Journal of Cancer Therapy
Vol.3 No.5(2012), Article ID:23454,11 pages DOI:10.4236/jct.2012.35077
A Pilot Trial Testing the Feasibility of Using Molecular-Guided Therapy in Patients with Recurrent Neuroblastoma*
![]()
1Center for Cancer Genomics and Computational Biology, Pediatric Oncology Translational Research Program at the Van Andel Research Institute in Grand Rapids, Grand Rapids, USA; 2Department of Pediatric Oncology, Helen DeVos Children’s Hospital, Michigan State University, Grand Rapids, USA; 3Cardinal Glennon Children’s Hospital, Saint Louis University School of Medicine, St. Louis University, St. Louis, USA; 4Vermont Cancer Center, Department of Medicine, University of Vermont, Burlington, USA; 5Rady Children’s Hospital San Diego, University of California San Diego School of Medicine at the University of California San Diego, San Diego, USA; 6Center for Children’s Cancer and Blood Disorders, Arnold Palmer Hospital for Children MD Anderson Cancer Center Orlando; Orlando, USA; 7Pediatric Hematology/Oncology, Medical University of South Carolina; Charleston, USA; 8Pediatric Oncology, Levine Children’s Hospital; Charlotte, USA; 9Division of Hematology/Oncology 2J, Connecticut Children’s Medical Center; Hartford, USA; 10Department of Pediatric Oncology, Oregon Health Science University; Portland, USA; 11Scientific Engineering Department, TransMed Systems, Cupertino, USA.
Email: #giselle.sholler@vai.org
Received July 27th, 2012; revised August 31st, 2012; accepted September 12th, 2012
Keywords: Neuroblastoma; Molecular Guided Therapy; Predictive Therapies; Molecular Profiling; Genomic Profiling; Pediatric Cancer
ABSTRACT
Background: Neuroblastoma is the most common extracranial solid tumor in children, and treatment options for recurrent neuroblastoma are limited. Using molecular profiling to target the molecular vulnerabilities of neuroblastoma with existing therapeutic agents may result in a rational, data-driven approach with potential to improve clinical outcomes. Methods: The primary objective of this pilot study was to evaluate the feasibility of supporting real-time treatment decisions through predictive modeling of genome-wide mRNA gene expression data from neuroblastoma tumor biopsies. Feasibility was defined as completion of tumor biopsy, histopathological evaluation, RNA extraction and quality control, gene expression profiling within a CLIA-certified laboratory, bioinformatic analysis, generation of a drug predicttion report, molecular tumor board review yielding a formulated treatment plan, and independent medical monitor review within a 2-week period. Results: Five patients with multiply relapsed or refractory neuroblastoma were enrolled between April and June 2010. All biopsies passed histopathology and RNA quality control. Generation of gene expression data and its analysis (3 - 7 days), reports which linked this data into medically actionable drug candidates (1 - 5 days), molecular tumor board (1 - 3 days) and independent medical monitor review (1 day) were all completed in real-time. The average time was 10.5 days for all patients. Conclusion: This study shows that it is feasible to create therapeutic treatment plans based on genomic profiling in less than 12 days. This warrants further testing in a Phase I study to determine safety of predicted treatments and evaluate whether the information obtained in these analyses would result in patient benefit.
1. Introduction
Neuroblastoma is the most common extracranial solid tumor in children and accounts for 15% of pediatric cancer deaths each year [1,2]. When children are diagnosed with neuroblastoma under one year of age, the prognosis is generally good. However, children diagnosed with neuroblastoma after 12 - 18 months of age have a poor prognosis and only 30% survive despite aggressive multimodal therapies [3,4]. Intensive treatments, including high dose chemotherapy, hematopoietic stem cell transplantation, immunotherapy, and maintenance therapy with retinoic acid, still result in a five-year event-free survival below 50% [5]. Furthermore, the long-term survival of patients who are treated with conventional therapies following relapse is less than 5%. Clearly, new therapies are needed for patients with refractory or recurrent neuroblastoma.
Neuroblastoma, like other cancers, results from a series of molecular changes that ultimately disrupt the molecular networks and alter cellular homeostasis. Several of the molecular pathways involved in tumor development and progression have been identified. Fluctuations in these networks can result from genetic or epigenetic cellular events and/or changes in the molecular constitution of the tumor microenvironment, which collectively dictate the phenotype of the biological system. The molecular networks engaged during tumor development and/or progression are complex leading tocellular plasticity [6] and genomic instability [7,8] of neoplasms. Genetic expression profiles can reflect these pathways involved in cancer cells. Recent genetic expression profiling of medulloblastoma tumors has been used to identify distinct subgroups within this cancer. This study identified the WNT pathway as predominant in one tumor group and the SHH pathway in another.
A fundamental challenge in targeted cancer treatment is how to identify optimal single and combinational treatments for molecularly heterogeneous tumors [9-14]. The approach must be able to utilize the expanding knowledge of molecular networks and the mechanisms of action of a growing pharmacopeia [15,16] in conjunction with standardized biomarker assessments to deliver targeted combinations of effective therapies to cancer patients. Individual biomarkers have recently been identified that can be used in the clinical setting to identify neuroblastoma patients most likely to respond to a specific therapy. For example, activation of the ALK gene through mutations has been identified in a subset of neuroblastoma patients, and small-molecule inhibition of the ALK-encoded receptor tyrosine kinase induced cytotoxicity in affected neuroblastoma cell lines [12]. Such studies, in conjunction with a large body of in vitro and in vivo data, have further demonstrated that the efficacy of specific treatments depends on the molecular constitution of the tumor [17,18]. Therefore, observed variations in tumor response to current therapies are attributable in large part to disease heterogeneity at the molecular level.
The determination of gene transcript abundance through gene expression profiling has been frequently exploited in biomarker research. Gene expression signatures have been used to create a connectivity map, in which the genomic consequences of drug exposure can be aligned with de-regulated genes within a tumor specimen to identify compounds that may reverse the tumorigenic genotype [19]. Screening of a large number of potential therapeutic agents in a panel of cancer cell lines with a baseline gene expression profile has permitted the association of a gene expression signature with predicted response to various drugs [11,20].
Algorithms for this pilot study were based in part on a knowledge base that uses the existing pharmacopeia’s postulated molecular mechanisms of action available from DrugBank [16] that contains 1359 molecular entities targeted by 1509 drugs. Included are several targets of particular relevance to neuroblastoma including VEGFA-VEGFR, TRK, mTOR, ALK, HDAC [12]. The transcriptional status of each of these targets can be reported relative to a biological reference composed of a set of normal controls.
