Pharmacology & Pharmacy
Vol.5 No.1(2014), Article ID:42388,12 pages DOI:10.4236/pp.2014.51017
Beat-to-Beat Variability in Field Potential Duration in Human Embryonic Stem Cell-Derived Cardiomyocyte Clusters for Assessment of Arrhythmogenic Risk, and a Case Study of Its Application
![]()
1Next Generation Systems CFU, Eisai Product Creation Systems, Eisai Co., Ltd., Tsukuba, Japan; 2Biomarkers and Personalized Medicine CFU, Eisai Product Creation Systems, Eisai Co., Ltd., Tsukuba, Japan; 3Biopharmaceutical Assessment CFU, Eisai Product Creation Systems, Eisai Co., Ltd., Tsukuba, Japan; 4Global Discovery Research Neuroscience and General Medicine PCU, Eisai Product Creation Systems, Eisai Co., Ltd., Tsukuba, Japan.
Email: *k5-yamazaki@hhc.eisai.co.jp
Copyright © 2014 Kazuto Yamazaki et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Kazuto Yamazaki et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received November 27th, 2013; revised December 29th, 2013; accepted January 12th, 2014
KEYWORDS
Human Embryonic Stem Cell-Derived Cardiomyocytes; Field Potential Duration; Short-Term Variability; QT Interval; Torsades de Pointes; Risk Assessment
ABSTRACT
We established a QT interval assessment system that uses human embryonic stem cell-derived cardiomyocyte clusters (hES-CMCs) in which the field potential duration (FPD) or corrected FPD (FPDc) was measured as an indicator of drug-induced QT interval prolongation. To investigate the applicability of the hES-CMC system to drug safety assessment, we investigated short-term variability in FPDc (STVFPDc) (beat rate rhythmicity) as a marker of torsadogenic risk. We investigated the FPDc and STVFPDc of hES-CMCs treated with hERG channel blockers (E-4031 or cisapride) or with our proprietary compounds X, Y, and Z. We also evaluated the electrocardiograms and hemodynamics of dogs treated with compound X, Y, or Z. The torsadogenic hERG channel blockers increased STVFPDc and prolonged FPDc. Compounds X, Y, and Z had hERG inhibitory activity. Compound X prolonged FPDc with increased STVFPDc, whereas compounds Y and Z tended to shorten FPDc in the hES-CMC system. In the in vivo canine study, compound X prolonged corrected QT (QTc), and compounds Y and Z tended to shorten QTc, showing a good correlation with the results in hES-CMCs. These findings suggest that combined assessment of FPDc and STVFPDc in the hES-CMC system increases the predictability of torsadogenic risk.
1. Introduction
Drug-induced QT prolongation is a critical clinical issue and leads to the withdrawal or use restriction of commercial drugs, because it increases the risk of ventricular tachycardia and torsades de pointes (TdP), which often degenerate into ventricular fibrillation. Therefore, every possible effort is taken in drug development to mitigate the risk of QT prolongation in humans. A target of most drugs that prolong the QT interval is the human ether-à-go-go-related gene (hERG) channel, which is important for repolarization. Therefore, hERG assays are performed as a first and indispensable step in drug safety assessment. However, hERG assays are not enough to forecast clinical QT prolongation reliably [1]. This may be because (1) the drug can affect a QT interval-prolonging channel other than the hERG channel [1,2], or (2) the drug can affect plural channels affecting QT intervals [3]. Accordingly, the best method of testing for QT prolongation risk is to use human cardiomyocytes that inherently express these multiple channels, but it has been difficult to obtain human primary cardiomyocytes. To overcome this hurdle, many researchers have applied human cardiomyocytes differentiated from embryonic stem cells or induced pluripotent stem cells to evaluate cardiotoxicity [4-7].
