Health
Vol.6 No.15(2014), Article
ID:49116,12
pages
DOI:10.4236/health.2014.615238
Oxygen Therapy in Critical Care: A Double Edged Sword
Isidro Prieto del Portillo, Susana Temprano Vázquez, Jesús Barea Mendoza, Rubén Viejo Moreno
Intensive Care Unit, Hospital 12 de Octubre, Madrid, Spain
Email: iprieto.hrc@salud.madrid.org
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 17 June 2014; revised 3 August 2014; accepted 17 August 2014
ABSTRACT
The oxygen therapy is a universal treatment in the hospital setting, especially in the critical care units. The purpose of this therapy is to avoid hypoxemia and to ensure an adequate supply of oxygen to the tissues. But often we overlook the potential adverse effects of oxygen therapy. Oxygen produces lung damage and induces apoptosis and cell death creating an imbalance between the production of reactive species of oxygen and the antioxidant mechanisms. The oxygen therapy inhibits systemic adaptive changes induced by hypoxia, disrupting compensatory mechanisms and causing deleterious effects. We are faced with a challenge in order to treat patients with respiratory failure, counterbalancing hypoxia with hyperoxia-induced damage and introducing therapy lines that are innovative but not risk-free as permissive hypoxemia. Currently many questions remain unresolved and there are not enough clinical studies that validate the therapeutic optimal oxygenation ranges. These ranges may differ depending on each patient and the underlying disease.
Keywords:Oxygen Therapy, Oxygen Toxicity, Hyperoxia, Reactive Oxygen Species, Intensive Care Unit

1. Introduction
Our metabolism is primarily aerobic, so oxygen is essential to keep alive and work, and in a certain way the cause of our death too. When it falls, mostly if it occurs acutely, causes a damage which may be irreversible.
Patients admitted in units of intensive care (ICU) need to maintain an adequate tissue distribution of oxygen (DO2) according to tissue consumption of oxygen (VO2). Thus, oxygen therapy, administered with or without positive pressure devices, is a key piece for treatment in ICU. Nevertheless, an inadequate oxygen delivery that produces hyperoxia may be even more pernicious than hypoxia itself to generate reactive species of oxygen (ROS) and induce cellular apoptosis. Traditionally, the objectives of resuscitation in ICU have obviated the excess oxygen-induced damage and they have focused on preventing hypoxia. Currently, we should avoid the hyperoxia much more than the hypoxia, although the limits are not clearly defined.
In the next revision, we will look over major indications of oxygen therapy, both hypoxia-induced damage as hyperoxia physiopathological bases and will look over recent published studies to try to describe the current situation and review future prospects.
2. Respiratory Failure
The main function of the respiratory system is to encourage the uptake of oxygen and remove carbon dioxide (CO2) excess product of cellular metabolism [1] . An acute respiratory failure (ARF) is the situation of disability to provide suitable oxygen delivery to tissues, associated or not with a failure in the elimination of CO2.
The elimination of CO2 is a process related to the ventilation (air effective alveolar volume) and it can be measured through blood partial pressure of CO2 (PaCO2), which may be altered by an imbalance between their production and removal ability. On the other hand, oxygenation is related to ventilation, to diffusion capacity of gases through the alveolar-capillary membrane and to capillary perfusion, and can be measured with oxygen blood partial pressure (PaO2). Therefore, the method of choice to assess the impact and severity of RF is the arterial blood gases.
ARF situations shows a PaO2 < 60 mmHg (hypoxemia) associated or not to an increase of the PaCO2 > 45 mmHg (hypercapnia), if we consider an ideal situation breathing ambient air at sea level [2] . Nevertheless, in situations of higher altitude, the normal value of PaCO2 is quietly lower, due to certain physiological hyperventilation. These parameters are the ones that have been accepted by international consensus, after observing that small descents of PaO2 below 60 mmHg is related to great drops in oxygen saturation according to the hemoglobin dissociation curve. By contrast, due to the large capacity of diffusion of CO2 in normal conditions, PaCO2 above 45 mm Hg determines a major failure of pulmonary ventilation.
In order to know more about the pathophysiology of ARF, we must understand the respiratory system as a whole, constituted by lung tissue and pump ventilation (thoracic cage and diaphragm). As a rule, the lung diseases causes hypoxemia while pump fails may cause hypoxemia with hypercapnia.
There are five main causes of hypoxemia: ventilation-perfusion mismatch, hypoventilation, oxygen diffusion alterations, shunting, and the reduction of delivered fraction of inspired oxygen (FiO2) [3] (Table 1).
2.1. Ventilation-Perfusion (V/Q) Mismatch
This is the most frequent cause of ARF4. Poor alveolar ventilation for any reason with a suitable capillary perfusion, cause lower concentration of oxygen (FiO2) in the alveoli and therefore hypoxemia.
Hypoxemia due to V/Q mismatch shows an increase in alveolar-arterial oxygen gradient P(A-a)O2 [4] . The gradient is the difference between alveolar pressure of oxygen (PAO2) and PaO2, and will allow us to distinguish between hypoxemia as a consequence of an alteration of the pulmonary parenchyma or extrapulmonary origin hypoxemia. The following equation is used to calculate the PAO2:

Table 1. Causes of hypoxemia.
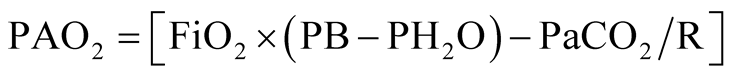
where PAO2 = partial pressure of oxygen in the alveoli; FiO2 = partial pressure of oxygen in the inspired air; PaCO2 = partial pressure of carbon dioxide in the alveoli (assumed equal to the blood, PaCO2) and R = ratio of respiratory exchange, which is equal to 0.8 in basal conditions.
Under normal conditions, P(A-a)O2 will be less than 20 mmHg. As age increases, the normal values of P(A-a)O2 may increase until 30 mmHg, since lung elastic capacity gradually diminishes and therefore the bronchioles collapse even earlier during exhalation [5] .