The algorithms described in this report were iteratively developed during a proof-of-feasibility study in which molecular profiling was performed on fifty late stage cancer patients, both adult and pediatric, and with a wide variety of tumor types [21]). This study of first generation predictive methodologies demonstrated the feasibility of this approach in the prospective clinical setting (CPW, personal communication). In a recently published study by Von Hoff et al. [22,23], molecular profiling including immunohistochemistry, in situ hybridization, and oligonucleotide microarray gene expression analysis was performed on tissue samples from 86 adult patients with refractory metastatic cancer to identify aberrant expression of molecular targets, which were used to choose a targeted treatment. They found that 27% of the patients who were treated according to their molecular target had a longer progression-free survival relative to the regimen on which the patient previously experienced progression (PFS ratio). Another phase I clinical trial program at MD Anderson evaluated the use of targeted therapy following tumor molecular analysis on advanced cancer patients. They found that patients matched with targeted drugs had higher rates of response, survival and time to treatment failure compared to those seen in patients treated without molecular matching [24,25].
Our pilot study was undertaken to evaluate the feasibility of using predictive modeling based on genomewide mRNA expression profiles from pediatric neuroblastoma tumor biopsies to make real-time treatment decisions. Feasibility was defined as the completion of the following sequential evaluations in a two-week time period: tumor biopsy, quality RNA extraction, mRNA Affymetrix U133 2.0 Plus GeneChip® hybridization, data analysis utilizing a series of predictive methodologies, the generation of an interactive report, molecular tumor board review yielding a formulated treatment plan, and an independent medical monitor review. Feasibility of genomic profiling through creation of individualized treatment plans was demonstrated in the five (100%) pilot study patients. This established that it is feasible to complete genomic profiling and create individualized treatment plans via a molecular tumor board in real-time (less than 12 days) for patients using the methods and logistical workflows described here.
2. Materials and Methods
2.1. Study Population
This was an open label pilot study in patients with refractory or recurrent neuroblastoma. Eligible patients were enrolled in this study at the University of Vermont following a registration process that included receipt of a signed patient consent form and a copy of the required baseline laboratory tests. Assent, when appropriate, was obtained according to institutional guidelines. Voluntary consent for optional biology studies was included. This trial was approved by the Institutional Review Board at the University of Vermont and the Protocol Review Committee at the Vermont Cancer Center (Clinical Trials. gov identifier: NCT01109238; Study ID NMTRC 001P).
2.2. Eligibility
Patients under 21 years old with refractory or relapsed neuroblastoma and measurable disease were eligible for this study. Inclusion criteria included current disease state for which there is currently no known curative therapy; Lansky Play Score more than 30; ANC > 750/μL and platelet count > 50,000/μL for patients without bone marrow metastases (no granulocyte or platelet criteria for patients with bone marrow metastases); adequate liver function, defined as total bilirubin ≤ 1.5 × upper limit of normal (ULN) for age and SGPT (ALT) < 10 × upper limit of normal (ULN) for age; no other significant organ toxicity defined as above Grade 2 by National Cancer Institute Common Toxicity Criteria for Adverse Events, version 3 (NCI-CTCAE V3.0 (http://ctep.cancer.gov/forms/CTCAEv3.pdf). Exclusion criteria included: patients who had received any chemotherapy within the last 21 days; patients receiving anti-tumor therapy for their disease or any investigational drug concurrently; patients with serious infection or a life-threatening illness (unrelated to tumor) that was above Grade 2 (NCI CTCAE V3.0); or patients with active, serious infections requiring parenteral antibiotic therapy within 2 weeks prior to screening. Screening procedures were performed no more than 5 days prior to biopsy. These included complete medical and surgical history; demographics; physical examination (including height and weight), baseline dermatologic and neurologic exam, and sites of palpable neoplastic disease; vital signs; baseline EKG; ECOG Performance status/Lansky Play status; CBC with differential; serum electrolytes (sodium, potassium, chloride, bicarbonate), blood urea nitrogen (BUN), creatinine, albumin, total protein, bilirubin, LDH, ALT, AST, and ferritin; urine catecholamines; and concomitant medications/therapies.
During the course of this study, 5 patients received therapy. All subjects were multiple relapsed/refractory neuroblastoma patients, with 4 males and 1 female, mean age 9, range (3 - 13).
