttranslational modifications such as phosphorylation, acetylation, and ubiquitination [8]. In studying the complex pattern of posttranslational modi- fications and its impact on cellular processes, it has been found that lysine acetylation, arginine/lysine methylation and serine/threonine phosphorylation will work together cooperatively for regulating the high mobility group proteins [9]. In the experiments with human cancer specimens, it has been found that the extent of acetyla- tion, formylation and methylation is higher in cultured cells [10]. It has also been found that proteins with mul- tiple posttranslational modifications may make contribu- tion to similar signalling functions [11]. In studying DNA repair, apoptosis and senescence, it has been found that the interplay between multiple protein modifications, including phosphorylation, ubiquitylation, acetylation and sumoylation is critical for properly propagating DNA damage signals [12] and the interplay between methylation and acetylation has been found important for activating p53 by responding to DNA damage signals SciRes Copyright © 2009  64 Z. R. Yang et al. / J. Biomedical Science and Engineering 2 (2009) 63-69 SciRes Copyright © 2009 JBiSE [13]. It is even found that there is crosstalk between dif- ferent posttranslational modifications [14]. In glycogen syntheses kinase-3, it has been found that O-GlcNAcy- lation O-phosphate is interplaying for cellular regulation [15]. The interplay has been also found in the steroid receptor coactivators [16]. In the study of transcriptional programming, it is found that interplay between post- translational modifications exists in H3 termini [17]. In histone, it has been found that there are multiple arginine posttranslational modifications which are critical for some disease development. Also in histone, the interplay between sumoylation and either acetylation or ubiquity- lation has been observed contributing to complex func- tions of proteins [18]. A recent study has used laboratory method to identify co-occurrence posttranslational modi- fications [19]. A computational method was proposed to predict the interplay between phosphorylation sites and O-GlcNAc sites based on peptides around modification residues [20,21]. The analysis was based on the predic- tion results from various PTM prediction tools and was based on peptide information only. Moreover, the method only focused on the competition mechanism between phosphorylation sites and O-GlcNAc modifications at the same residues. The patterns of co-occurrence of posttranslational modifications are so far unclear or have not yet emerged through large scale studies. Bioinformatics study will help revealing those patterns and will benefit many de- sirable cellular engineering processes, i.e. disease con- trol and prevention based on handling signalling path- ways subjectively. This study is aimed to analyse the patterns of co-occurrence of multiple posttranslational modifications and visualizing their relationship through a probabilistic analysis. Computational approaches, such as structural bioin- formatics [22], molecular docking [23], molecular pack- ing [24,25], pharmacophore modelling [26], Mote Carlo simulated annealing approach [27], diffusion-controlled reaction simulation [28], bio-macromolecular internal collective motion simulation [29], QSAR [30,31], pro- tein subcellular location prediction [32,33], identifica- tion of membrane proteins and their types [34], identifi- cation of enzymes and their functional classes [35], identification of GPCR and their types [36], identifica- tion of proteases and their types [37], protein cleavage site prediction [38,39,40], and signal peptide prediction [41,42], can timely provide very useful information and insights for both basic research and drug design and are widely welcome by science community. The present study is devoted to develop a computational approach for studying co-occurrence of posttranslational modifi- cations. 