In this context, we have recently established an in vitro system that uses human embryonic stem cell-derived cardiomyocyte clusters (hES-CMCs) to assess the potential of drugs to influence the QT interval [8]. In this hES-CMC system, we record the field potential duration (FPD)—as QT-like intervals—of clusters treated with a compound; we then calculate the corrected FPD (FPDc) and examine whether or not the compound prolongs FPDc. However, there have been reports that the QT interval is not a good surrogate marker of TdP and ventricular fibrillation [9-18]; the beat-to-beat variability of QT or QTc is suggested to be a better parameter [14,15]. It would therefore be of interest to investigate whether short-term variability in FPDc (STVFPDc), as beat-to-beat variability, is increased along with FPDc prolongation in hES-CMCs treated with torsadogenic agents.
In our previous paper, we used well-known reference compounds, including E-4031 as a hERG channel blocker, to validate our in vitro QT assessment system. We found that changes in FPDc were well matched with in vivo QT prolongation or with the results expected from the compounds’ mechanisms of action [8]. However, needless to say, the assessment system must be applicable to the safety assessment of drug candidates of which the effects on channels involved in QT intervals have not been clarified. There have been cases in which drug candidates give in vivo QT results that are inconsistent with the findings in hERG assays.
In this study, we used representative hERG channel blockers, class I antiarrhythmic drugs, and a calcium channel blocker to examine the possibility of using STVFPDc as an additional parameter to improve TdP risk prediction in the hES-CMC system. Moreover, we used our proprietary compounds to assess the applicability of FPDc and STVFPDc and compared our in vitro and in vivo observations.
2. Materials and Methods
2.1. Compounds
E-4031 (N-[4-[1-[2-(6-methylpyridin-2-yl)ethyl]piperidine- 4-carbonyl]phenyl]methanesulfonamide dihydrochloride dihydrate) was purchased from Wako Pure Chemical Industries, Ltd. (Osaka, Japan). Cisapride monohydrateflecainide acetate, mexiletine hydrochloride, quinidine hydrochloride monohydrate, and verapamil were purchased from Sigma-Aldrich (St. Louis, MO). Three proprietary compounds (compounds X, Y, and Z) were synthesized by Eisai Co., Ltd. (Tsukuba, Japan). Compounds Y and Z were synthesized as derivatives of compound X to reduce its hERG inhibitory activity. All compounds were dissolved in dimethyl sulfoxide (DMSO; Wako Pure Chemical Industries, Ltd. Osaka, Japan) to prepare stock solutions at 10 mM for in vitro experiments.
2.2. Cell Lines
Generation of a hERG channel stably expressing the Chinese hamster ovary (CHO) K1 cell line was reported by Taniguchi et al. [19]. A Cav1.2-expressing cell line (human Cav1.2/β2/α2δ1 calcium channel cell line, catalog number CT6004) was purchased from ChanTest (Cleveland, OH).
2.3. Cell Culture
CHO cells stably expressing the hERG channel were maintained in a 5% CO2 incubator at 37˚C in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS; Invitrogen) and 1× antibiotics-antimycotics (Invitrogen). Cav1.2-expressing cells were cultured in a 5% CO2 incubator at 37˚C in Ham’s F12 (Invitrogen) supplemented with 10% FBS (Invitrogen), 1× penicillin-streptomycin (Invitrogen), 0.01 mg/mL blasticidin (Kaken Pharmaceutical Co., Ltd., Tokyo, Japan), 0.25 mg/mL Geneticin (Invitrogen), 0.25 mg/mL hygromycin (Invitrogen), and 0.4 mg/mL Zeocin (Invitrogen). Before use, the Cav1.2- expressing cells were treated with 0.8 μg/mL tetracycline (Invitrogen) for 24 h at 37˚C and then cultured for an additional 24 h at 27˚C in a 5% CO2 incubator. During tetracycline induction of the channel, verapamil was added at 1 μM to keep the cells viable [20].