2.3. Study Design
2.3.1. Sample Procurement and Gene Expression Profiling
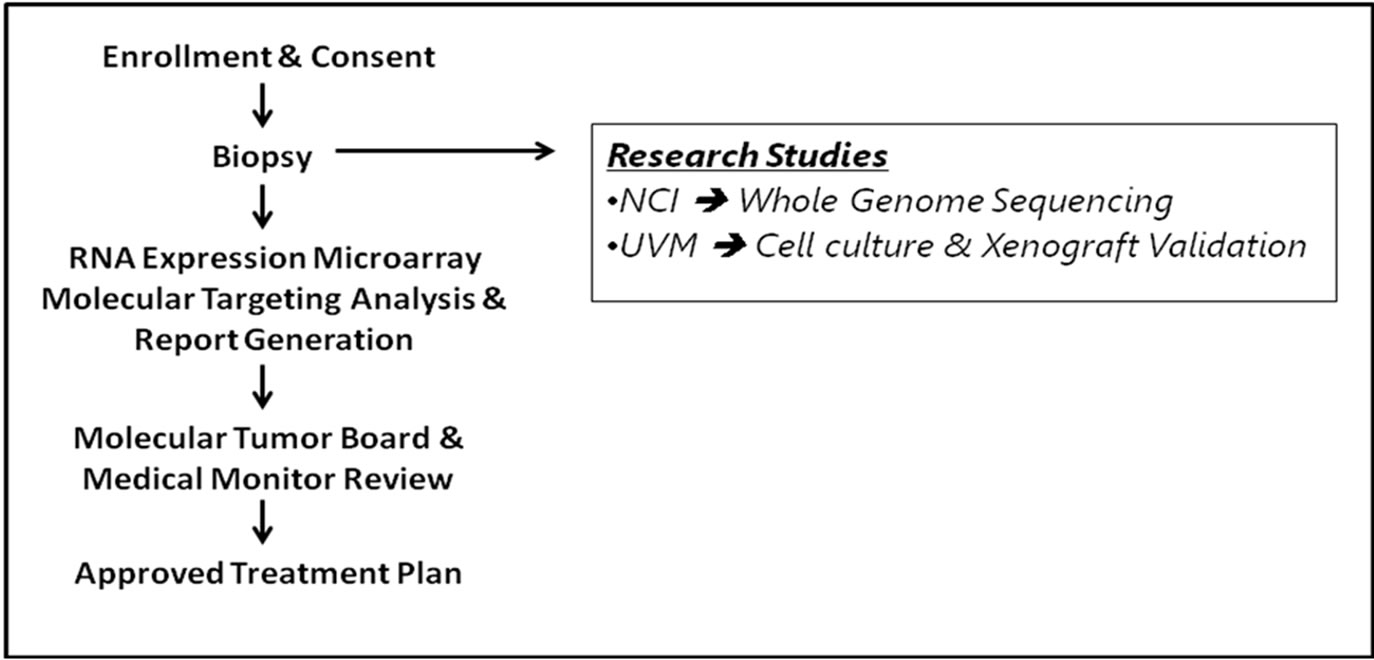
Patients underwent a scheduled surgical resection and/or diagnostic biopsy procedure. Common sample identifiers were provided by the Lead Study Coordinator and used by all parties throughout the project to ensure sample, data and report alignment between participating organizations. At the time of tissue resection or bone marrow aspirate, a fresh tumor sample was committed for this specific research study and prepared immediately. Viable, fresh tumor over 0.2 g from a tumor biopsy, 19-gauge needle core biopsy, or 4 cc from bone marrow aspirate was obtained. A clinical specimen was submitted for pathological assessment to ensure appropriate cellularity. De-identified subject samples were sent to various sites for assessments with the following order of priority; A single tumor biopsy in RNAlater (Qiagen) was shipped overnight to Clinical Reference Laboratory, a CLIAcertified laboratory (CRL; Lenexa, KS), for mRNA gene expression profiling; a sample was sent to Spectrum Health (Grand Rapids, MI) for flow cytometric analysis and sorting (2 cc of bone marrow); another sample was sent to the University of Vermont for in vitro/in vivo correlative biology studies (2 cc of bone marrow and solid tumor biopsy as available); another biopsy sample was sent to the NCI for next generation sequence analysis (2 cc of bone marrow and solid tumor biopsy as available); in addition, 10 - 20 mL of blood in a PAXgene blood DNA tube was sent to NCI for next generation sequence analysis of germline DNA (Figure 1(a)).
Affymetrix gene expression profiling was carried out using standard procedures. The RNA extraction, amplification (using NuGEN Ovation® PICO kit), Affymetrix U133 2.0 Plus GeneChip® hybridization, and scanning procedures utilized standard protocols and adhered to strict standard operating procedures that have been CLIA-certified by CRL to ensure accuracy and reproducibility of results. Pass criteria included; 1) RIN > 6.5 using the Agilent 2100 BioAnalyzer; 2) RNA 260/280 and 260/230 absorbance ratios > 1.8 by NanoDrop; 3) total cDNA yield ≥ 5 µg/30 µL; 4) cDNA 260/280 and 260/230 absorbance ratios ≥ 1.8 by NanoDrop. Data files were processed using the Affymetrix Expression Console™ and the MAS5.0 statistical algorithm. Pass
 (a)
(a)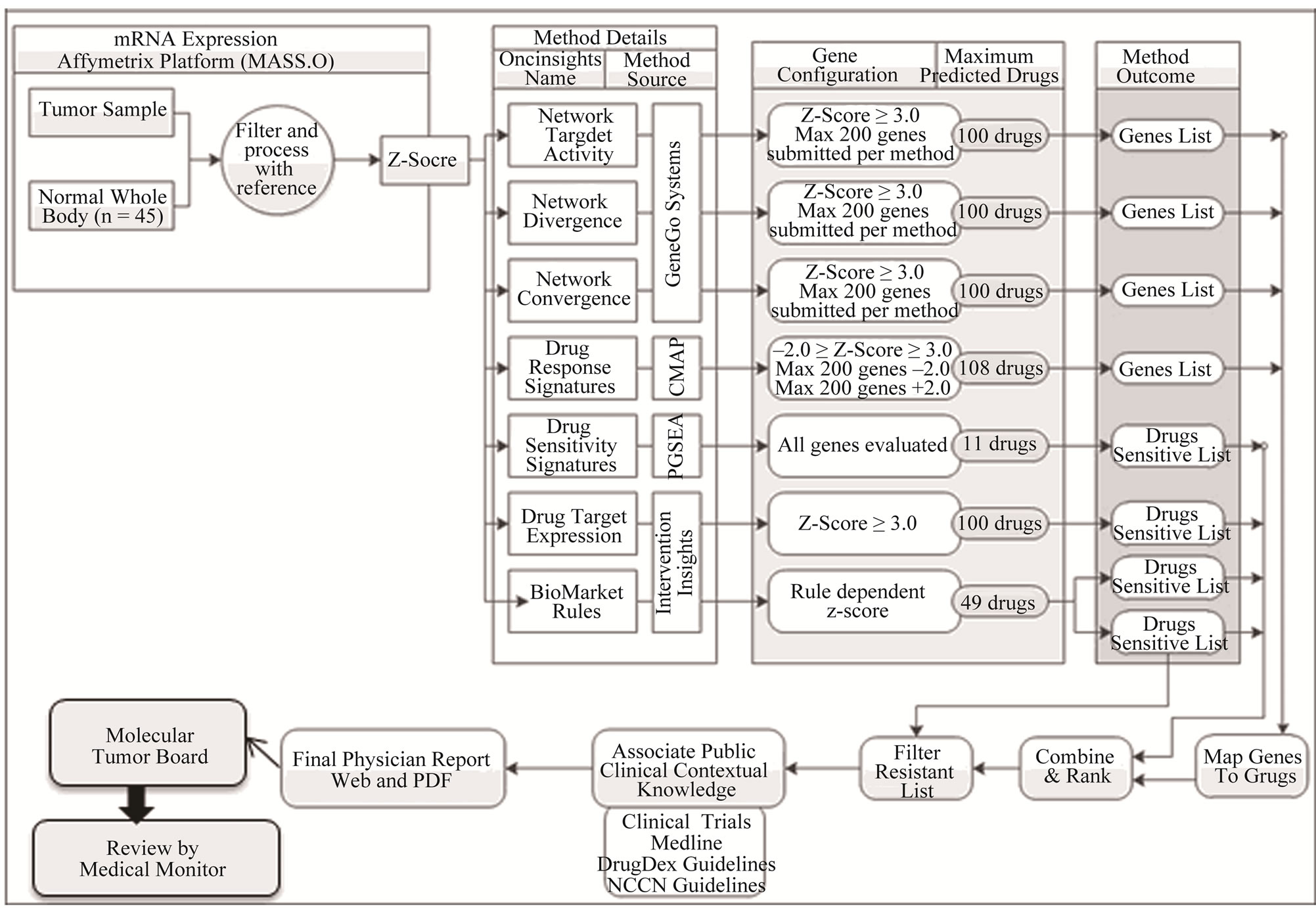 (b)
(b)
Figure 1. (a) General study workflow schema. The biopsy from the patient was used for microarray analysis at the CLIA-certified laboratory CRL and research studies carried out at various institutions. CRL: Clinical Reference Laboratory; NCI: National Cancer Institute; UVM: University of Vermont; (b) Generation of drug prediction report. Gene expression data was compared to a series of normal biological controls and Z-scores were run through the different predictive algorithms to create a medically actionable drug prediction report as described in Materials and Methods.