2. APPROACH The first thing we need to do is to scan all the sequences in a data set to find all neighbouring modifications. We use frequency as the joint probability to measure the quantita- tive property of co-occurrence of modifications first. Through analyzing the frequency of two modifications, the likelihood that a pair of modifications occurs can be quan- tified. However, the frequency analysis only shows how likely two modifications can occur simultaneously in the same sequence. For instance, we may observe that the probability for hydroxyproline and hydroxylysine to occur simultaneously in the same sequence is 18.6%. But this does not indicate how likely hydroxylysine de- pends on hydroxyproline. In other words, if we have ob- served a hydroxyproline in a sequence, how likely can we find a hydroxylysine in the same sequence as the neighbouring modification? We first define the joint prob- ability of two different modification residues as the fre- quency defined as onsmodificati ngneighbouri ofnumber the slysimutaneouoccur and t number tha the ),( YX YXP = (1) below Here, X and Yare two different modification residues. We then define the marginal probability as the frequency for one modification residue to occur in a data set onsmodificati allofnumber the onmodificati ofnumber the )( X XP = (2) Using the product theory in probability, we have Here )()|()()|(),( XPXYPYPYXPYXP = = (3) )|( YXP reads out as the conditional probability for X to occur given that Y has happened. Based on the above calculation, we will have two conditional prob- abilities, either the probability of observing X if Y has been observed or the probability of observing Y if X has been observed. Based on the conditional prob- abilities and the marginal probabilities, we can use the Bayes rule to determine the posterior probabilities which are commonly used for decision-making. The Bayes rule is defined as ∑ = mmm ii iXPXYP XPXYP YXP )()|( )()|( )|( (4) Here we treat Y as a target modification, for instance a hyrdoxyproline residue. i X is the ith potential contrib- uting modification for the target modification Y. The posterior probability indicates if Y has been observed, what is the probability that it has a neighbour i X. It quantifies quantitatively how possible i X is most probable neighbouring modification of Y . Note that 1)|( = ∑ iiYXP . The posterior probabilities are then used to draw networks to illustrate the relationship be- tween a target modification residue and other modifica- tion residues. This probabilistic network here is a simple 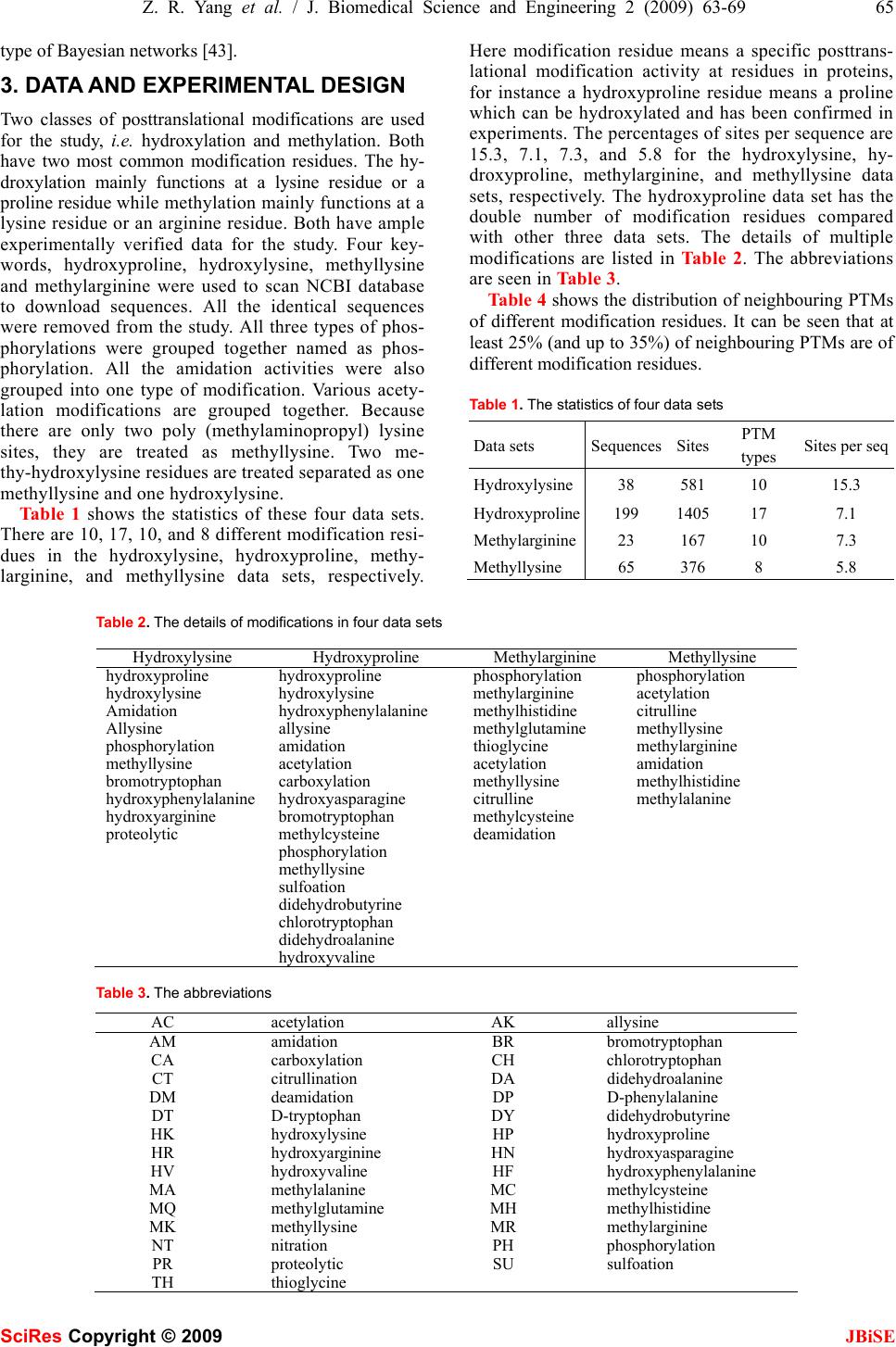 Z. R. Yang et al. / J. Biomedical Science and Engineering 2 (2009) 63-69 65 SciRes Copyright © 2009 JBiSE type of Bayesian networks [43]. 3. DATA AND EXPERIMENTAL DESIGN Two classes of posttranslational modifications are used for the study, i.e. hydroxylation and methylation. Both have two most common modification residues. The hy- droxylation mainly functions at a lysine residue or a proline residue while methylation mainly functions at a lysine residue or an arginine residue. Both have ample experimentally verified data for the study. Four key- words, hydroxyproline, hydroxylysine, methyllysine and methylarginine were used to scan NCBI database to download sequences. All the identical sequences were removed from the study. All three types of