2.2. Hypoventilation
The alveoli ventilation allows the replacement in blood of the oxygen consumed by the tissues as well as the removal of the CO2 produced by the cellular metabolism. The situation where the alveolar ventilation is insufficient to maintain the homeostasis is known as hypoventilation and it presents characteristically with hypoxemia and hypercapnia. Severe and at times acute hypercapnia induces the appearance of respiratory acidosis and hence, a decrease in arterial pH (<7.35) due to the limited capacity of renal compensation. In these situations, P(A-a)O2 is usually normal.
2.3. Diffusion Alterations
The exchange of gases through the alveolar-capillary membrane occurs by passive diffusion which is proportional to the surface of exchange and inversely proportional to the thickness of the alveolar-capillary membrane. Due to high reserve of gases diffusion across the membrane, it is infrequent to prove hypoxemia just by this single factor. However, with another element added to the thickening of the wall, for example, a decrease in the exposure time of the red blood cells by pulmonary alveolar capillary as it happens during the exercise, the presence of hypoxemia may be expected [6] .
2.4. Shunting
This is the situation in which the alveoli are adequately perfused but poorly ventilated (e.g. areas of atelectasis or alveolar collapse). In these areas, blood passes through pulmonary capillaries without receiving oxygen and gets to the arterial circulation. Characteristically, these patients do not improve their PaO2 even if FiO2 delivered is raised, because the blood that passes through the collapsed alveoli is not exposed to high oxygen concentration [7] . Although erythrocytes are exposed to ventilated alveoli and take contact with high FiO2, but the hemoglobin is nearly saturated and just dissolved oxygen in plasma increases [8] .
2.5. Reduction of Delivered Fraction of Inspired Oxygen (FiO2)
This situation is typical in low oxygen concentration environments. Here, the oxygen concentration in the alveoli which participates in gas exchange is low, so this causes hypoxemia. In a situation where we are situated at three thousand meters above sea level, there will be a decrease of 30% in atmospheric PaO2 [9] , so if we ascend, altitude sickness may occur. Symptoms are dyspnea, nausea, agitation, pulmonary and cerebral edema and even cardiorespiratory arrest. Equally this could appear in overcrowding environments, caves or polluted areas by other gases other than oxygen where FiO2 is less than 21%.
3. Classification of Acute Respiratory Failure
3.1. ARF Due to Failure in Oxygenation, Hypoxemic or Type I [10]
The characteristic data is PO2 in arterial blood less than 60 mmHg, with normal or even descended (due to compensatory hyperventilation) PaCO2 values. The P (A-a)O2 is typically elevated (>20 mmHg). It is cause by intrinsic pulmonary pathology (e.g. pneumonia), chronic pulmonary obstructive disease (COPD), asthma, disorders of pulmonary capillaries, congenital heart disease or heart failure [11] . In severe cases, administration of oxygen at high concentrations is an essential part of the treatment although it will just lightly increases PaO2 it is possibly due to V/P mismatch or shunting (Table 2).
3.2. ARF Due to Ventilation Dysfunction, Mixed, Global or Type II
The characteristic laboratory data is an increase of arterial PaCO2 with hypoxemia. It occurs when either there is an excessive production associated with inappropriate elimination of CO2 or in a primary disorder in which alveolar ventilation is decreased so there is not effective elimination of CO2 [11] . In both cases, oxygen therapy is not usually enough and it may be necessary non-invasive or invasive mechanical ventilation. The P(A-a)O2 is normal (<20 mmHg) (Table 3).
Table 4 shows the main ranges target of arterial oxygen saturation in different types of ARF.
4. Hypoxia
Hypoxia triggers some physiological and physiopathological responses to keep balance between supply and
Table 2 . Most frequent causes of ARF type I.
Table 3. Most frequent causes of ARF type II.
Table 4. Main ranges target of arterial oxygen saturation (SatO2) in different situations.
need of oxygen in tissues. When these mechanisms fail or are not enough to keep an adequate aerobic metabolism tissue, hypoxia or dysoxia occur [12] .
Hypoxia has a principal role in many acute and chronic diseases. It plays a central role in cardiovascular, lung diseases or even cancer [13] .
4.1. Response to Hypoxia
4.1.1. Systemic Hypoxia Response
During the first minutes in hypoxia, respiratory and cardiovascular changes occur resulting with an increment of cardiac output and alveolar ventilation.
In tissues, it is especially important the response of the smooth muscle cells of arterioles. The types of responses are different depending on the vascular bed which they belong. At a systemic level, there is a relaxation of the arterioles secundary to opening potassium channels ATP dependent. Otherwise, myocytes of the pulmonary vasculature react with intense vasoconstriction in hypoxia.
There are two elements with an important role in changes relates PO2. The carotid bodies perform this function in the systemic circulation, they are located in the bifurcation of the common carotid arteries and are very vascularized They contains cells rich in potassium channels which are inactivated with hypoxemia conditioning cell depolarization and subsequent delivery of neuroendocrine afferent signals. In the bronchial tree, the neuroepithelial bodies have a similar response to changes in environmental oxygen content [12] .
4.1.2. Cellular Response
Cardiovascular and respiratory systems work together to keep tissue oxygen input. Oxygen is necessary to obtain ATP, who is essential for maintaining cells working. ATP is principally used to molecular synthesis (mainly proteins) and to keep homeostasis by cellular ionic pumps (Na+/K + ATPase).
Failure in pumps running by ATP deficit causes ionic imbalance, ionic changes in plasmatic membrane, cellular calcium intake, proteases and lipases Ca-dependent activation. The final result is cellular edema, hydrolysis of cellular components, and necrosis.