2.4. Patch-Clamp Recordings
For the hERG assay, electrophysiological recordings were conducted with a QPatch 16 automated patch-clamp system (Sophion Bioscience A/S, Ballerup, Denmark). Its basic protocol has been described previously [21]. The external solution consisted of (in mM) 145 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES (2-[4- (2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid) (pH adjusted to 7.4 with 1 M NaOH, and osmolarity to 305 mOsm with 1 M sucrose), and the internal solution consisted of (in mM) 120 KCl, 5.374 CaCl2, 1.75 MgCl2, 10 EGTA (ethylene glycol-bis(2-aminoethylether)-N, N, N’, N’- tetraacetic acid), and 10 HEPES (pH adjusted to 7.2 with 1 M KOH, and osmolarity to 300 mOsm with 1 M sucrose). hERG currents were evoked by a 5-s depolarizing potential of +20 mV from a holding potential of -80 mV, followed by a 5-s repolarizing step to −50 mV to measure tail currents.
For the Cav1.2 current, electrophysiological recordings were conducted with a manual patch-clamp. HEPESbuffered physiological saline with the following composition was used as the external solution (in mM): 137 NaCl, 4 KCl, 1.8 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose (pH adjusted to 7.4 with 1 M NaOH). Glass patch pipettes were fire-polished and filled with the following internal solution (in mM): 120 Cs-aspartate, 10 CsCl, 10 EGTA, 5 Mg-ATP, 0.4 Tris-GTP, and 10 HEPES (pH adjusted to 7.2 with 50% CsOH, and osmolarity to 290 mOsm with 1 M sucrose). All recordings were performed with an Axopatch 200B patch-clamp amplifier (Axon Instruments, Foster City, CA), filtered at 1 kHz with a low-pass Bessel filter, digitized at 0.05 ms/point, and stored on a Dell computer (Dell Inc., Round Rock, TX) using a DIGIDATA 1322A (Axon Instruments). Cav1.2 currents were evoked by applying 200-ms voltage steps to 0 mV from a holding potential of −40 mV at a frequency of 0.1 Hz. Peak inward currents at 0-mV step pulses were measured, and IC50 values for inhibition of the Cav1.2 current were calculated from the concentration-response curves.
2.5. Use of hES-CMCs to Assess Potential to Influence QT Intervals
This method has been described in detail by Yamazaki et al. [8]. Briefly, the procedures were as follows. hESCMCs were obtained from Cellartis AB (Göteborg, Sweden). The clusters were adhered to the electrodes of the probes (MED-P515A; Alpha MED Sciences, Osaka, Japan) of a MED64 System, a multielectrode device for recording field potentials. The clusters were treated with cisapride (a hERG channel blocker) at 100 nM, and their FPDs, inter-spike intervals (ISIs), and beat frequencies were recorded with the MED64 System with its own software (Mobius QT; Alpha MED Sciences). The preparations were placed on a hot plate and kept at 37˚C. For evaluation of compounds, a cluster was first treated with vehicle (0.1% DMSO), and then treated with a compound in stepwise concentration increments. The FPD for the vehicle and each concentration of the compound was measured for 10 min. The last 30 FPDs and ISIs of the 10-min recordings were extracted for analysis. The FPDc was calculated according to Fridericia’s formula: FPDc = FPD/ISI1/3. Clusters with 5 to 20% FPDc prolongation were chosen. After 7 days as a washout period, the selected clusters were used to assess the target and control compounds. In this assay, we maintained the clusters in DMEM (Sigma-Aldrich) supplemented with 20% FBS (Equitech-Bio, Kerrville, TX), 1× penicillin-streptomycin, 1× non-essential amino acids (Invitrogen), 0.1 mM β-mercaptoethanol (Invitrogen), 50 mM NaCl, and 15 mM NaHCO3, adjusted to pH 7.2 with 1 N NaOH.