phos- phorylations were grouped together named as phos- phorylation. All the amidation activities were also grouped into one type of modification. Various acety- lation modifications are grouped together. Because there are only two poly (methylaminopropyl) lysine sites, they are treated as methyllysine. Two me- thy-hydroxylysine residues are treated separated as one methyllysine and one hydroxylysine. Table 1 shows the statistics of these four data sets. There are 10, 17, 10, and 8 different modification resi- dues in the hydroxylysine, hydroxyproline, methy- larginine, and methyllysine data sets, respectively. Here modification residue means a specific posttrans- lational modification activity at residues in proteins, for instance a hydroxyproline residue means a proline which can be hydroxylated and has been confirmed in experiments. The percentages of sites per sequence are 15.3, 7.1, 7.3, and 5.8 for the hydroxylysine, hy- droxyproline, methylarginine, and methyllysine data sets, respectively. The hydroxyproline data set has the double number of modification residues compared with other three data sets. The details of multiple modifications are listed in Table 2. The abbreviations are seen in Table 3. Table 4 shows the distribution of neighbouring PTMs of different modification residues. It can be seen that at least 25% (and up to 35%) of neighbouring PTMs are of different modification residues. Table 1. The statistics of four data sets Data sets SequencesSites PTM types Sites per seq Hydroxylysine38 581 10 15.3 Hydroxyproline199 1405 17 7.1 Methylarginine23 167 10 7.3 Methyllysine 65 376 8 5.8 Table 2. The details of modifications in four data sets Hydroxylysine Hydroxyproline Methylarginine Methyllysine hydroxyproline hydroxyproline phosphorylation phosphorylation hydroxylysine hydroxylysine methylarginine acetylation Amidation hydroxyphenylalanine methylhistidine citrulline Allysine allysine methylglutamine methyllysine phosphorylation amidation thioglycine methylarginine methyllysine acetylation acetylation amidation bromotryptophan carboxylation methyllysine methylhistidine hydroxyphenylalanine hydroxyasparagine citrulline methylalanine hydroxyarginine bromotryptophan methylcysteine proteolytic methylcysteine deamidation phosphorylation methyllysine sulfoation didehydrobutyrine chlorotryptophan didehydroalanine hydroxyvaline Table 3. The abbreviations AC acetylation AK allysine AM amidation BR bromotryptophan CA carboxylation CH chlorotryptophan CT citrullination DA didehydroalanine DM deamidation DP D-phenylalanine DT D-tryptophan DY didehydrobutyrine HK hydroxylysine HP hydroxyproline HR hydroxyarginine HN hydroxyasparagine HV hydroxyvaline HF hydroxyphenylalanine MA methylalanine MC methylcysteine MQ methylglutamine MH methylhistidine MK methyllysine MR methylarginine NT nitration PH phosphorylation PR proteolytic SU sulfoation TH thioglycine  66 Z. R. Yang et al. / J. Biomedical Science and Engineering 2 (2009) 63-69 SciRes Copyright © 2009 JBiSE Among these neighbouring PTMs of different modi- fication residues, 33%, 68%, 78%, 84% have the dis- tance less than 10 residues for the methylarginine, the methyllysine, the hydroxylysine, and the hydroxyproline data sets, respectively as seen in Table 5. Because of this, two PTMs of different modification residues may likely share similar structure (at least an overlapped local structure) for binding. This suggests that the cooperative activities of PTMs of different modification residues are critical to cellular signalling/functioning. Based on the collected sequences, we produce a pro- gram to search for all the posttranslational modification residues in four data