Changes that lead to cell death are complex. When major descents (anoxia, 0.1% oxygen) occurs, proapoptotic proteins are released such as Bax, cytochrome C or activated caspase. On the other hand, in mild hypoxemia cellular, the responses are not clearly defined. Some authors even conclude that hypoxemia could have protective effect against apoptosis in some cell types [12] [14] .
4.1.3. Metabolic Changes Induced by Hypoxia
As a result of hypoxia, cells undergo through changes with the result of an increment an anaerobic glycolysis and decrease of higher energy consumption processes.
Glucose is metabolized to pyruvate by glycolysis. In an aerobic environment, pyruvate dehydrogenase (PDH) turns acetyl coenzyme A to piruvate. Piruvate is oxidized into mitochondria and produces ATP and H2O. In hypoxemic environment, PDH is inhibited so the lactate dehydrogenase (LDHA) activity is stimulated; therefore pyruvate is metabolized mainly into lactic acid.
Initially, cell does without anabolism and the synthesis of new molecules. Other processes related to the ionic pumps maintenance, membrane potential and calcium metabolism become of first importance. In fact, some authors propose that electrical activity in different tissues plays a role in the susceptibility against hypoxia (e.g. increased susceptibility of central nervous system to hypoxia) [12] .
4.1.4. Changes in Gene Expression: The Role of Hypoxia Inducible Factor (HIF-1)
During hypoxia an adaptive change cascade begins; some of the changes will remain through several months if initial condition is maintained. It was noted that hypoxia triggers changes in mRNAs transcription pattern. Over the last years, the HIF-1 has been defined as the main molecule in this adaptive process [14] .
The HIF-1 contains a constitutive subunit (HIF-1B) and an oxygen regulated subunit (HIF-1A). In aerobic conditions, HIF-1A is hydroxylated, which allows binding to Von Hippel-Lindau protein (VHL). This complex joins to ubiquitins and it degrades into 26S proteasome. So in normoxia conditions there are low citoplasmatics levels of HIF-1A due to continuous degradation.
In hypoxic conditions, the degradation process is inhibited because HIF-1A hydroxylation doesn’t occur. HIF 1-A gets increased, which promotes binding to HIF-1B. The complex will initiate multiple gene transcription so RNA polymerase II activity is activated [15] [16] .
Therefore, HIF-1 causes an increment of the expression of multiple factors: placental growth factor (PLGF), vascular endotelial growth factor (VEGF), platelet derived growth factor (PDGF) or erythropoietin (EPO). These factors have a main role in adaptation to hypoxia processes such as carbohydrate metabolism, endothelial proliferation or vascular modeling [14] (Figure 1).
5. Hyperoxia
High content of oxygen (FIO2 > 90%) air breathing for a long time, leading or not to hyperoxia, causes frequently acute lung damage. Severity of injury has been correlated with time of exposure, oxygen concentration and other adjuvant factors (mechanical ventilation, bacterial superinfection…). Several mechanisms have been proposed.
5.1. Reactive Oxygen Species and Hyperoxia-Induced Lung Injury
ROS are a group of highly reactive molecules. Its main characteristic is the presence of unbound electrons in its electric formation. Therefore, they have high tendency to react with other molecules around them.
Aerobic metabolism needs to turn oxygen into ATP and H2O. During this process, ROS acts as intermediate metabolites such as superoxide anion, hydrogen peroxide, and hydroxyl radical.
Under standard conditions there is balance between ROS production and antioxidant mechanisms. Main protection mechanisms include enzymes (superoxide dismutase, catalase and glutathione peroxidase) and non-enzymatic factors (C and E vitamin, beta carotene...). Under hyperoxia conditions a linear increase in ROS production occurs and as a result, an imbalance (Figure 2).
These molecules, given its electrical instability, react quickly with other macromolecules in the cell generating a primary damage. Proteins and lipids conditioning lesions in membranes, mitochondrial dysfunction, DNA damage and eventually death (Figure 3).