Poincaré plots were drawn by plotting FPDc against the previous intervals for 30 consecutive beats. STV, defined as the mean orthogonal distance from the plot coordinates to the line y = x, was calculated as follows: STVD = Σ|Dn+1 − Dn|/(30 × 21/2), where D represents the FPDc of beat n [14,15]. A simple moving average with a 10-ms window length was used to filter out noise that would have affected the STV analysis of FPD.
2.6. Animals
Purpose-bred male beagle dogs were purchased from NARC Corp. (Sanmu, Japan). The dogs were provided with a commercial diet (DS-A; Oriental Yeast, Tokyo, Japan) and water ad libitum. They were kept under conventional conditions of controlled temperature, humidity, and lighting (23 ± 3˚C, 55 ± 15%, and a 12-h light-dark cycle with lights on at 07:00 a.m.). All experimental procedures were conducted according to the Eisai Animal Care Committee’s guidelines.
2.7. Measurement of Electrocardiogram and Hemodynamic Parameters of Anesthetized Dogs Treated with Compound X, Y, or Z
Three dogs were used per group. Each dog was intravenously anaesthetized with 15 to 20 mg/kg body weight sodium thiopental. Anesthesia was maintained with inhalation of a mixture of isoflurane (1.5 to 2.0%) and oxygen via an anesthetic machine (FO-20S and MKIIIai; Acoma Medical Industry Co., Ltd., Tokyo, Japan) under artificial ventilation with a ventilator (ARF-900EII; Acoma Medical Industry) to maintain a constant depth of anesthesia. Artificial ventilation was performed at a tidal volume of 20 to 22 mL/kg and a rate of 12 respirations per minute.
A heparin-coated polyurethane catheter (7Fr; Instech Laboratories, Inc., Plymouth Meeting, PA) was inserted into the carotid artery for measurement of aortic pressure. The other end of the catheter was connected to a micro-tip catheter transducer (MPC-500; Miller Instruments, Burnaby, Canada) attached to a pressure amplifier (AP- 601G, Nihon Koden Corp., Tokyo, Japan). In addition, the maximum upstroke velocity of the left ventricular pressure (LVdP/dtmax) was measured. Standard lead II electrocardiograms were obtained with a bioelectric amplifier (S-1516; Nihon Koden). Electrocardiograms were recorded on thermal array recorders (WS-682G; Nihon Koden).
The animals were left for 30 to 60 min for hemodynamic stabilization. Stepwise incremental doses of 0.1, 0.3, and 1 mg/kg of compound X, or 1, 3, or 10 mg/kg of compound Y or Z, or corresponding vehicle (90% saline, 5% 1 N HCl, and 5% DMSO for compound X; 50 mM HCl and 5% DMSO for compounds Y and Z) were intravenously infused for 10 min per dose. Observations were made throughout each 10-min dosing period and for 10 min after the last period. QT and RR intervals were measured for two or three consecutive beats on a standard lead II electrocardiogram and expressed as means. QTcF intervals were calculated according to Fridericia’s formula: QTcF = QT/RR1/3.
2.8. Determination of Plasma Total Concentrations of Compounds X, Y, and Z in Dogs
Blood samples (approximately 0.3 mL) were collected via the cephalic vein into heparinized syringes at the end of each 10-min infusion. The blood samples were immersed in an ice bath and then centrifuged at 10,000 rpm for 2 min at 4˚C to obtain plasma. The plasma samples were stored at −20˚C. Twenty microlitres of 0.1 M HCl were added to each 20-µL plasma sample. Four hundred microlitres of internal standard solution (acetonitrile containing 100 ng/mL of propranolol) was added to each sample; the sample was then mixed. The mixture was centrifuged at 3000 rpm for 10 min at 4˚C. The supernatant was placed onto a centrifugal filter device (MultiScreen Solvinert, hydrophilic PTFE membrane 0.45 µm; Millipore Co., Billerica, MA) and then centrifuged at 2000 rpm for 5 min at 4˚C. The resulting filtered samples were used for liquid chromatography—tandem mass spectrometry.