sets. The sites must have the nota- tion as </site_type=”modified”>, </experiment=”ex- perimental evidence, …”>, and </note_type=X>. Here X can be various types, for instance, 4-hydroxyproline, 3-hydroxyproline, 5-hydroxyproline, etc. For each of four posttranslational modifications, we find all the in- volved posttranslational modification sites. The se- quences in different data sets are analyzed separately although there are some overlaps among them. 4. RESULTS Table 6 shows the frequencies as the joint probabilities of nine types of modifications for the hydroxylysine data set. Proteolytic is removed as there is only one such site in the data set. The highest joint probability is 60.2% for two hydroxyproline residues to be neighbours. How- ever, the joint probability for two hydroxylysine residues to be neighbours is only 6.81%. The co-occurrence probability for these two types of hydroxylation is 18.8%. These two probabilities indicate that for every hydroxylysine residue, the probability for it to have a hydroxyproline as the neighbour is three times higher than the probability for it to have the same hy- droxylysine residue as the neighbour. The joint prob- abilities for a hydroxylysine to have an amidation, allysine, phosphorylation residue, bromotryptophan, hydroxyphenylalanine, and hydroxyarginine residue as the neighbour are 0.55%, 0.55%, 0.18%, 0.74%, and 0.18%, respectively. This means that except for the same type of modification, Table 4. The distribution of neighbouring PTMs of different modi- fication residues sites Percentage over total Hydroxylysine 147 25% Hydroxyproline 434 31% Methylarginine 40 24% Methyllysine 132 35% Table 5. The times for different modifications to occur in one sequence 1-2 3-4 5-6 7-8 9-10 >11 Sum Hydroxylysine 32 32 33 3 14 33147 Hydroxyproline 175 97 46 18 29 69434 Methylarginine 3 2 2 2 4 2740 Methyllysine 48 24 5 11 2 42132 Table 6. The joint probability as frequency of co-occurred modi- fications for the hydroxylysine data set HPHKAMAKPH MK BR HFHR HP60.218.80 0 0 0 0.18 0 0 HK18.86.810.550.550.18 0 0.18 0.740.18 AM0 0.550 0 0 0 0 0 0 AK0 0.550 0 0 0 0 0 0 PH0 0.180 0 4.05 5.52 0 0 0 MK0 0 0 0 5.52 1.47 0 0 0 BR0.180.180 0 0 0 0 0 0.18 HF0 0.740 0 0 0 0 0.370 HR0 0.180 0 0 0 0.18 0 0 hydroxylysine has a high correlation with allysine, ami- dation, and hydroxyphenylalanine modifications. Figure 1 visualises the probabilistic relationship among different types of modifications in the hy- droxylysine data set using the posterior probabilities, where all the posterior probabilities less than 10% are omitted for simplicity. The network demonstrates that hydroxylysine only depends on hydroxyproline (24%). However, hydroxylysine has great impacts on five modi- fication residues, allysine (100%), amidation (100%), hydroxyphenylalanine (67%), hydroxyarginine (50%), and bromotryptophan (33%). Phosphorylation and me- thyllysine modification residues are independent from the hydroxylysine block. They are mutually correlated to each other. Table 7 shows the joint probabilities for the hy- droxyproline data set. It can be seen that the probability for two hydroxyproline residues to be neighbours is 57.7%. The likelihood for a hydroxyproline to have a hydroxylysine Figure 1. The probabilistic network as the relationship among modification residues in the hydroxylysine data set. The values represent the posterior probabilities. The arcs mean the directions. For instance, the arc from HK to AM with a number 100 means P(HK|AM)=100%. In other words, the probability of observing a hydroxylysine residue nearby an observed amidation residue is almost certain  Z. R. Yang et al. / J. Biomedical