In addition the hyperoxia is able to trigger a secondary inflammatory response through the activation of macrophages, neutrophils and platelets; generating a secondary damage [17] [18] .
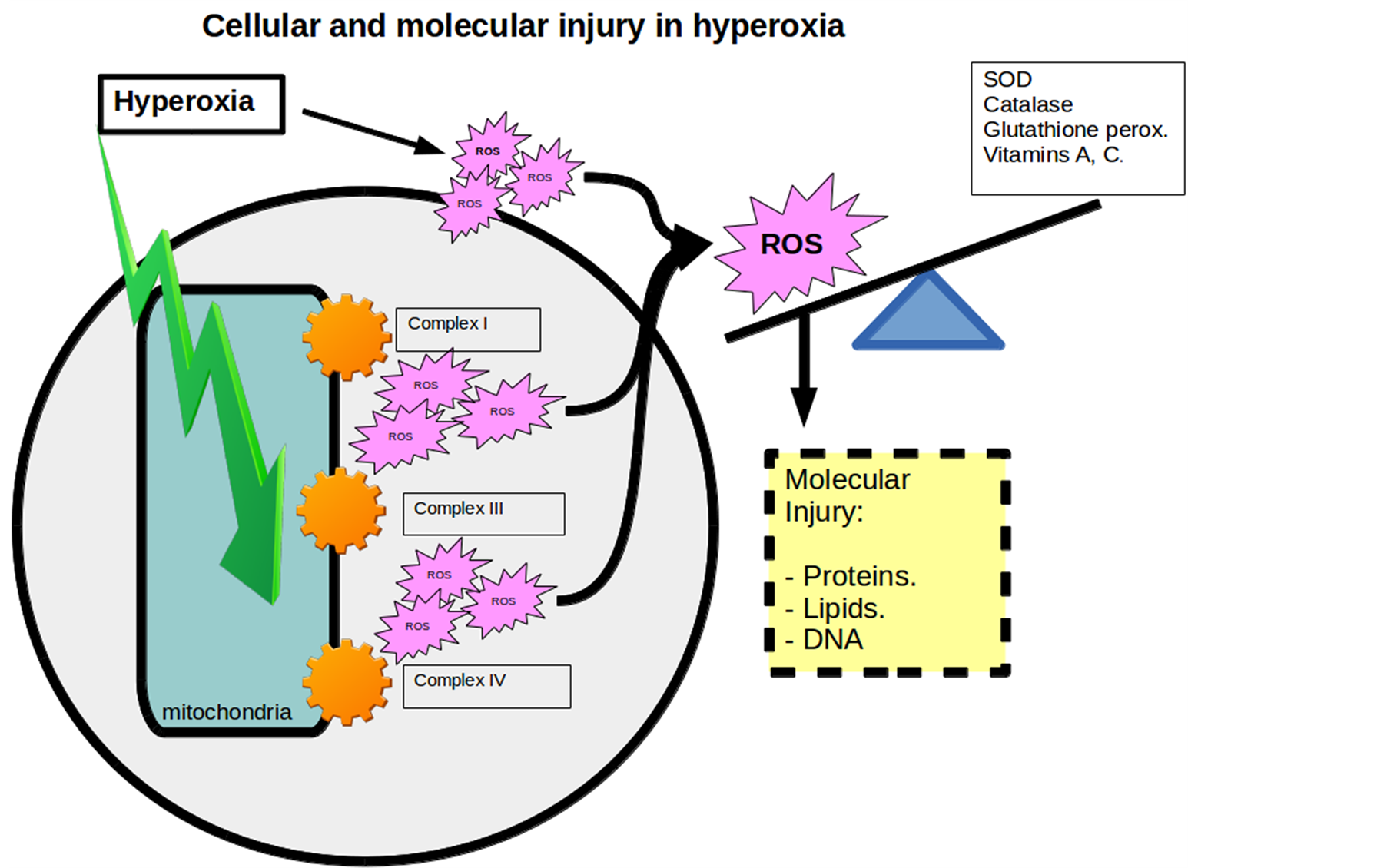
Figure 1. Cellular and molecular injury in hyperoxia.

Figure 2. Direct and secondary damage in hyperoxia.

Figure 3. Hypoxic inducible factor-1 (HIF-1): Regulations and pathways.
5.2. Mechanisms of Cell Death and Hyperoxia-Induced Pulmonary Damage
As a consequence of hyperoxia, phenomena of apoptosis and pulmonary necrosis which involve multiple pathways occur. Apoptosis is due both to mitochondrial activation mediated by the caspase-9 group and to extrinsic pathways activation (caspase-8).
The activation of mitochondrial pathway has been associated with proapoptotics elements of BCL-2 family. These trigger the release of cytochrome C, increasing mitochondrial membrane permeability and finally apoptosis. Extrinsic pathway starts with Fas/Fas-L complex and the subsequent activation of caspase-8 pathway.
At the same time, some molecular pathways which can counteract the activation of proapoptotic waterfalls in the hyperoxia have been described. As an example, TLR-4, which starts an antiapoptotic signals like Bcl-XL, c-Jun or JNK.
Hyperoxia-induced tissue damage occurs in endothelial cells, macrophages and respiratory epithelium. During the first hours damage occurs on pulmonary endothelium. Endothelium, as a highly active tissue, can activate platelet aggregation and inflammatory cells chemotaxis. So, an inflammatory response and a secondary damage by the formation of a new generation of ROS starts (Figure 4).
Hypothesis about “primary vascular disorder” is based on its high susceptibility due to his very active metabolism that regularizes large amount of vasoactive and fibrinolytic substances (serotonin, noradrenaline, bradikinin, angiotensin...).
Multiple cytokines and growth factors participate in hypoxia-induced damage. The most important is the IL-1 who activates others as IL-6, IL-8 and TNF-α. These molecules play a key role in activation of inflammatory cells after primary endothelial damage [17] -[19] .
5.3. Effect of Hyperoxia on Hypoxia Compensating Mechanisms
Recently it has been proposed a hypoxemia classification in four categories depending on the time of establishment: acute hypoxemia (<6 hr), subacute hypoxemia (6 - 7 days), prolonged hypoxemia (7 - 90 days) and chronic hypoxemia (>90 days) [20] [21] .
Healthy individuals exposed to prolonged hypoxemia develop multiple compensating mechanisms (increase in cardiac output, alveolar ventilation and erythrocyte mass, etc). However, ill subjects usually are not able to develop this type of adaptive responses due to the underlying disease.
Other changes exist at cellular level like changes in mitochondrial metabolism by unbounding multiple metabolic processes and thus achieving greater efficiency in ATP production.
Under experimental conditions, cell groups exposed to prolonged hypoxia decreases oxygen intake up to 40% - 60% by down-regulation processes who are not essential for cell viability. This regulatory mechanism is reversible and do not establish a definitive damage.
Traditionally, management of respiratory failure has been to avoid hypoxemia maintaining hyperoxia at significant levels. Several authors have hypothesized that hyperoxia in critical patients with subacute hypoxemia (ADRS, pneumonia) could interrupt the establishment of compensating mechanisms (mitochondrial, gene related to HIF-1...) with the consequent deleterious effect. Therefore, they report how important is to determine safe and well tolerated PO2 levels for our patients [18] [20] -[22] .
6. Clinical and Experimental Studies
6.1. Lung/Acute Respiratory Distress Syndrome (ARDS)
The gaseous exchange surface is the lung, and it is as well one of the higher risk tissues to be damaged by high concentrations of oxygen. Oxygen toxicity is rare with FiO2 < 0.5 [23] . Patients with ARDS often require FiO2 > 0.5 and have an increased risk of worsening the underlying lesion. Positive pressure ventilation with high FiO2 (0.6 - 0.9) results as a specific pathological changes regardless of deleterious effect of ventilator. Bailey et al.