2.9. Statistical Analysis
All values are expressed as means ± S.E.M. Regression curves of inhibition of channel currents by compounds were generated with GraphPad Prism Version 5.04 (GraphPad Software, Inc., San Diego, CA), and the IC50 values were calculated. Constrained linear regression was made by using GraphPad Prism, and 95% confidence was calculated. The two-tailed paired Student’s t-test was used to analyze the changes in FPDc, and STVFPDc, compared with those under vehicle treatment. In the comparison of in vivo data between the vehicleand compound X-, Y-, or Z-treated groups, two-way repeated measures analysis of variance was performed, followed by Bonferroni’s test as a post hoc test, using GraphPad Prism. We considered p values less than 0.05 to be significant.
3. Results
3.1. Effects of hERG Channel Blockers, Class I Antiarrhythmic Drugs, and Calcium Channel Blocker on FPDc and STVFPDc of hES-CMCs
E-4031 induced concentration-dependent FPDc prolongation in hES-CMCs (Table 1); significant prolongation was recognized at 30 (p < 0.05), 100 (p < 0.01) and 300 (p < 0.01) nM. With the prolongation of FPDc, significant increases in STVFPDc were observed at 100 (p < 0.05) and 300 (p < 0.05) nM. In a similar way, cisapride treatment prolonged FPDc concentration-dependently, accompanied by increased STVFPDc (Table 1): although this STVFPDc increase was not statistically significant, three of six clusters showed >70% increases in STVFPDc with a cisapride dose of 100 nM or higher (p = 0.051 at 300 nM for actual values).
The following class I antiarrhythmic drugs were investigated: quinidine (class Ia), mexiletine (class Ib), and flecainide (class Ic). Quinidine significantly prolonged FPDc in a concentration-dependent manner and tended to increase STVFPDc. Neither mexiletine nor flecainide caused significant changes in FPDc or STVFPDc. In agreement with known pharmacology, verapamil treatment significantly shortened FPDc in a concentration-dependent fashion, without significant effects on STVFPDc. These results are described in Table 1.
3.2. Effects of Compounds X, Y, and Z on FPDc and STVFPDc of hES-CMCs
Table 2 shows the changes in FPDc and STVFPDc of hESCMCs treated with compound X, Y, or Z. Compound X at 1, 3, and 10 μM prolonged FPDc by 12.1% (p < 0.01), 21.1% (p < 0.01), and 26.7% (p < 0.05), respectively—in a concentration-dependent manner—compared with pretreatment. This compound also gave concentration-dependent increases in STVFPDc, with significant increases at 3 (p < 0.01) and 10 (p < 0.05) μM. Typical early afterdepolarization (EAD) was induced by compound X in one of five clusters. EAD occurred about 3.5 min after the addition of compound X at 10 μM (Figure 1(a)). Waveforms at EAD are illustrated in Figure 1(b) compared with that just before EAD occurrence. In contrast, compound Y led to a tendency toward shortening of FPDc concentration-dependently, with 10% shortening at 30 μM (p = 0.080 for actual values). In the case of compound Z, one of four clusters showed >20% shortening at 30 μM, although there was no significant shortening of FPDc. Neither compound Y nor Z gave significant changes in actual values of STVFPDc.