Science and Engineering 2 (2009) 63-69 67 SciRes Copyright © 2009 JBiSE as the neighbour is 8.46%. However, the likelihood for a hydroxyproline to have a hydroxyphenylalanine as the neighbour is 18.2%. This means that a hydroxyproline is more likely to have a hydroxyphenylalanine to co-occur rather than hydroxylysine. The other two important modifications for hydroxyproline are carboxylation and amidation. The probability for a hydroxyproline residue and an amidation residue to be neighbours is 4.06% and the probability for a hydroxyproline residue and a car- boxylation residue to be neighbours is 1.91%. The other co-occurred modifications with joint probabilities larger than 0.2% are acetylation and bromotryptophan. The probabilistic relationship among different modi- fications shown in Figure 2 is built for the hy- droxyproline data set using the posterior probabilities. All the posterior probabilities less than 10% are not shown. In the probabilistic network, it can be seen that the most contributing modifications for hydroxyproline is hydroxyphenylalanine (20%). However, hydroxyproline has contributed to 11 other modification residues. For instance, the posterior probability P(HP|AC) is 100% meaning that whenever we have found an acetylation residue, it is certain that there is a hydroxyproline resi- due nearby. The posterior probability P(HP|HP) is 63% while P(HK|HP)=9%, P(AK|HP)=1%, P(AM|HP)=4%, P(AC|HP)=1%, P(CA|HP)=2%, P(BR|HP)=1%. Table 7. The joint probability (larger than 0.2%) as frequency of co-occurred modifications for the hydroxyproline data set HP HK HF AM AC CABR HP 57.7 8.46 18.2 4.06 0.41 1.910.5 HK 8.46 1.58 0 0.08 0 0 0.08 HF 18.2 0 0.25 0 0 0 0 AM 4.06 0.08 0 0.17 0 0.170.41 AC 0.41 0 0 0 0 0 0 CA 1.91 0 0 0.17 0 2.240.33 BR 0.5 0.08 0 0.41 0 0.33 0 Figure 2. The probabilistic network as the relationship among modifications in the hydroxyproline data set. The values represent the posterior probabilities Table 8 shows the frequencies of modifications in the methylarginine data set. The likelihood for two methy- larginine residues to co-occur as neighbours is 40.3%. The interesting phenomenon is that the co-occurrence probability between methylarginine and methyllysine residues (as neighbours) is 0%. The contributing modi- fications to methylargine residues are phosphorylation (11.1%), acetylation (2.78%), methylhistidine (1.39%), and citrulline (1.39%). This is not as expected as it is thought that both two dominant methylation modifica- tions should be highly correlated. Shown in Figure 3 is the probabilistic relationship among the modifications derived from the methylargin- ine data set. The network shows that the most contribut- ing modifications to methylarginine modification is phosphorylation (19%), i.e. P(PH|MR)=19%. For any observed methylarginine residue in a protein, the prob- ability of observing a phosphorylation residue is 19%. Meanwhile, methylarginine residue can be an important pre-request for other modification residues. For instance, the probability of observing a methylarginine residue if a methylhistidine residue has been observed is 100% and the probability that a methylarginine residue is standing near an observed acetylation residue is 33%.100%. Table 8. The joint probability as frequency of co-occurred modi- fications for the methylarginine data set PHMRMHMG TH AC MKCT PH26.411.10 0 0 2.78 1.390.69 MR11.140.31.390.69 0.69 2.78 0 1.39 MH0 1.390 0 0 0 0 0 MG0 0.690 0 0.69 0 0 0 TH0 0.690 0.69 0 0 0 0 AC2.782.780 0 0 2.08 0.690 MK1.390 0 0 0 0.69 0.690 CT0.691.390 0 0 0 0 2.78 Figure 3. The probabilistic network as the relationship among modifications in the methylarginine data