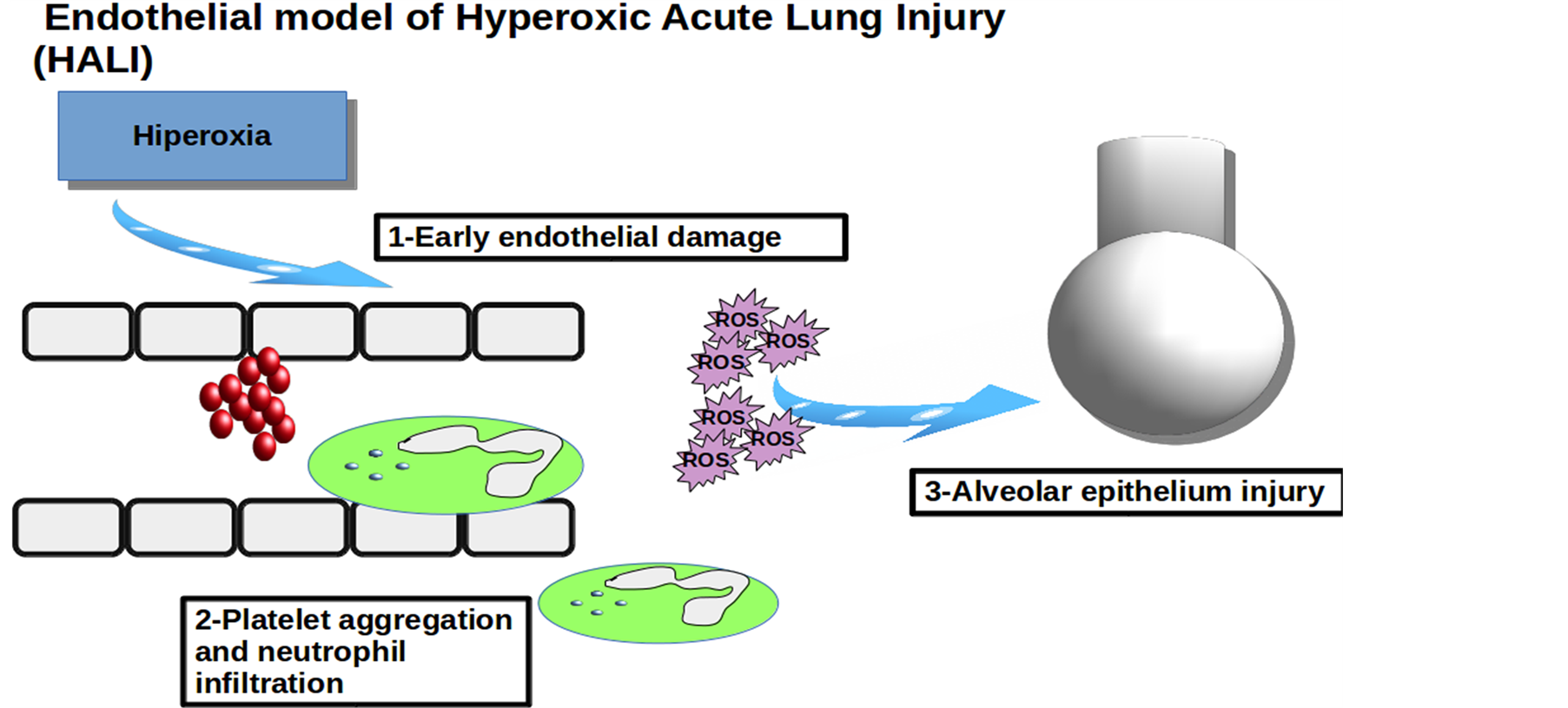
Figure 4. Endotelial model of Hyperoxic Acute Lung Injury (HALI).
studied mechanically ventilated mices exposing them to oxygen concentrations > 90% over 24 hours [24] . They observed an increased susceptibility to injury induced by the ventilator than those exposed to ambient air. Sinclair et al. [25] recently published a study with mechanically ventilated rabbits with high tidal volumes and moderate oxygen concentration-exposed (FiO2 = 0.5). They observed more significantly serious histological lesions in those in comparison with the ones with air environment ventilation. Ventilated animals with high volumes and moderate oxygen concentration developed alveolar-capillary membrane injury, significantly, compared with those with the same parameters of ventilation but with normoxia [26] .
There are no clinical trials in humans that evaluate both the effectiveness and the deleterious effect of therapeutic high FiO2 (e.g. FiO2 over necessary to assure suitable DO2). The ARDS Network study [27] compared higher positive end expiratory pressure (PEEP) levels against lower ones in patients with ARDS, using a protocol based on oxygenation to reach approximately 5 cm H2O PEEP differences among the two groups. Although significantly differences nearly 20% FiO2 among both groups were found, there were no differences in mortality, days on mechanical ventilation and ICU stay; so they concluded that higher peep levels did not increment mortality in this patients. In this study, it is difficult to reach a conclusion principally due to the fact that the study was not designed to evaluate this purpose and it is not clear if differences of 0.1 in FiO2 among experimental groups represent a clinically significant difference. An analysis of 101 studies in ARDS concludes that the PO2/FiO2 ratio is not a reliable outcome predictor. The British Thoracic Society Guidelines recommend SatO2 between 94% - 98% for a large majority of acute patients but there is not clinical evidence that support these recommendations [28] .
Gilbert-Kawai et al. (The Cochrane) has recently published a review about the permissive hypoxemia against normoxia in critical patients receiving mechanical ventilation, focused on mortality and morbidity. They did not find evidence that low levels of oxygen in blood could be advantageous [29] . However, Suzuki et al. in a study performed in 105 ICU patients with mechanical ventilation for >48 hours showed advantages with conservative oxygen therapy, maintaining an objective of SatO2 90% - 92%, comparing to a conventional treatment, causing less clinical and physiological adverse effects and avoid exposure to high levels of oxygen [22] .