criteria at this stage included 1) Background < 100; 2) Percent present call ≥ 30; and 3) Scale factor < 100. Upon passing all criteria, MAS5.0 processed. CEL and normalized pivot. TXT files were extracted and deposited on a secure FTP site for subsequent analysis.
The research sample was logged and immediately culture, xenograft models and RNA extraction. Tumor cells were grown to 70% confluency in neurobasal media (RPMI with EGF and FGF). Cells were injected into the inguinal fat pad of NOD SCID mice for generation of patient xenografts. Bone marrow samples were sorted for tumor cells by flow cytometry at Spectrum Health using the immunophenotyping six-color, ganglioside GD2, analysis and monoclonal antibodies to membrane antigen expression of ganglioside GD2FITC dye, CD81FITCdye, CD45PerCPdye leukocytic, CD56APC NCAM antigen, CD9, and/or CD34 antigens. Within the Pediatric Oncology Branch of the NCI, isolation of DNA and RNA from patient samples was performed. These research samples were used for comparative genomics hybridization, exome sequencing of tumor, and germline DNA using Next Generation sequencing.
2.3.2. Generation of Drug Prediction Report
The reporting of drugs with predicted efficacy was performed using methodologies and reporting tools developed at the Van Andel Research Institute and in collaboration with Intervention Insights. Normalized gene expression data from each patient’s tumor was compared to a normal control reference sample set to obtain a relative gene expression profile where each gene probe set was represented by a Z-score. The Z-score is measure of the relative expression in the tumor with respect to the normal reference. It is calculated as the number of standard deviations that the intensity of a probe set in the tumor is over or under expressed relative to the mean of the same probe set intensity in the normal reference. After this pre-processing step, data were submitted to the following collection of methodologies to identify agents for consideration (summarized in Figure 1(b)).
Biomarker rules: This method employed predefined and published rules maintained in a drug-biomarker knowledge base in which the efficacy of a specific drug has been associated with the expression of a specific molecular marker [22]. Unlike the other methods described, this method has rules that predict both drug sensitivity and drug resistance based on the expression of biomarkers.
Drug target expression: Genes over expressed in the tumor (Z-scores ≥ +3) that represented an established therapeutic target were submitted and therapeutic compounds that met the rule requirement based upon their confirmed mechanism of action (MOA) were selected and displayed in the report. The mechanism of action of drugs and the alignment to therapeutic targets was performed using a variety of public and commercial knowledge bases including DrugBank [16], PharmGKB [23], GeneGo-Thomson Reuters (www.genego.com), UptoDate (www.uptodate.com), MedTrack (www.medtrack.com) and DrugDex (http://thomsonreuters.com/products_services/healthcare/healthcare_products/a-z/drugdex_system/) as well as extensive literature searches to confirm the drug-target evidence.
Network-Based Methods: The underlying method predicted activity of drug targets based on topological analysis and was developed in partnership with GeneGoThomson Reuters [26-28] various derivatives of this tool (referred to as the “hidden nodes” algorithms) are described in detail and freely available at http://www.genego.com/hidden_nodes.php. In brief, these systems biology based methodologies were developed to identify key regulators of the observed transcriptional profile after constructing molecular networks on the basis of prior protein-protein interaction knowledge. The key nodes (putative targets) within the identified and topologically enriched networks may be “hidden” since they do not necessarily represent genes differentially expressed in the patient’s tumor. Derivatives of this methodology included the analysis of target genes that represent key points of information convergence and divergence, which can be considered putative effectors and drivers respectively. After these respective analyses, the overlay of the drug-target knowledge base with topologically significant nodes provided a method to predict drug efficacy.
Drug response signatures: This method reproduced the Connectivity Map concept initially developed by the Broad Institute [20] in which the genomic consequence of drug exposure is used to connect drug effect to disease signatures. The hypothesis underlying this method is that drugs that reverse the disease genotype (gene expression profile) towards normalcy have the potential to reverse the disease phenotype. Up to 500 of the most overand under-expressed genes in the patient’s tumor (Z-scores ≥ +1.5 or ≤–1.5 respectively) were submitted to this method. Rank-based statistics were used to identify drugs with a significant inverse connectivity to the disease genotype.
Drug sensitivity signatures: This implemented the Parametric Gene Set Enrichment Analysis (PGSEA) method to align NCI-60 cell line sensitivity signatures that are predictive across at least two independent cell contexts with the patient’s differentially expressed genes. All genes that passed the pre-processing thresholds were evaluated. The NCI-60 drug signature mapped over and under expressed genes (determined by pre-drug treatment) to the observed in vitro drug sensitivity as measured by the half maximal inhibitory concentration (IC50) of the various cell lines studied [11,29].
2.3.3. Report Generation
Upon execution of these analyses, a compiled report was generated. The report allowed the molecular tumor board to quickly navigate to the underlying knowledge and evidence at multiple levels, including the molecular predictions and inferring methodologies, and any evidence from published literature and clinical trials that may support the use of the predicted agent in the patient’s disease context. While the total pharmacopeia coverage available at the time of this study was 182 FDAapproved drugs, only those with established pediatric dosing (n = 108) were used.
