American Journal of Plant Sciences
Vol.4 No.5(2013), Article ID:32152,7 pages DOI:10.4236/ajps.2013.45134
Genealogy of Cucumber mosaic virus Isolated from Ornamental Species
![]()
1Laboratório de Fitovirologia e Fisiopatologia, São Paulo, Brazil; 2Laboratório de Bioquímica Fitopatológica, Centro de Pesquisa e Desenvolvimento de Sanidade Vegetal, Instituto Biológico, São Paulo, SP, Brazil; 3ClonAgri, Artur Nogueira, SP, Brazil.
Email: *duarte@biologico.sp.gov.br
Copyright © 2013 Ligia Maria Lembo Duarte et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received March 25th, 2013; revised April 26th, 2013; accepted May 18th, 2013
Keywords: Phylogeny; Maximum Likelihood; Molecular Characterization; CMV; Ornamentals
ABSTRACT
Cucumber mosaic virus (CMV) has the broadest host range, infecting more than 1300 species in more than 500 genera from over 100 botanical families. In ornamental plants, CMV can cause mosaic and distortion of leaves, stunting, color break, and malformation of flowers. CMV coat protein (CP) sequences obtained from seven ornamental plants and other homologous sequences available in GenBank were compared, and phylogenetic relationships were established. Total RNA from virus-infected ornamental species were extracted, submitted to RT-PCR with specific primers, and amplicons obtained were sequenced. A nucleotide substitution model and phylogenetic analyses were carried out using the PAUP program. The seven sequences of CMV CP obtained showed similar identity percentages and close relationships with subgroup I isolates from other countries and hosts. CMV isolates from different regions of São Paulo state, Brazil (Salvia splendens, Catharanthus roseus, Nematanthus nervosus (=Hypocyrta nervosa), Impatiens walleriana, Eucharis grandiflora and Commelina sp.) formed a monophyletic group, indicating a possible common origin. It was found that when lily sequences of CMV only from different geographic regions were compared, Brazilian isolates shared the same common ancestor with those from Poland and Taiwan. Furthermore, this monophyletic group presented a quite basal position.
1. Introduction
The Cucumber mosaic virus (CMV), one of the four species in the Cucumovirus genus, Bromoviridae family [1], is characterized by isometric particles, three capped plus-sense single stranded RNAs and five open reading frames (ORFs) [2]. CMV is a typical generalist found worldwide, with the broadest host range among plant viruses, including more than 1300 species in more than 500 genera of over 100 families. New CMV hosts are reported every year [3,4], including both cultivated and spontaneous plants [5]. Whether alone or in combination with other viruses CMV affects food crops, medicinal and aromatic plants, and weeds [6,7]. CMV also infects many ornamentals, frequently causing flower breaks which may either enhance or detract from the aesthetic qualities of the flower, depending in part on the flower color [8]. Moreover, it induces symptoms such as mosaic, stunting, chlorosis, necrosis and leaf deformation.
Isolates of CMV have been classified into subgroups I and II, based on their biological and serological properties and nucleic acid hybridization tests [6]. A further division of subgroup I, into IA and IB, has been proposed based on phylogeny estimates, with the CP ORF and rearrangements in the 5’ nontranslated region (NTR) of RNA 3 [2,9].
In Brazil, CMV is the only species of Cucumovirus detected so far, and all isolates described belong to subgroup I [10]. CMV is transmitted by almost 60 aphid species in a non-persistent manner [11], which increases virus dissemination among different plant species, with low genetic variability among isolates. Additionally, the virus is also transmitted by seed, as well as by mechanical inoculation.
The present study draws an overview of CMV, and compares it with other Brazilian and world isolates. ORF sequences corresponding to coat protein (CP) from seven ornamental isolates were aligned with sequences available on GenBank, and phylogenetic analyses were carried out.
2. Material and Methods
2.1. Virus Material and Mechanical Transmission
Ornamental plants (Table 1) showing virus-like symptoms from different regions of São Paulo State, Brazil were inspected by electron microscopy and mechanically inoculated. Infected leaf tissues were homogenized in 0.05 M phosphate buffer plus 0.5% sodium sulfite and mechanically applied onto leaves of indicator plants using carborundum. Different herbaceous plants were used: Gomphrena globosa, Chenopodium amaranticolor, C. murale, C. quinoa, Datura stramonium, Nicotiana benthamiana, N. debneyi, N. glutinosa, N. rustica and N. tabacum “Xanthi”. Inoculated plants were kept in an insect-proof greenhouse for up to 30 days to allow the progression of symptoms.