set. The values represent the posterior probabilities  68 Z. R. Yang et al. / J. Biomedical Science and Engineering 2 (2009) 63-69 SciRes Copyright © 2009 JBiSE Table 9 shows the frequencies for the methyllysine data set. The probability for two methyllysine residues to be neighbours is 28.3%, which is not dominantly high. Interestingly, we have found the contributing modifica- tions for methyllysine are acetylation (15.11%) and phosphorylation (10.61%). It is again difficult to find the evidence that two methylation modifications are highly correlated. Figure 4 shows the probabilistic network as the rela- tionship among the modifications in the methyllysine data set. Here, the most contributing modifications to methyllysine are phosphorylation (22%) and acetylation (31%). The methyllysine residue has a high correlation with methylarginine, methyhistine, and methylalanine. All have the posterior probabilities as 100%. 5. CONCLUSION This paper has studied the co-occurrence pattern of two types of posttranslational modifications with four modi- fication residues. The study aims to reveal how post- translational modifications are correlated to each other, i.e. how one posttranslational modification contributes to the others. It has been found that the hydroxylysine and hydroxyproline residues are not the most mutually de- pendent modification residues, so are the methylarginine Table 9. The joint probability as frequency of co-occurred modi- fications for the methyllysine data set PH AC CT MKMR AM MHMA PH 8.04 11.58 0 10.61 0 0.32 0 0 AC 11.58 28.3 0.96 15.11 1.93 0 0 0 CT 0 0.96 0 0.960 0 0 0 MK 10.61 15.11 0.96 21.22 0 0 0.320.64 MR 0 1.93 0 0 0 0 0 0 AM 0.32 0 0 0 0 0 0 0 MH 0 0 0 0.320 0 0 0 MA 0 0 0 0.640 0 0 0 Figure 4. The probabilistic network as the relationship among modifications in the methyllysine data set. The values represent the posterior probabilities and methyllysine residues. We have found that the hy- droxyllysine residues depend on the hydroxyproline resi- dues with a posterior probability 24% and the hy- droxyproline residues are the unique major contributing modification residue for the hydroxylysine residues. However, we have found the hydroxyproline residues nearly do not depend on the hydroxylysine residues. Instead, the hydroxyphenylalanine residues are the con- tributing modification residue to the the hydroxyproline residues with a posterior probability 20%. Among the methylarginine residues and the methyllysine residues, we have found that the phosphorylation residues are the main player for both of these two modification residues. In addition, the acetylation residues are needed for the methyllysine residues as well. Surprisingly, two different methylation residues also do not rely on each other. Al- though the study is limited to two modification classes with four modification residues, it is expected that this method can be generalized to a wide range of multiple posttranslational modification pattern discovery. REFERENCES [1] E. Lai, T. Teodoro, and A. Volchuk, (2007) Endoplasmic reticu- lum stress: signaling the unfolded protein response. Physiology (Bethesda), 22, 193-201. [2] J. F. Haince, S. Kozlov, V. L. Dawson, T. M. Dawson, M. J. Hendzel, M.F. Lavin, and G. G. Poirier, (2007) Ataxia telangiec- tasia mutated (ATM) signaling network is modulated by a novel poly (ADP-ribose)-dependent pathway in the early response to DNA- damaging agents. J Biol Chem., 282, 16441-16453. [3] S. Lutsenko, A. Gupta, , J. L. Burkhead, and V. Zuzel, (2008) Cellular multitasking: The dual role of human Cu-ATPases in cofactor delivery and intracellular copper balance. Arch Bio- chem Biophys., (in