2.3.4. Molecular Tumor Board Review and Formation of Treatment Plan
The molecular tumor board consisted of 6 - 11 pediatric oncologists, 1 - 3 bioinformatics specialists, and a pediatric pharmacist. The generated report was submitted to the board, and a molecular tumor board meeting was held to discuss each patient and formulate a treatment plan. Primary and secondary reviewers generated a synopsis with literature review prior to the meeting. Decision rules for the molecular tumor board included molecular-based predictions of drug efficacy supplemented with evidence gathered from automated searching of the literature clinical trials, and the Internet; FDA-approved drugs with established standard and safe dosing schedules; analysis of drug combinations with regards to safety, mechanism of action, availability, and cost; preference toward lowtoxicity, targeted therapies; and drug combinations of up to a maximum of four agents. Literature searches of previously established and tested regimens were reviewed and such regimens were given priority. The pharmacist completed an analysis of potential drug interactions between the guided agents and the subject’s routine medications and supplements. The final molecular tumor board treatment decisions were presented to the medical monitor, who had the opportunity to reject the proposed regimen (which would have required reconvening of the molecular tumor board). The final treatment regimen, the decision of the medical monitor, and the minutes of the molecular tumor board meeting were documented on the Post Report Form.
3. Results
Five patients were enrolled in this study within three months between April 2010 and June 2010 (Table 1). Of the five patients, three had relapsed neuroblastoma, two had refractory neuroblastoma, and four out of five were Stage 4 at diagnosis (one was Stage 2B). The patients ranged in age from 3 to 8 years old at diagnosis, and the time since diagnosis was 2 - 6.5 years. All patients had received 2 - 13 prior relapsed therapies. At study entry, three patients had progressive disease and two had stable disease.
The primary objective of this study was to evaluate the feasibility of using predictive modeling to make real-time treatment decisions based on genome-wide mRNA expression profiles of neuroblastoma tumor biopsies. Feasibility was defined as the completion of a specific set of events within a 14-day time period. To calculate feasibility, we measured the number of days between each of the following events: tumor biopsy, RNA extraction and gene chip hybridization at CRL, genomic analysis and generation of report, molecular tumor board review with formulated treatment plan, and medical monitor review.
Tumor biopsies were evaluated by clinical pathology and processed by CRL. The sample handling, RNA extraction, amplification, Affymetrix U133 2.0 Plus GeneChip hybridization, and scanning procedures used CLIA-certified CRL standard protocols. After the tumor biopsy, the RNA extraction, amplification, hybridization to Affymetrix U133 2.0 Plus GeneChip, and scanning procedures were completed in 3 - 7 days (Table 2). The RNA RINs were greater than 6.5 in 5/5 patients, so the chip analysis was completed for all subjects (Table 3).
The gene expression data from the tumor biopsy was then compared to a reference sample to obtain a relative gene expression profile and gene probe set represented by a Z-score (Figure 1(b)). After this step, data were submitted for analysis using the algorithms described in Methods, and a compiled report was generated. After completion of the gene expression data, the number of days to generate this report ranged from the same day to 5 days (Table 2).
Next, a molecular tumor board meeting using the information contained in the report was held. Treatment protocols were able to be generated from the molecular tumor board meeting for all patients. This occurred within 1 - 3 days after the report was received (Table 2). Specific treatment details consisted of a regimen chosen from a guided list of agents implicated in critical molecular signaling pathways and/or from signature-based
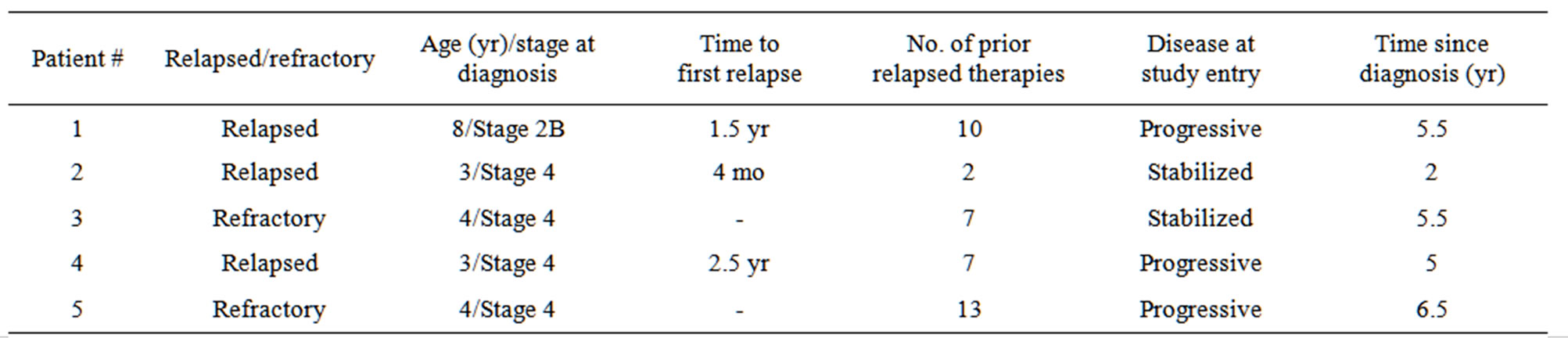
Table 1. Medical history of study population.

Table 2. Results of feasibility.
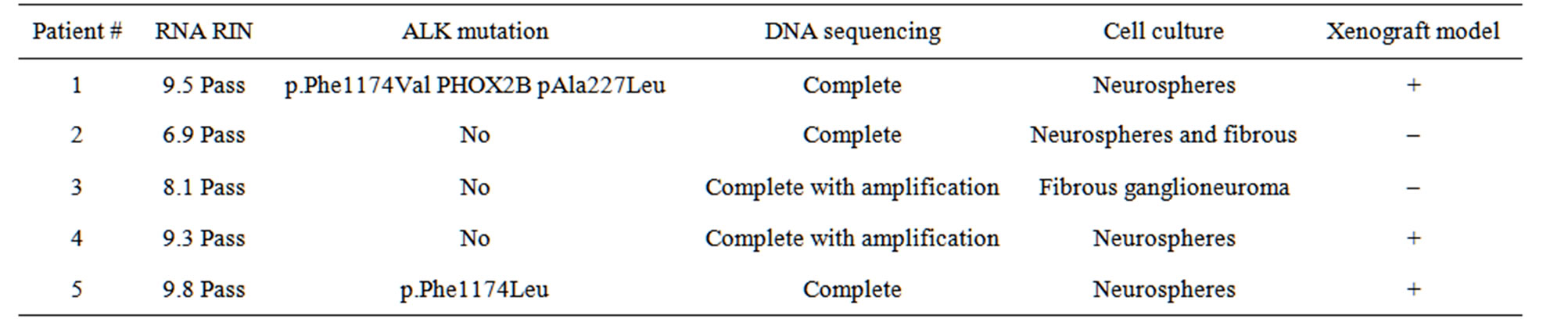
Table 3. Patient results for pilot study steps.