2.2. Serological Tests
The leaves of each sample were submitted to DASELISA using antisera against CMV subgroups I and II (Agdia Inc., Elkhart, IN, USA). Reactions were considered positive when the absorbance value was three times as high as that of the corresponding control. Positive and negative controls (Agdia Inc., Elkhart, IN, USA) were used.
2.3. RNA Extraction, RT-PCR, Cloning and Sequencing
CMV isolates from the ornamental plants were propagated in N. glutinosa and their total RNA was extracted according to Chomczynski and Sacchi [12].
Anti-sense primer (5’GCCGTAAGCTGGATGGAC AA3’) for the 3’-terminus of CP [13] and sense primer (5’CATCGACCATGGACAAATCTGAATCAAC3’)
corresponding to the 3’-terminus of ORF movement protein (MP) were used for the RT-PCR. PCR was carried out with an initial heating cycle at 94˚C/2 min, followed by 35 cycles at 94˚C/1 min, 50˚C/1 min and 72˚C/1 min and then one cycle of elongation at 72˚C/7 min. Purified amplicons (Concert Rapid Gel Extraction Kit, GibcoBRL) were cloned (Escherichia coli XL-1 Blue competent cells) and sequenced using automated sequencing (ABI Prism Big Dye Terminator System, PE Applied Biosystems).
2.4. Sequence and Phylogenetic Analyses
Four sets of phylogenetic analyses were carried out with CP CMV sequences obtained from seven ornamental species (Table 1) and other homologous sequences of CMV isolates obtained from GenBank: 1) 20 sequences of serogroup I from different geographic regions, two from serogroup II and one sequence of Tomato aspermy virus (TAV), used as outgroup; 2) 20 sequences of ornamental species-CMV isolates from different geographic regions and one of serogroup II used as outgroup (S 70105); 3) 12 CP sequences from different regions of Brazil; 4) 19 sequences of CMV-lily isolates. The sequences were aligned visually, using the Se-Al program.
Phylogenetic reconstructions were obtained by maximum parsimony (MP) (PAUP 3.1.1) [14] and maximum likelihood (ML) method (PAUP 3.1.1) [15]. The MP reconstruction took into account all molecular characters assessed as independent, unordered and equally weighted, ACCTRAN (accelerated transformation) algorithms with heuristic search by stepwise addition, simple addition of the sequences and branch-swapping by the TBR (tree bisection and reconnection) algorithm. Robustness of the nodes of the phylogenetic tree was assessed by bootstrap, bootstrap percentages computed after 1,000 resamplings using the branch-and-bound method [16]. Consistency (CI) and retention (RI) indexes [17] were measured using the MacClade 3.03 program [18]. The ML analysis was

Table 1. Isolates of Cucumber mosaic virus (CMV) from ornamental species from São Paulo state, Brazil.
based on the assessment of the evolutionary models for substitutions, by way of a test of the rate of similarity using the Modeltest program [19]. Once the model was found, the tree topology was constructed.
3. Results
All isolates induced mosaic symptoms on N. glutinosa, except the lily CMV isolate (CMV-Lil), which affected systemically only N. tabacum “Xanthi” and N. benthamiana. CMV-Sal did not induce symptoms in D. stramonium.
Positive reaction with antiserum against CMV-I only was observed by DAS-ELISA performed with leaf extract from the 7 ornamental species.
Products of 657 bp in size were obtained by RT-PCR using total RNA extracts from ornamental plants infected by CMV as template. DNA amplicons corresponding to the complete CP gene were sequenced. The sequences were submitted to GenBank, and the accession numbers can be seen in Table 1.
Comparisons among nucleotide sequences of CMVCat, CMV-Euch, CMV-Eus, CMV-Nem, CMV-Imp, CMV-Lil and CMV-Sal CP showed similar identity percentages and close relationships with subgroup I isolates from other countries and hosts.
Alignment of the nt CP from the CMV ornamental Brazilian isolates revealed a high level of sequence identity (96% - 98%), and a identity percentage that ranged from 90 to 96, compared with the other isolates. The CP from these isolates showed a strong conservation of nucleotide sequences, varying between one (CMV-Imp and CMV-Euch) and 17 changes (CMV-Lil). Thirty-eight variable sites were observed, of which 22 were silent nucleotide changes, i.e. those that do not result in an amino acid substitution.