6.2. Acute Myocardial Infarction (AMI)
Over one hundred years oxygen therapy has been administered for treatment of AMI and angina with little evidence regarding efficacy or deleterious effect of this practice. Mechanism caused by oxygen can be harmful, including the paradoxical effect of oxygen in reducing coronary blood flow and increasing systemic vascular resistance [30] .
Wijesinghe et al. [31] examined the impact of oxygen on infarction size of patients with AMI and concludes that there is little evidence to determine the efficacy and safety of treatment with high flow oxygen in this patients. There is no evidence that treatment with oxygen when compared with ambient air, has a benefit in this situation. Moreover, it could be harmful resulting with a larger size of AMI and increased mortality. An analyzed review in 2013 from the Cochrane results in the same conclusion: there is no evidence from randomized trials that support the routine use of oxygen in patients with AMI. AVOID study (air versus oxygen in myocardial infarction) [32] , in press, randomizes patients with AMI receiving treatment as usual with oxygen therapy or no treatment unless oxygen saturation is <94%, analyzing size of infarction, complications and survival. Maybe it will contribute to clarify questions about this issue. The guidelines of the European Society of Cardiology 2012 and the AHA/ACCF 2013 for the AMI with S-T elevation just recommend oxygen therapy in hypoxemic patients (SpO2 < 90%) [33] .
6.3. Acute Ischemic Stroke
Oxygen supply should be provided to maintain SpO2 > 94% (Class I, level of evidence C) [34] . Although administration of supplemental oxygen seems rather intuitively and there is little data that support it. The ischemic stroke causes a decrease of oxygen in the brain, resulting in tissue hypoxia and cell death. Therefore, raising oxygen levels in an ischemic tissue can produce neuroprotection. A study observed that high flow oxygen within 14 hours from the onset of stroke might be beneficial in selected patients. They found a trend in lowering the mortality and decreasing comorbilities in patients with stroke in middle cerebral artery territory treated with oxygen 40% facial mask compared with a flow of oxygen of 2 liters per minute (lpm) by nasal cannula [35] . Results of a great controlled trial in stroke patients found no significant difference in mortality over one year or neurological deficits among patients receiving oxygen at 3 lpm by nasal cannula for 24 hours after income and those not receiving it. Therefore, based on these data, routine supplement of oxygen in not hypoxic patients with moderate mild stroke is not recommended [36] .
6.4. COPD
Current literature provides overwhelming evidence, in COPD patient, on the use of oxygen therapy to keep SatO2 88% - 92% cause it reduces the risk of death due to respiratory failure, especially in those patients susceptible to hypercapnia [37] . A prospective study published in 2010 [38] compared high flow oxygen with conventional oxygen therapy via nasal cannula to keep SatO2 between 89% - 92%. This study showed that adjust oxygen therapy maintaining SatO2 89 - 92% reduces the risk of death by hypercapnia and respiratory failure in a 58% (all subjects) and 78% (those with confirmed COPD).
6.5. Resuscitation Following Cardiac Arrest
A retrospective study carried out by Kilgannon et al. [39] included over 6.000 patients who had required resuscitation maneuvers after cardiorespiratory arrest. They assessed the effect of the hyperoxemia (defined as PaO2 > 300 mmHg) compared to normoxemia (PaO2 60 - 300 mmHg) and hypoxemia (<60 mmHg). Results of this study show that hyperoxia group had a higher mortality rate (63%) than patients in normoxia (45%) and hypoxic (57%) groups. They conclude that hyperoxia in these patients is associated with increased hospital mortality compared to normoxia or hypoxia. The ANZ study [40] (12.108 patients included) showed that hyperoxia was relatively uncommon. There was not a consistently reproducible relation with mortality. In a recent review about the impact of oxygen and carbon dioxide after suffering a cardiac arrest, Bellomo et al. suggest that hyperoxia and hypocapnia may be deleterious and harmful and must be avoided and that these patients should be treated like the rest of critical patients requiring mechanical ventilation.
6.6. Prevention of Surgical Infections Sites
Pryor et al. [41] studied use of hyperoxia in the prevention of infection in surgical wound. 165 patients undergoing into a major abdominal surgery were randomized to receive FiO2 0.8 vs 0.35 during surgery or in the first two postoperative hours. The incidence of infection was significantly greater in the group receiving FiO2 of 0.8 than in the group of 0.35 (25% vs. 1.3%, p = 0.02). FiO2 remained as a significant predictor of surgical wound infection in a multivariate analysis. These findings contradict an earlier study [42] that reported a 50% reduction in the incidence of infection in open colorectal surgery patients who received O2 0.8 against those who received 0.3.
7. Conclusions
Oxygen therapy is one of the most commonly treatment used in the hospital environment. Its use, as with any other treatment may result in adverse events. So it should be always prescribed if a clear indication exists. The primary objective is to correct hypoxemia. Its use in the prevention of hypoxemia in susceptible patient or as treatment in relieving work of breathing, is not clearly defined. But the administration of oxygen may produce adverse events such as carbonic acid retention, atelectasis and alveolar and pulmonary endothelium damage. This side effects multiplies in case of hyperoxia. The hyperoxia-induce tissue damage is due to ROS production, by altering the immune response, and by inducing cellular apoptosis.
It seems necessary to establish limits of oxygenation that prevent the deleterious effects of hypoxia and hyperoxia, particularly in pathologies in which prevails a mechanism of ischemia-reperfusion as the cardiac arrest, stroke or the postoperative period of cardiac surgery. These limits are not yet well defined, but certainly should be lower than those used in routine practice. Although in this context, the permissive hypoxemia may be a promising therapy, there is currently no scientific evidence that justifies it and setting too low oxygenation can lead to uncontrolled episodes of hypoxemia and disoxia.
A significant number of patients in the ICU required mechanical ventilation [43] . The damage induced by this over a healthy lung increases significantly in diseased lungs. Several mechanisms have been described to explain this phenomenon as the opening and closing of alveolar units (atelectrauma), the over-stretching and pulmonary break (volutrauma and barotrauma) or the induction of self-damage inflammatory response (biotrauma). Probably in this last section we have to take into account the damage caused by oxygen by itself (oxygentrauma) and assess ways to modulate it.