press). [4] J. M. Souza, G. Peluffo, and R. Radi, (2008) Protein tyrosine nitration-Functional alteration or just a biomarker? Free Radic Biol Med., (in press). [5] J. Z. Wang, and F. Liu, (2008) Microtubule-associated protein tau in development, degeneration and protection of neurons. Prog Neurobiol., 85, 148-175. [6] V. V. Sumbayev and I. M. Yasinska, (2008) Protein S-nitrosation in signal transduction: assays for specific qualitative and quanti- tative analysis. Methods Enzymol, 440, 209-219. [7] T. Obsil, and V. Obsilova, (2008) Structure/function relation- ships underlying regulation of FOXO transcription factors. On- cogene, 27, 2263-2275. [8] A. Scoumanne and X. Chen, (2008) Protein methylation: a new mechanism of p53 tumor suppressor regulation. Histol Histopa- thol., 23, 1143-1149. [9] Q. Zhang, and Y. Wang, (2008) High mobility group proteins and their post-translational modifications. Biochim Biophys Acta., (in press). [10] J. R. Wisniewski, A. Zougman, S. Kruger, P. Ziołkowski, M. Pudełko, M. Bębenek and M. Mann, (2008) Constitutive and dynamic phosphorylation and acetylation sites on NUCKS, a hypermodified nuclear protein, studied by quantitative pro- teomics. Proteins, (in press). [11] C. A. Galea, A. Nourse, Y. Wang, S. G. Sivakolundu, W. T. Heller and R. W. Kriwacki, (2008) Role of intrinsic flexibility in signal transduction mediated by the cell cycle regulator, p27 Kip1. J Mol Biol. 376, 827-838. [12] M. S. Huen and J. Chen, (2008) The DNA damage response pathways: at the crossroad of protein modifications. Cell Res., 18, 8-16. [13] M. S. Huen and J. Chen, (2008) The DNA damage response pathways: at the crossroad of protein modifications. Cell Res.,  Z. R. Yang et al. / J. Biomedical Science and Engineering 2 (2009) 63-69 69 SciRes Copyright © 2009 JBiSE 18, 8-16. [14] T. Hunter, (2007) The age of crosstalk: phosphorylation, ubiq- uitination, and beyond. Mol Cell., 28, 730-738. [15] Z. Wang, A. Pandey and G. W. Hart, (2007) Dynamic interplay between O-linked N-acetylglucosaminylation and glycogen syn- thase kinase-3-dependent phosphorylation. Mol Cell Proteomics, 6, 1365-1379. [16] S. Li and Y. Shang (2007) Regulation of SRC family coactivators by post-translational modifications. Cell Signal, 19, 1101- 1112. [17] P. R. Thompson and W. Fast, (2006) Histone citrullination by protein arginine deiminase: is arginine methylation a green light or a roadblock? ACS Chem Biol., 21, 433-41. [18] D. Nathan, K. Ingvarsdottir, D. E. Sterner, G. R. Bylebyl, M. Dokmanovic, J. A. Dorsey, K. A. Whelan, M. Krsmanovic, W. S. Lane, P. B. Meluh, E. S. Johnson, and S. L.Berger, (2006) His- tone sumoylation is a negative regulator in Saccharomyces cere- visiae and shows dynamic interplay with positive-acting histone modifications. Genes Dev., 20, 966-976. [19] J. Seo, J. Jeong, Y. M. Kim, N. Hwang, E. Paek and K. J. Lee, (2008) Strategy for comprehensive identification of post-trans- lational modifications in cellular proteins, including low abun- dant modifications: application to glyceraldehyde-3-phosphate dehydrogenase. J Proteome Res., 7, 587-602. [20] A. Ahmad, T. S. Khan, D. C. Hoessli, E. Walker-Nasir, A. Ka- leem, A. R. Shakoori and S. Nasir-ud-Din, (2008) In silico modulation of HMGN-1 binding to histones and gene expres- sion by interplay of phosphorylation and O-GlcNAc modifica- tion. Protein Pept Lett., 15, 193-199. [21] A. Kaleem, D. C. Hoessli, I. Ahmad, E. Walker-Nasir, A. Nasim, A. R. Shakoori and S. Nasir-ud-Din, (2008) Immediate-early gene regulation by interplay between different post-translational modifications on human histone H3. J Cell Biochem., 103, 835-851. [22] K. C. Chou, (2004) Review: Structural bioinformatics