predictions of drug efficacy summarized in the guided therapy report. The final molecular tumor board treatment decision was presented to the medical monitor who had the opportunity to reject the proposed regimen. The time between the molecular tumor board meeting and the medical monitor sign-off was 1 - 2 days (Table 2). Therefore, the total number of days from biopsy to medical monitor sign-off was 10 - 12 days for 5/5 patients, which met our objective of 14 days or less (Table 2).
The genes targets that were seen in these patients included expected genes TOP2A, MTOR and HDAC as well as new targets such as ERBB2, KIT and RAF1. These will undergo further validation. To accomplish this, tumor cells were maintained for additional biological studies. All tumor cells were able to be grown in culture. The tumor cells injected in mice were able to engraft to generate mouse models for 3/5 patients (Table 3). NextGen sequencing was completed at the NCI in 5/5 patients. DNA was adequate in 3/5 patients, while 2/5 patients required DNA amplification for completion of sequencing. Samples were sent for ALK testing at a CLIA certified lab, which determined the ALK mutation status of all patients. Of the five patients, two had a mutation of the ALK gene (Table 3).
4. Discussion
This is the first study to demonstrate the feasibility of using molecular profiling on tumors from pediatric patients in real time in which the critical steps of tumor board and independent medical monitor review were incorporated. Enrollment of five pediatric patients with refractory neuroblastoma was achieved in four months. Acceptable mRNA quality and microarray analysis was completed in all five patients. Most notably, the timeline for report generation, molecular tumor board review, individualized treatment plan formulation, and medical monitor review was completed in 12 days or less, which surpassed the goal of 14 days. This timeline confirms that the implementation of individualized treatment on the basis of real time genomic profiling and bioinformatics analysis is feasible. In addition, our correlative analysis studies demonstrated that NextGen sequencing was possible for all patients; tumor cells from each patient were successfully grown in cell culture permitting ongoing drug testing studies; and 3/5 tumor cell lines were successfully engrafted into mouse models permitting in vivo evaluation of predicted agents.
Collaboration between pediatric oncologists, pharmacists, geneticist and bioinformatics specialists, and the medical monitor was achieved in a time-efficient manner. The interdisciplinary make-up of the molecular tumor board was critical to creating an individual treatment plan that accounted for understanding the genomic predictions, the therapeutic decisions, and drug interactions for safety. As technologies such as next generation sequencing dramatically increase the extent of genomic data available, formation of skilled multidisciplinary teams across institutions will be critical to ensure the appropriate interpretation and extraction of actionable knowledge.
The report generated for each patient uses a novel approach by combining analytical methods including biomarker and drug target rules, systems biology approaches to predict key drivers of disease, and signature-based methods that utilize empirical drug screening data. This bioinformatics platform matches specific molecular markers, changes in gene expression, and molecular networks with drugs for those molecular targets based on the scientific literature and clinical trial data. By doing so, it takes into account the molecular heterogeneity of neuroblastoma between patients allowing each to be treated according to their genomic make-up. Since this study used one tumor biopsy from each patient, it may not be able to account for the tumor heterogeneity that exists within each patient at any one time or the tumor heterogeneity within one patient over time. To account for this intra-patient variability, future studies will include profiling of multiple tumor biopsies from the same patient from different anatomical sites where possible to assess temporal and spatial heterogeneity. Tumor biopsies performed over time will begin to address the adaptability and acquired drug resistance of the tumor to specific treatment regimens for each patient, and thereby identify combinational targeting strategies to treat and prevent rapid onset of refractory disease.
This pilot study provides evidence that genome-wide mRNA expression profiling from neuroblastoma tumor samples can be used to formulate individualized predictive treatment plans for pediatric patients in a feasible and time-efficient manner. The cell lines and mouse models generated from the patients’ tumor cells provide an opportunity to validate these predictive treatment regimens and to compare them with treatments that were predicted to not be clinically beneficial. The results from the NextGen sequencing will be used to assess correlation between identified mutations and expression data from the microarray. The tumor resource created can also be used to evaluate the potential predictive value of other molecular technologies such as single nucleotide polymorphisms, RNA sequencing, gene amplifications/ deletions, gene promoter methylation, protein modifications, or epigenetic changes [13,30-31].
5. Conclusion
This study shows that it is feasible to create therapeutic treatment plans based on genomic profiling in less than 12 days. The next step in this process is the implementation and validation of the predictive treatment plans. A Phase I study (NCT01355679) is currently underway to determine the feasibility and safety of treatment using individualized therapies and to measure benefit assessing response rate and progression-free survival ratio of pediatric patients with refractory neuroblastoma treated using these integrated precision methodologies.
6. Acknowledgements
We would like to acknowledge Intervention Insights for support and collaboration in developing the predictive reports and tumor board participation; and Pam Kidd and John Roys from Spectrum Health for flow cytometric analysis of neuroblastoma in bone marrow, and Javed Khan from the Pediatric Oncology Branch, National Cancer Institute for DNA sequencing.
This work was made possible by generous funding suport by the Charles and Meryl Witmer Foundation; Friends of Will; the Ishan Gala Foundation; Lillie’s Friends Foundation; Owen Moscone Foundation; NB Alliance; Max’s Ring of Fire. Intervention Insights and Affymetrix provided in kind services, supplies and reagents.
REFERENCES
- American Cancer Society, “Cancer Facts and Figures 2010,” American Cancer Society, Atlanta, 2010.
- M. L. Bernstein, J. M. Leclerc, G. Bunin, et al., “A Population-Based Study of Neuroblastoma Incidence, Survival, and Mortality in North America,” Journal of Clinical Oncology, Vol. 10, No. 2, 1992, pp. 323-329.