They are necessary new clinical studies that evaluate the more conservative strategies of oxygen therapy, especially in critical patients with acute lung damage or pathologies with ischemia-reperfusion injury. Until that time, we must avoid the administration of high concentrations of oxygen, preventing the hyperoxia and maintaining of PaO2 levels that ensure an adequate tissue perfusion.
References
- West, J.B. (2007) Pulmonary Pathophysiology: The Essentials. 7th Edition, Lippincott Williams and Wilkins, Philadelphia.
- Campbell, E.J.M. (1965) Respiratory Failure. BMJ, 1, 1451-1460. http://dx.doi.org/10.1136/bmj.1.5448.1451
- De Lucas, P., Güell, R.R., Rodríguez, J.M. and Antón, A.A. (2007) Tratado de Insuficiencia Respiratoria. Ergón Ltd., Madrid.
- Cabrera Navarro, P. and Rodríguez de Castro, F. (2005) Manual de Enfermedades Respiratorias. 2a Ed. Union Internacional Contra la Tuberculosis y Enfermedades Respiratorias ED, 605-611.
- George, R.B., Light, R.W., Matthay, M.A. and Matthay, R.A. (2005) Chest Medicine: Essentials of Pulmonary and Critical Care Medicine. 5th Edition, Lippincott-Williams and Wilkins, Philadelphia.
- Nunn, J.F. (1993) Diffusion and Alveolar/Capillary Permeability. In: Nunn’s Applied Respiratory Physiology, 4th Editon, Butterworth-Heinemann Ltd, Edinburgh, 198-218. http://dx.doi.org/10.1016/B978-0-7506-1336-1.50015-2
- Greene, K.E. and Peters, J.I. (1994) Pathophysiology of Acute Respiratory Failure. Clinics in Chest Medicine, 15, 1-12.
- Mackenzie, I. (2008) Core Topics in Mechanical Ventilation. Cambridge University Press, Cambridge, 12-17.
- Carpio, C., Romera, D. and Fernández-Bujarrabal, J. (2010) Insuficiencia Respiratoria Aguda. Medicine, 10, 4332-4338.
- Roussos, C. and Koutsoukou, A. (2003) Respiratory Failure. European Respiratory Journal, 22, 3S-14S.http://dx.doi.org/10.1183/09031936.03.00038503
- Wood, L.D.H. (1998) The Pathophysiology and Differential Diagnosis of Acute Respiratory Failure. In: Hall, J.B., Schmidt, G.A. and Wood, L.D.H., Eds., Principles of Critical Care, McGraw-Hill, New York, 449-508.
- Michiels, C. (2004) Physiological and Pathological Responses to Hypoxia. The American Journal of Pathology, 164, 1875-1882. http://dx.doi.org/10.1016/S0002-9440(10)63747-9
- Eltzschig, H.K. and Carmeliet, P. (2011) Hypoxia and Inflammation. The New England Journal of Medicine, 364, 656-665. http://dx.doi.org/10.1056/NEJMra0910283
- Semenza, G.L. (2011) Oxygen Sensing, Homeostasis and Disease. The New England Journal of Medicine, 365, 537-547. http://dx.doi.org/10.1056/NEJMra1011165
- Semenza, G.L. (2006) Regulation of Physiological Responses to Continuous and Intermittent Hypoxia by Hypoxia-Inducible Factor 1. Experimental Physiology, 91, 803-806. http://dx.doi.org/10.1113/expphysiol.2006.033498
- Benizri, E., Ginouvès, A. and Berra, E. (2008) The Magic of the Hypoxia-Signaling Cascade. Cellular and Molecular Life Sciences, 65, 1133-1149.
- Kallet, R.H. and Matthay, M.A. (2013) Hyperoxic Acute Injury Lung. Respiratory Care, 58, 123-141. http://dx.doi.org/10.4187/respcare.01963
- Altemeier, W.A. and Sinclair, S.E. (2007) Hyperoxia in the Intensive Care Unit: Why More Is Not Always Better. Current Opinion in Critical Care, 13, 73-78. http://dx.doi.org/10.1097/MCC.0b013e32801162cb
- Horowitz, S. (1999) Pathways to Cell Death in Hyperoxia. Chest, 116, 64S-67S. http://dx.doi.org/10.1378/chest.116.suppl_1.64S
- Martin, D.S. and Grocott, M.P.W. (2013) Oxygen Therapy in Critical Illness: Precise Control of Arterial Oxygenation and Permissive Hypoxemia. Critical Care Medicine, 41, 423-432. http://dx.doi.org/10.1097/CCM.0b013e31826a44f6
- Martin, D.S., Khosravi, M., Grocott, M.P. and Mythen, M.G. (2010) Concepts in Hypoxia Reborn. Critical Care, 14, 315. http://dx.doi.org/10.1186/cc9078
- Suzuki, S., Eastwood, G.M., Glassford, N.J., Peck, L., Young, H., Garcia-Alvarez, M., et al. (2014) Conservative Oxygen Therapy in Mechanically Ventilated Patients: A Pilot Before-and-After Trial. Critical Care Medicine, 42, 1414-1422.
- Clark, J.M. and Lambertsen, C.J. (1971) Pulmonary Oxygen Toxicity: A Review. Pharmacological Reviews, 23, 37-133.
- Nash, G., Blennerhassett, J.B. and Pontoppidan, H. (1967) Pulmonary Lesions Associated with Oxygen Therapy and Artificial Ventilation. The New England Journal of Medicine, 276, 368-374. http://dx.doi.org/10.1056/NEJM196702162760702
- Bailey, T.C., Martin, E.L., Zhao, L. and Veldhuizen, R.A. (2003) High Oxygen Concentrations Predispose Mouse Lungs to the Deleterious Effects of High Stretch Ventilation. Journal of Applied Physiology, 94, 975-982.