The most parsimonious of the 13,080 MP phylograms reconstructed from 657 nucleotides (nt) of the CP of 29 CMV isolates, with 293 steps, was used to compute the strict consensus tree. The values of the CI (excluding uninformative characters) and RI were 0.83 and 0.79, respectively.
Phylogenetic analyses (MP and MV) using Tomato aspermy virus as outgroup showed that seven CMV isolates share a common ancestor with CMV-I (100% bootstrap value), since they fell within a cluster distant from CMV-II. The division into two subgroups was also very clear, since the WL (S70105) and Alstroemeria (AJ 131622) strains belonging to subgroup II formed a monophyletic group and sister group of other subgroup I isolates (data not shown).
The phylogenetic tree proposed under ML condition, using CP sequence of CMVII WL strain as outgroup and CMV isolated from ornamental plants, revealed a monophyletic group formed by Brazilian ornamental CMV isolates supported by a high bootstrap value, except for CMV-Lily (Figure 1). A very consistent monophyletic group (100% bootstrap) formed by CMV-lily from Brazil, China and Taiwan was also recognized. In addition, the lily CMV clade formed a monophyletic group with CMV from Gladiolus (AJ131623) and Chrysanthemum (AF 316362) from The Netherlands and Korea, respectively.
Among the CMV isolates from the same species (Impatiens sp., Lilium sp., Eustoma grandiflora and Catharanthus roseus) from Brazil, China, India and Taiwan, only those from lily plants shared the same clade.
CMV-Lil (Brazil) shared the same common ancestor of CMV isolated from lily plants from Poland and Taiwan. In addition the Indian lily sequence (AJ831578) presented a quite basal position in the phylogenetic tree proposed (Figure 2).
4. Discussion
No clear differences were observed in host range for the isolates obtained, which could easily be transmitted by mechanical inoculation showing similar symptoms. CMVLily affected systemically only N. tabacum “Xanthi” and N. benthamiana. These results are consistent with those obtained by [20], suggesting that the CMV from lily is host-selective, compared to other CMV strains. Among the Brazilian isolates studied, CMV-Sal did not induce symptoms in D. stramonium, unlike the isolate described in India [7].
The results obtained for symptoms, host range and DAS-ELISA showed that viruses isolated from the seven ornamental species are CMV-I, confirming the prevalence of the CMV subgroup in Brazil [10].
With respect sequence analyses the overall higher degree of homology at amino acid level between all CMV strains might indicate the constraints imposed on the virus: variation in the CP to maintain the structural and functional role presumably for virion stability, transmission by aphids and the movement within the host plants [21].
It is worth mentioning that in Brazil the occurrence of CMV subgroup II has not been described so far, though the prevalence of CMV subgroup IA was reported [10]. According to [22], the CMV strains of subgroup I are considered more virulent than those of subgroup II. Subgroup IB is more common in Asia, with the parental lineage from Southeast Asia appearing to be the origin of CMV. The less virulent strains belong to subgroup II and were found to occur in the USA, Australia, and Africa.
The lineages that gave rise to CMV from Brazilian Eustoma seem to have a more basal divergence in comparison with the other lineages, when CP sequences from ornamental species only were analyzed. CMV from the ornamental species Alstroemeria sp. (AJ131622), Tagetes erecta (AM396983) and Impatiens sp. (DQ 018289) belongs to subgroup II.
CMV could not be separated in terms of its hosts. However, a tendency of isolates from the same geographical region to share the same common ancestor was observed.