- G. M. Brodeur, J. Pritchard, F. Berthold, et al., “Revisions of the International Criteria for Neuroblastoma Diagnosis, Staging, and Response to Treatment,” Journal of Clinical Oncology, Vol. 11, No. 8, 1993, pp. 1466-1477.
- J. R. Park, A. Eggert and H. Caron, “Neuroblastoma: Biology, Prognosis, and Treatment,” Pediatric Clinics of North America, Vol. 55, No.1, 2008, pp. 97-120.
- K. K. Matthay, J. G. Villablanca, R. C. Seeger, et al., “Treatment of High-Risk Neuroblastoma with Intensive Chemotherapy, Radiotherapy, Autologous Bone Marrow Transplantation, and 13-cis-Retinoic Acid, Children’s Cancer Group,” The New England Journal of Medicine, Vol. 341, No. 16, 1999, pp. 1165-1173. doi:10.1056/NEJM199910143411601
- S. Huang, “Back to the Biology in Systems Biology: What Can We Learn from Biomolecular Networks?” Briefings in Functional Genomics Proteomic, Vol. 2, No. 4, 2004, pp. 279-297. doi:10.1093/bfgp/2.4.279
- A. Aranda-Anzaldo, “Cancer Development and Progression: A Non-Adaptive Process Driven by Genetic Drift. Acta Biotheoretica, Vol. 49, No. 2, 2001, pp. 89-108. doi:10.1023/A:1010215424196
- E. Wang, A. Lenferink and M. O’Connor-McCourt, “Cancer Systems Biology: Exploring Cancer-Associated Genes on Cellular Networks,” Cellular and Molecular Life Sciences, Vol. 64, No. 14, 2007, pp. 1752-1762. doi:10.1007/s00018-007-7054-6
- A. Balakrishnan, F. E. Bleeker, S. Lamba, et al. “Novel Somatic and Germline Mutations in Cancer Candidate Genes in Glioblastoma, Melanoma, and Pancreatic Carcinoma,” Cancer Reseaech, Vol. 67, No. 8, 2007, pp. 3545-3450. doi:10.1158/0008-5472.CAN-07-0065
- H. H. Heng, “Cancer Genome Sequencing: The Challenges Ahead,” BioEssays, Vol. 29, No. 8, 2007, pp. 783- 794. doi:10.1002/bies.20610
- J. K. Lee, D. M. Havaleshko, H. Cho, et al., “A Strategy for Predicting the Chemosensitivity of Human Cancers and Its Application to Drug Discovery,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 104, No. 32, 2007, pp. 13086-13091. doi:10.1073/pnas.0610292104
- Y. P. Mosse, M. Laudenslager, L. Longo, et al., “Identification of ALK as a Major Familial Neuroblastoma Predisposition Gene,” Nature, Vol. 455, No. 7251, 2008, pp. 930-935. doi:10.1038/nature07261
- R. E. George, T. Sanda, M. Hanna, et al., “Activating Mutations in ALK Provide a Therapeutic Target in Neuroblastoma,” Nature, Vol. 455, No. 7251, 2008, pp. 975- 978. doi:10.1038/nature07397
- Network CGAR, “Comprehensive Genomic Characterization Defines Human Glioblastoma Genes and Core Pathways,” Nature, Vol. 455, No. 7216, 2008, pp. 1061- 1068.
- T. Sjoblom, S. Jones, L. D. Wood, et al., “The Consensus Coding Sequences of Human Breast and Colorectal Cancers,” Science, Vol. 314, No. 5797, 2006, pp. 268-274. doi:10.1126/science.1133427
- J. P. Overington, B. Al-Lazikani, A. L. Hopkins, “How Many Drug Targets Are There?” Nature Reviews Drug Discovery, Vol. 5, No. 12, 2006, pp. 993-996. doi:10.1038/nrd2199
- D. S. Wishart, C. Knox, A. C. Guo, et al., “DrugBank: A Knowledgebase for Drugs, Drug Actions and Drug Targets,” Nucleic Acids Research, Vol. 36, 2008, pp. D901- D906. doi:10.1093/nar/gkm958
- G. Bollag, P. Hirth, J. Tsai, et al., “Clinical Efficacy of a RAF Inhibitor Needs Broad Target Blockade in BRAFMutant Melanoma,” Nature, Vol. 467, No. 7315, 2010, pp. 596-599. doi:10.1038/nature09454
- E. L. Kwak, Y. J. Bang, D. R. Camidge, et al., “Anaplastic Lymphoma Kinase Inhibition in Non-Small-Cell Lung Cancer,” The New England Journal of Medicine, Vol. 363, No. 18, 2010, pp. 1693-1703. doi:10.1056/NEJMoa1006448
- J. Lamb, E. D. Crawford, D. Peck, et al., “The Connectivity Map: Using Gene-Expression Signatures to Connect Small Molecules, Genes, and Disease, Science, Vol. 313, No. 5795, 2006, pp. 1929-1935.
- J. E. Staunton, D. K. Slonim, H. A. Coller, et al., “Chemosensitivity prediction by Transcriptional Profiling,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 98, No. 19, 2001, pp. 10787-10792. doi:10.1073/pnas.191368598
- C. P. Webb, “Personalized Medicine: The Need for System Integration in the Design of Targeted Therapies,” In: R. Head, Ed., Computational and Systems Biology: Applications and Methods, Research Signpost, Kerala, 2009.
- D. D. Von Hoff, J. J. Stephenson Jr., P. Rosen, et al., “Pilot Study Using Molecular Profiling of Patients’ Tumors to Find Potential Targets and Select Treatments for Their Refractory Cancers,” Journal of Clinical Oncology, Vol. 28, No. 33, 2010, pp. 4877-4883. doi:10.1200/JCO.2009.26.5983
- A. Tsimberidou, N. G. Iskander, D. S. Hong, J. J. Wheler, S. Fu, S. A. Piha-Paul, A. Naing, G. S. Falchook, F. Janku, R. Luthra, S. Wen and R. Kurzrock, “Personalized Medicine in a Phase I Clinical Trials Program: The M.D. Anderson Cancer Center Initiative,” Journal of Clinical Oncology, Vol. 29, 2011, pp. CRA2500.