- Sinclair, S.E., Altemeier, W.A., Matute-Bello, G. and Chi, E.Y. (2004) Augmented Lung Injury Due to Interaction between Hyperoxia and Mechanical Ventilation. Critical Care Medicine, 32, 2496-2501. http://dx.doi.org/10.1097/01.CCM.0000148231.04642.8D
- The Acute Respiratory Distress Syndrome Network (2000) Ventilation with Lower Tidal Volumes as Compared with Traditional Tidal Volumes for Acute Lung Injury and the Acute Respiratory Distress Syndrome. The New England Journal of Medicine, 342, 1301-1308. http://dx.doi.org/10.1056/NEJM200005043421801
- O’Driscoll, B.R., Howard, L.S. and Davison, A.G. (2008) BTS Guideline for Emergency Oxygen Use in Adult Patients. Thorax, 63, vi1-vi68. http://dx.doi.org/10.1136/thx.2008.102947
- Gilbert-Kawai, E.T., Mitchell, K., Martin, D., Carlisle, J. and Grocott, M.P. (2014) Permissive Hypoxaemia versus Normoxaemia for Mechanically Ventilated Critically Ill Patients. Cochrane Database of Systematic Reviews, No. 5.
- Farquhar, H., Weatherall, M., Wijesinghe, M., Perrin, K., Ranchord, A., Simmonds, M. and Beasley, R. (2009) Systematic Review of Studies of the Effect of Hyperoxia on Coronary Blood Flow. American Heart Journal, 158, 371-377. http://dx.doi.org/10.1016/j.ahj.2009.05.037
- Wijesinghe, M., Perrin, K., Ranchon, A., et al. (2009) Routine Use of Oxygen in the Treatment of Myocardial Infarction: Systematic Review. Heart, 95, 198-202. http://dx.doi.org/10.1136/hrt.2008.148742
- Stub, D., Smith, K., Bernard, S., et al. (2012) A Randomized Controlled Trial of Oxygen Therapy in Acute Myocardial Infarction Air Verses Oxygen in Myocardial Infarction Study (AVOID Study). American Heart Journal, 163, 339-345.e1.
- ACCF/AHA Task Force on Practice Guidelines (2013) Manual for ACCF/AHA Guideline Writing Committees: Methodologies and Policies from the ACCF/AHA Task Force on Practice Guidelines. American College of Cardiology and American Heart Association. http://assets.cardiosource.com/Methodology_Manual_for_ACC_AHA_Writing_Committees.pdf
- Jauch, E.C., Saver, J.L., Adams Jr., H.P., Bruno, A., Connors, J.J., Demaerschalk, B.M., Khatri, P., McMullan Jr., P.W., Qureshi, A.I., Rosenfield, K., Scott, P.A., Summers, D.R., Wang, D.Z., Wintermark, M., Yonas, H., on Behalf of the American Heart Association Stroke Council, Council on Cardiovascular Nursing, Council on Peripheral Vascular Disease and Council on Clinical Cardiology (2013) Guidelines for the Early Management of Patients with Acute Ischemic Stroke: A Guideline for Healthcare Professionals from the American Heart Association/American Stroke Association. Stroke, 44, 870-947. http://dx.doi.org/10.1161/STR.0b013e318284056a
- Elley, H., Chiu, H., Liu, C.-S., Tan, T.-Y. and Chang, K.-C. (2006) Venturi Mask Adjuvant Oxygen Therapy in Severe Ischemic Stroke. Archives of Neurology, 63, 741-744.
- Ronning, O.M. and Guldvog, B. (1999) Should Stroke Victims Routinely Receive Supplemental Oxygen? A Quasi-Randomized Controlled Trial. Stroke, 30, 2033-2037. http://dx.doi.org/10.1161/01.STR.30.10.2033
- Plant, P.K., Owen, J.L. and Elliott, M.W. (2000) One Year Period Prevalence Study of Respiratory Acidosis in Acute Exacerbations of COPD: Implications for the Provision of Non-Invasive Ventilation and Oxygen Administration. Thorax, 55, 550-554. http://dx.doi.org/10.1136/thorax.55.7.550
- Austin, M.A., Wills, K.E., Blizzard, L., Walters, E.H. and Wood-Baker, R. (2010) Effect of High Flow Oxygen on Mortality in Chronic Obstructive Pulmonary Disease Patients in Prehospital Setting: Randomised Controlled Trial. BMJ, 341, c5462. http://dx.doi.org/10.1136/bmj.c5462
- Bellomo, R., Bailey, M., Eastwood, G.M., et al. (2011) Arterial Hy-peroxia and In-Hospital Mortality after Resuscitation from Cardiac Arrest. Critical Care, 15, R90. http://dx.doi.org/10.1186/cc10090
- Eastwood, G.M., Young, P.J. and Bellomo, R. (2014) The Impact of Oxygen and Carbon Dioxide Management on Outcome after Cardiac Arrest. Current Opinion in Critical Care, 20, 266-272. http://dx.doi.org/10.1097/MCC.0000000000000084
- Pryor, K.O., Fahey 3rd, T.J., Lien, C.A. and Goldstein, P.A. (2004) Surgical Site Infection and the Routine Use of Perioperative Hyperoxia in a General Surgical Population: A Randomized Controlled Trial. JAMA, 291, 79-87. http://dx.doi.org/10.1001/jama.291.1.79
- Greif, R., Akca, O., Horn, E.P., et al., for the Outcomes Research Group (2000) Supplemental Perioperative Oxygen to Reduce the Incidence of Surgical-Wound Infection. The New England Journal of Medicine, 342, 161-167. http://dx.doi.org/10.1056/NEJM200001203420303
- Chang, D. (2014) Clinical Application of Mechanical Ventilation. 4th Edition, Delmar, New York.