The lineages that gave rise to CMV from Brazilian Eustoma seem to have a more basal divergence in comparison with the other lineages, when CP sequences from ornamental species only were analyzed. CMV from Due to different environmental constraints on the evolution of new CMV strains, it is important to study the phylogenetic relationship of viruses at local level [21]. When phylogenetic analysis was performed using CP sequences of CMV isolates from different hosts from Brazil only, ornamental CMV isolates from São Paulo state [Salvia (CMV-Sal), Catharanthus (CMV-Cat), Nematanthus (CMV-Nem), Impatiens (CMV-Imp), Eucharis (CMVEuch) and Commelina] formed a monophyletic group,

Figure 1. Maximum likelihood tree constructed with coat protein sequences of Cucumber mosaic virus (CMV) isolates from ornamental plants and comparable CMV sequences retrieved from GenBank using PAUP program with K80+I+G substitution model, invariable site (I = 0.39) and gamma-distributed rate (G = 0.64). CMV II WL strain (S70105) was used as outgroup. * (= Hypocyrta nervosa). Bootstrap values are indicated near the branches
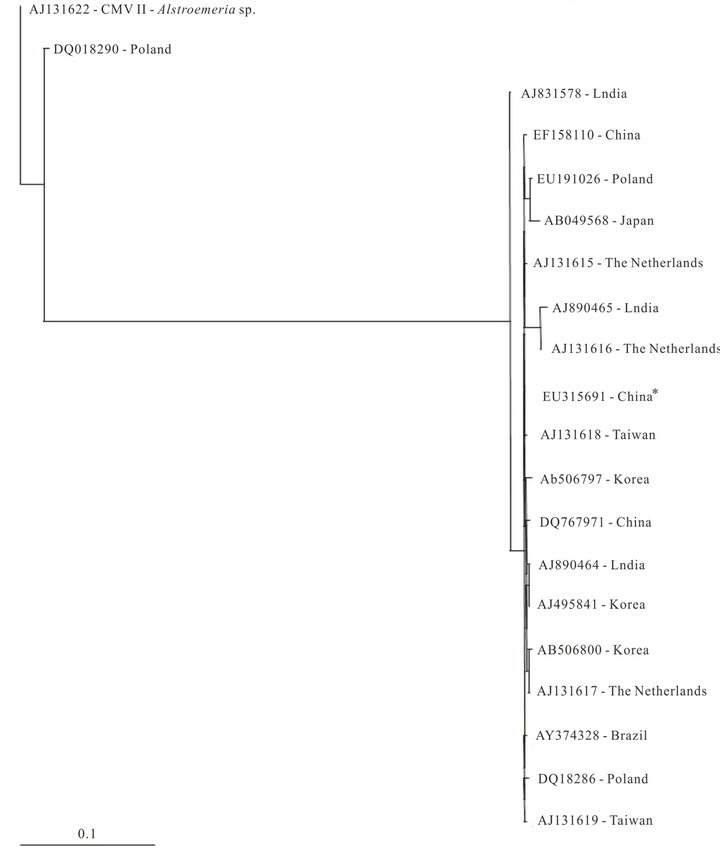
Figure 2. Maximum likelihood tree constructed with coat protein sequences of Cucumber mosaic virus (CMV) isolates from Lilium sp. plants from Brazil (highlighted) and from different geographic regions retrieved from GenBank using PAUP program with K80 substitution model and gamma-distributed rate (G = 0.45). CMV II Astroemeria strain (AJ 131622) was used as outgroup. * CMV from wild lily.
indicating a possible common origin (data not shown). Similar results were observed when phylogenetic analysis was performed with CP sequences of CMV isolates from different countries from ornamentals only. In a study on CMV infecting Gladiolus, Dubey et al. [21] suggested that the formation of one cluster among all Indian strains may indicate their common origin.
Although CMV-Eus and CMV-Lily isolates did not cluster with those isolated from ornamental plants, they shared the same common ancestor of isolates from the same geographic region (data not shown).
The comparison of CP sequences of CMV-Lily with sequences of CMV isolated from lily plants deposited in GenBank revealed that the Indian lily sequence presented a quite basal position in the phylogenetic tree proposed. The other isolates were divided into two groups, and the Brazilian isolate shared the same common ancestor with Poland and Taiwan isolates. In addition, the monophyletic group formed by CMV-Lil also presented a quite basal position. It should be noted that the majority of commercially available lilies distributed worldwide originate from Asia. The wild lilies were taken to Europe 100 years ago, and bred as commercial lily varieties [20].
Chen et al. [23] observed that lily isolates of distinct geographic origins (France, The Netherlands and Taiwan), as well as those isolated from other horticultural varieties were extremely homologous to each other, sharing a common ancestor. Interestingly, the Brazilian isolate of lily showed the same behavior. The authors suggested two possible reasons: recent dispersal of contaminated bulbs through trade or genuine host specificity, since lily plants mechanically inoculated with several CMV isolates, only lily isolates were able to re-infect lily plants successfully. A hypothesis was then proposed, suggesting that CMV lily isolates constitute a unique pathological population, evolutionary adapted to lily plants [24]. It should be noted that a CMV isolate from wild lily of China remained at the most basal position within the monophyletic group composed by Asian and Dutch isolates. This fact supports the idea of the origin of lily CMV isolate in Asia and its dispersal of contaminated bulbs through trade [23]. Because lily CMVs are normally transmitted through the bulb, once they infect a certain lily they can exist for generations in that lily and its vegetative offspring [20]. Davino et al. [11], studying population genetics of CMV infecting medicinal, aromatic and ornamental plants, suggested that several events of long-distance migration (gene flow) have occurred between Italy and other countries. Nucleotide diversities between different subpopulations were of the same order as within subpopulations diversities, supporting the hypothesis of gene flow between distant geographical regions.