- C. F. Thorn, T. E. Klein, R. B. Altman, “Pharmacogenomics and Bioinformatics: PharmGKB,” The Pharmacogenomics Journal, Vol. 11, No. 4, 2010, pp. 501-505. doi:10.2217/pgs.10.15
- Z. Dezso, Y. Nikolsky, T. Nikolskaya, et al., “Identifying Disease-Specific Genes Based on Their Topological Significance in Protein Networks,” BMC Systems Biology, Vol. 3, 2009, p. 36. doi:10.1186/1752-0509-3-36
- W. Shi, M. Bessarabova, D. Dosymbekov, et al., “Functional Analysis of Multiple Genomic Signatures Demonstrates That Classification Algorithms Choose Phenotype-Related Genes,” The Pharmacogenomics Journal, Vol. 10, No. 4, 2010, pp. 310-323. doi:10.1038/tpj.2010.35
- A. Vellaichamy, Z. Dezso, L. JeBailey, et al., “Topological Significance Analysis of Gene Expression and Proteomic Profiles from Prostate Cancer Cells Reveals Key Mechanisms of Androgen Response,” PLoS One, Vol. 5, No. 6, 2010, p. e10936.
- J. S. Gheeya, Q. R. Chen, C. D. Benjamin, et al., “Screening a Panel of Drugs with Diverse Mechanisms of Action Yields Potential Therapeutic Agents Against Neuroblastoma,” Cancer Biology & Therapy, Vol. 8, No. 24, 2009, pp. 2386-2395. doi:10.4161/cbt.8.24.10184
- S. Jones, X. Zhang, D. W. Parsons, et al., “Core Signaling Pathways in Human Pancreatic Cancers Revealed by Global Genomic Analyses,” Science, Vol. 321, No. 5897, 2008, pp. 1801-1806. doi:10.1126/science.1164368
- P. Northcott, A. Korshunov, H. Witt, et al., “Medulloblastoma Comprises Four Distinct Molecular Variants,” Clinical Oncology, Vol. 29, No. 11, 2011, pp. 1408-1414.
Abbreviations and Acronyms
NCI-National Cancer Institute VARI-Van Andel Research Institute CLIA-Clinical Laboratory Improvement Amendments CRL-Clinical Reference Laboratory ALK-Anaplastic Lymphoma Kinase VEGF-Vascular Endothelial Growth Factor TRK-Tropomyosin-Related Kinase mTOR-Mammalian Target of Rapamycin HDAC-Histone Deacetylase PFS-Progression-Free Survival ULN-Upper Limit of Normal SGPTSerum Gluamic Pyruvic Transaminase ALTAlanine Aminotransferase NCI CTCAE-National Cancer Institute Common Terminology Criteria for Adverse Events EKG-Electrocardiogram ECOG-Electrocochleography CBC-Complete Blood Count BUN-Blood Urea Nitrogen LDH-Lactate Dehydrogenase AST-Aspartate Aminotransferase FTP-File Transfer Protocol NOD SCID-Nonobese Diabetes Severe Combined Immunodeficiency FDAFood and Drug Administration
Authors and Affiliations
Corresponding Author:
Giselle L. Saulnier Sholler Helen DeVos Children’s HospitalVan Andel Research Institute 333 Bostw-ick Ave Grand Rapids, MI 49503 Giselle.sholler@vai.org 616-243-5465 William Ferguson Cardinal Glennon Children’s HospitalSt. Louis University 1465 S. Grand Blvd.
Saint Louis, MO 63104 fergusws@slu.edu Genevieve Bergendahl Van Andel Research Institute 333 Bostwick Ave Grand Rapids, MI 49503 Genevieve.bergendahl@vai.org Erika Currier University of Vermont 149 Beaumont Ave Burlington, VT 05406 ecurrier@uvm.edu Shannon R. Lenox University of Vermont 149 Beaumont Ave Burlington, VT 05406 slenox@uvm.edu Jeffrey Bond University of Vermont 149 Beaumont Ave Burlington, VT 05406 Jeffrey.bond@uvm.edu Marni Slavik University of Vermont 149 Beaumont Ave Burlington, VT 05406 marnislavik@yahoo.com William Roberts Rady Children’s Hospital San Diego, UCSD 3020 Children’s Way MC5035 San Diego, CA 92123-4282 wroberts@chsd.org Deanna Mitchell Helen DeVos Children’s Hospital 100 Michigan NE Grand Rapids, MI 49503 Deanna.Mitchell@helendevoschildrens.org Don Eslin MD Anderson Cancer Center Orlando 92 West Miller St., MP318 Orlando, FL 32806 Don.Eslin@orlandohealth.com Jacqueline Kraveka Medical University of South Carolina 135 Rutledge Avenue, Room 480 Charleston, SC 29425 kravekjm@musc.edu Joel Kaplan Levine Children’s Hospital 1000 Blythe Blvd Charlotte, NC 28203 Joel.Kaplan@carolinashealthcare.org Nehal S. Parikh Connecticut Children’s Medical Center 282 Washington Street Hartford, CT 06106 Nparikh@ccmckids.org Suman Malempati Oregon Health Science University 3181 SW Sam Jackson Park Rd., CDRC-P Portland, OR 97239-3098 malempat@ohsu.edu Gina Hanna MD Anderson Cancer Center Orlando 92 West Miller St., MP318 Orlando, FL 32806 Gina.hanna@gmail.com Emily Eugster Van Andel Research Institute 333 Bostwick Ave Grand Rapids, MI 49503 Emily.eugster@vai.org David Cherba Van Andel Research Institute 333 Bostwick Ave Grand Rapids, MI 49503 David.cherba@vai.org Jeremy Miller TransMed Systems 21170 Canyon Oak Way Cupertino, CA 95014 jmiller@xbtransmed.com Craig Webb Van Andel Research Institute 333 Bostwick Ave Grand Rapids, MI 49503 Craig.webb@vai.org
Authors’ Contributions
GS contributed to the conception and design of the protocol, acquisition of data, analysis and interpretation of data. GB contributed to design of the protocol, regulatory process, and analysis of data. WF, WR, DM, DE, JK, JK, NP, SM, GH all contributed to design of the protocol, analysis and interpretation of data. EC, SL contributed to acquisition of data. JB contributed to genomic analysis and drafting of the manuscript. MS contributed to drafting of the manuscript. EE, DC, JM contributed to genomic analysis. CW contributed to design of the protocol, interpretation and analysis of data. All authors read and approved the final manuscript.
NOTES
*All author(s) declare that they have no competing interests except: Craig Webb, Jeremy Miller.
#Corresponding author.