and its impact to biomedical science. Current Medicinal Chemistry, 11, 2105-2134. [23] K. C. Chou, D. Q. Wei and W. Z. Zhong (2003) Binding mecha- nism of coronavirus main proteinase with ligands and its impli- cation to drug design against SARS. (Erratum: ibid., 2003, Vol.310, 675). Biochem Biophys Res Comm, 308, 148-151. [24] K. C. Chou, G. Nemethy and H. A. Scheraga, (1984) Energetic approach to packing of a-helices: 2. General treatment of non- equivalent and nonregular helices. Journal of American Chemi- cal Society, 106, 3161-3170. [25] K. C. Chou, G. M. Maggiora, G. Nemethy and H. A. Scheraga, (1988) Energetics of the structure of the four-alpha-helix bundle in proteins. Proceedings of National Academy of Sciences, USA, 85, 4295-4299. [26] S. Sirois, D. Q. Wei, Q. S. Du and K. C. Chou, (2004) Virtual- Screening for SARS-CoV Protease Based on KZ7088 Pharma- cophore Points. J Chem Inf Comput Sci, 44, 1111-1122. [27] K. C. Chou, (1992) Energy-optimized structure of antifreeze protein and its binding mechanism. Journal of Molecular Biol- ogy, 223, 509-517. [28] K. C. Chou and G. P. Zhou, (1982) Role of the protein outside active site on the diffusion-controlled reaction of enzyme. Jour- nal of American Chemical Society, 104, 1409-1413. [29] K. C. Chou, (1988) Review: Low-frequency collective motion in biomacromolecules and its biological functions. Biophysical Chemistry, 30, 3-48. [30] Q. S. Du, R. B. Huang, Y. T. Wei, L. Q. Du and K. C. Chou, (2008) Multiple Field Three Dimensional Quantitative Struc- ture- Activity Relationship (MF-3D-QSAR). Journal of Compu- tational Chemistry, 29, 211-219. [31] H. Gonzalez-Díaz, Y. Gonzalez-Díaz, L. Santana, F. M. Ubeira and E. Uriarte, (2008) Proteomics, networks, and connectivity indices. Proteomics, 8, 750–778. [32] K. C. Chou and H. B. Shen, (2007) Review: Recent progresses in protein subcellular location prediction. Analytical Biochemis- try, 370, 1-16. [33] K. C. Chou and H. B. Shen, (2008) Cell-PLoc: A package of web-servers for predicting subcellular localization of proteins in various organisms. Nature Protocols, 3, 153-162. [34] K. C. Chou and H. B. Shen, (2007) MemType-2L: A Web server for predicting membrane proteins and their types by incorporat- ing evolution information through Pse-PSSM. Biochem Biophys Res Comm, 360, 339-345. [35] H. B. Shen, and K. C. Chou, (2007) EzyPred: A top-down ap- proach for predicting enzyme functional classes and subclasses. Biochem Biophys Res Comm, 364, 53-59. [36] K. C. Chou, (2005) Prediction of G-protein-coupled receptor classes. Journal of Proteome Research, 4, 1413-1418. [37] K. C. Chou and H. B. Shen, (2008) ProtIdent: A web server for identifying proteases and their types by fusing functional do- main and sequential evolution information. Biochem Biophys Res Comm, 376, 321-325. [38] K. C. Chou, (1993) A vectorized sequence-coupling model for predicting HIV protease cleavage sites in proteins. J Biol Chem, 268, 16938-16948. [39] K. C. Chou, (1996) Review: Prediction of HIV protease cleav- age sites in proteins. Analytical Biochemistry, 233, 1-14. [40] H. B. Shen and K. C. Chou, (2008) HIVcleave: a web-server for predicting HIV protease cleavage sites in proteins. Analytical Biochemistry, 375, 388-390. [41] K. C. Chou and H. B. Shen, (2007) Signal-CF: a subsite-coupled and window-fusing approach for predicting signal peptides. Bio- chem Biophys Res Comm, 357, 633-640. [42] H. B. Shen and K. C. Chou, (2007) Signal-3L: a 3-layer ap- proach for predicting signal peptide. Biochem Biophys Res Comm, 363, 297-303. [43] P. Pourret and B. Naim, (2008) Marcot, Bayesian Networks: A Practical Guide to Applications. Chichester, UK: Wiley. |