The occurrence of CMV in different crops and regions of the world shows the success of this virus species. In this sense, phylogenetic analysis is a useful tool to important information about CMV evolutionary history. Studies by Roossinck [2] showed that phylogenetic trees estimated by ORFs located in different RNAs were not congruent, indicating that they had independent evolutionary histories. The authors also report that rearrangements may have led to genetic diversity among isolates of CMV and contributed to its enormous evolutionary success.
New isolates in new hosts, consistently reported both in Brazil and in other parts of the world, can lead to colonization of other susceptible species and unexpected synergism with other viruses [25], causing considerable losses in economically important crops.
The occurrence of novel viral isolates in an array of new different hosts, which has been repeatedly reported both in Brazil and other countries, may be seen as evidence of the colonization of susceptible plant species in the near future, as well as of the emergence of synergy of CMV with other virus species harmful to plants. In this scenario, further studies are required to investigate this phenomenon, shedding new light on the development of innovative management strategies against viral infections in order to prevent the substantial losses they cause in economically important crops worldwide.
REFERENCES
- A. M. Q. King, M. J. Adams, E. B. Carstens and E. J. Lefkowitz, “Virus Taxonomy,” Ninth Report of the International Committee on Taxonomy of Viruses, Elsevier/ Academic Press, San Diego, 2011.
- M. J. Roossinck, “Evolutionary History of Cucumber mosaic virus Deduced by Phylogenetic Analyses,” Journal of Virology, Vol. 76, No. 7, 2002, pp. 3382-3387. doi:10.1128/JVI.76.7.3382-3387.2002
- F. García-Arenal and P. Palukaitis, “Cucumber mosaic virus,” In: B. W. J. Mahy and M. H. V. van Regenmortel, Eds., Encyclopedia of Virology, Academic Press, Oxford, 2008, pp. 614-619. doi:10.1016/B978-012374410-4.00640-3
- I. Pagán, A. Fraile, E. Fernandez-Fueyo, N. Montes, C. Alonso-Blanco and F. García-Arenal, “Arabidopsis thaliana as a Model for the Study of Plant-Irus Covolution,” Philosophical Transactions of the Royal Society of London Series B-Biological Sciences, Vol. 365, No. 1548, 2010, pp. 1983-1995. doi:10.1098/rstb.2010.0062
- S. Flasinski, S. W. Scott, O. W. Barnett and S. Sun, “Diseases of Peperomia, Impatiens, and Hibbertia Caused by Cucumber mosaic virus,” Plant Disease, Vol. 79, 1995, pp. 843-848. doi:10.1094/PD-79-0843
- P. Palukaitis, M. J. Roossinck, R. G. Dietzgen and R. I. B. “Francki, Cucumber mosaic virus,” Advances in Virus Research, Vol. 41, 1992, pp. 281-348. doi:10.1016/S0065-3527(08)60039-1
- A. Samad, P. V. Ajayakumar, M. K. Gupta, A. K. Shukla, M. P. Darokar, B. Somkuwar and M. Alam, “Natural Infection of Periwinkle (Catharanthus roseus) with Cucumber mosaic virus, subgroup IB,” Australasian Plant Disease Notes, Vol. 3, 2008, pp. 30-34. doi:10.1071/DN08013
- R. A. Valverde, S. Sabanadzovic and J. Hammond, “Viruses That Enhance the Aesthetics of Some Ornamental Plants: Beauty or Beast?” Plant Disease, Vol. 96, No. 5, 2012, pp. 600-611. doi:10.1094/PDIS-11-11-0928-FE
- M. J. Roossinck, L. Zhang and K. Hellwald, “Rearranments in the 5’ Nontranslated Region and Phylogenetic Analyses of Cucumber Mosaic Virus RNA 3 Indicate Radial Evolution of Three Subgroups,” Journal of Virology, Vol. 73, No. 8, 1999, pp. 6752-758.
- M. Eiras, A. J. Boari, A. Colariccio, A. L. R. Chaves, M. R. S. Briones, A. R. Figueira and R. Harakava, “Characterization of Isolates of the Cucumovirus Cucumber mosaic virus Present in Brazil,” Journal of Plant Pathology, Vol. 86, No. 1, 2004, pp. 59-67.
- S. Davino, S. Panno, E. A. Rangel, M. Davino, M. G. Bellardi and L. Rubio, “Population Genetics of Cucumber Mosaic Virus Infecting Medicinal, Aromatic and Ornamental Plants from Northern Italy,” Archives of Virology, Vol. 157, No. 4, 2012, pp. 739-745. doi:10.1007/s00705-011-1216-4
- P. Chomczynski and N. Sacchi, “Single Step Method of RNA Isolation by Acid Guanidium Thiocianate-HenolChloroform Extraction,” Analytical Biochemistry, Vol. 162, No. 1, 1987, pp. 156-159. doi:10.1016/0003-2697(87)90021-2
- S. Wylie, C. R. Wilson, R. A. C. Jones and M. G. K. Jones, “A Polymerase Chain Reaction Assay for Cucumber Mosaic Virus in Lupin Seeds,” Australian Journal of Agricultural Research, Vol. 44, No. 1, 1993, pp. 41-51. doi:10.1071/AR9930041
- D. L. Swofford, “PAUP*: Phylogenetic Analysis Using Parsimony (* and Related Methods), Version 4.0,” Sinauer Associates, Sunderland, 2002.
- J. Felsenstein, “Evolutionary Trees from DNA Sequences: A Maximum Likehood Approach,” Journal of Molecular Evolution, Vol. 17, No. 6, 1981, pp. 368-376. doi:10.1007/BF01734359
- J. D. Thompson, T. J. Gibson, F. Plewniak, F. Jeanmougin and G. Higgins, “The Clustal X Windows Interface: Flexible Strategies for Multiple Sequence Alignment Aided by Quality Tools,” Nucleic Acids Research, Vol. 25, No. 24, 1997, pp. 4876-4882.
- J. Felsenstein, “The Number of Evolutionary Trees,” Systematc Zoology, Vol. 27, No. 4, 1978, pp. 401-410. doi:10.2307/2412923
- W. P. Maddison and D. R. Maddison, “MacClade, v. 3.03,”
- Sinauer Associates, Inc. Publishers, Sunderland, 1992.
- D. Posada and K. A. Crandall, “A Model Test: Testing the Model of DNA Substitution,” Bioinformatics, Vol. 14, No. 9, 1998, pp. 817-818. doi:10.1093/bioinformatics/14.9.817
- C. Masuta, Y. Seshimo, M. Mukohara, H. J. Jung, S. Uedas, K. H. Ryu and J. K. Choi, “Evolutionary Characterization of Two Isolates of Cucumber mosaic virus Isolated from Japan and Korea,” Journal of General Plant Pathology, Vol. 68, No. 2, 2002, pp. 163-168. doi:10.1007/PL00013070
- V. K. Dubey, Aminuddin, and V. P. Singh, “Molecular Characterization of Cucumber mosaic virus Infecting Gladiolus, Revealing Its Phylogeny Distinct from the Indian Isolate and Alike the Fny Strain of CMV,” Virus Gene, Vol. 41, No. 1, 2010, pp. 126-134. doi:10.1007/s11262-010-0483-6
- V. Koundal, Q. M. R. Hao and S. Praveen, “Characterization, Genetic Diversity, and Evolutionary Link of Cucumber mosaic virus Strain New Delhi from India,” Biochemical Genetics, Vol. 49, No. 1-2, 2011, pp. 25-38. doi:10.1007/s10528-010-9382-8
- Y. K. Chen, A. F. L. M. Derks, S. Langeveld, R. Goldbach and M. Prins, “High Sequence Conservation among Cucumber Mosaic Virus Isolates from Lily,” Archives of Virology, Vol. 146, No. 8, 2001, pp. 1631-1636. doi:10.1007/s007050170085
- H. Berniak, M. Kamińska and T. Malinowski, “Cucumber mosaic virus Groups IA and II Are Represented among Isolates from Naturally Infected Lilies,” European Journal of Plant Pathology, Vol. 127, No. 3, 2010, pp. 305- 309. doi:10.1007/s10658-010-9600-6
- D. Gallitelli, “The Ecology of Cucumber mosaic virus and Sustainable Agriculture,” Virus Research, Vol. 71, No. 1-2, 2000, pp. 9-21. doi:10.1016/S0168-1702(00)00184-2
NOTES
*Corresponding author.