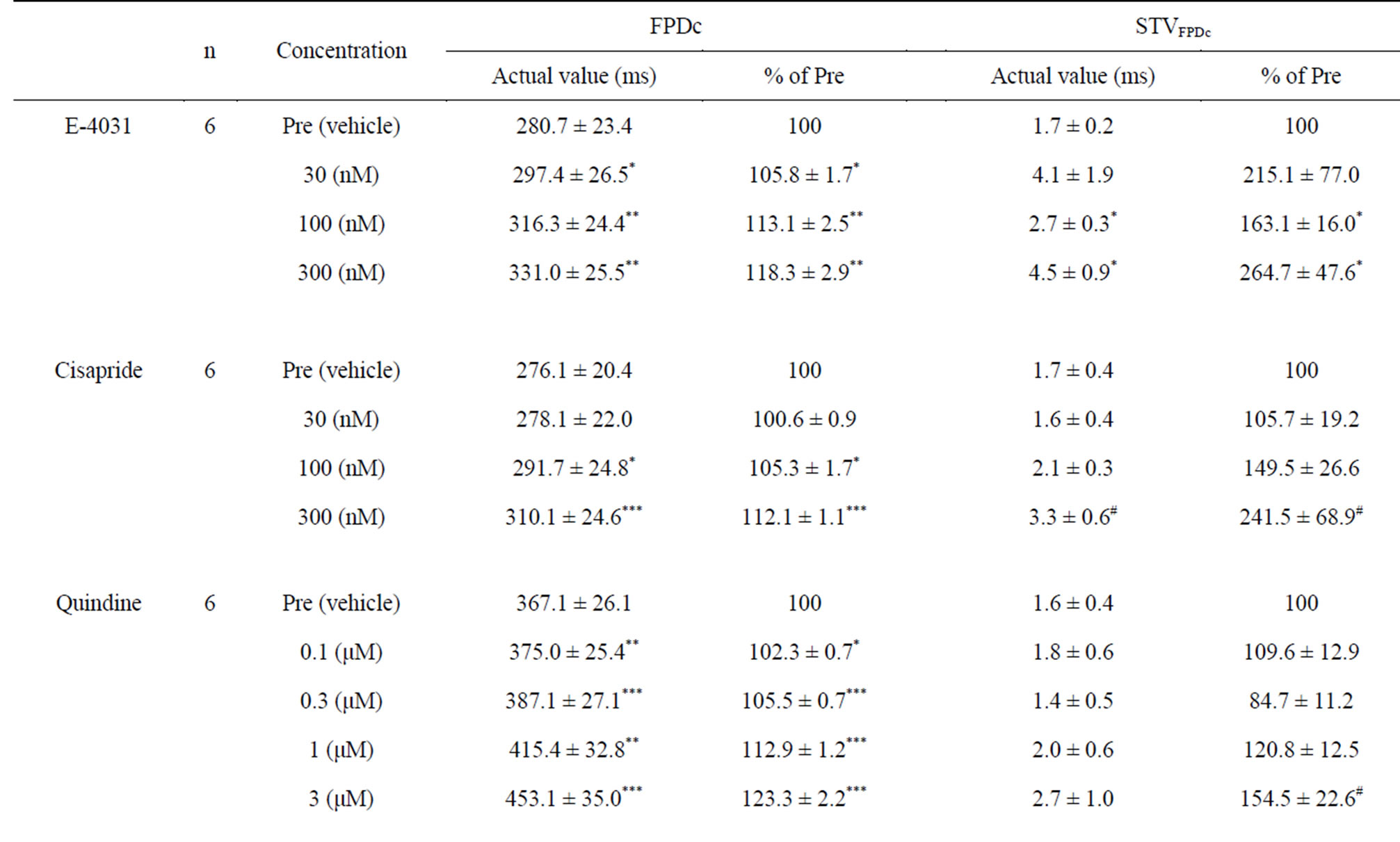
Table 1. Poincaré plot analysis of the corrected field potential duration (FPDc) of human embryonic stem cell-derived cardiomyocyte clusters treated with E-4031, cisapride, quinidine, mexiletine, flecainide, or verapamil. Calculations include the short-term variability of FPDc (STVFPDc).

Table 2. Poincaré plot analysis of the corrected field potential duration (FPDc) of human embryonic stem cell-derived cardiomyocyte clusters treated with compound X, Y, or Z. Calculations include the short-term variability of FPDc (STVFPDc).
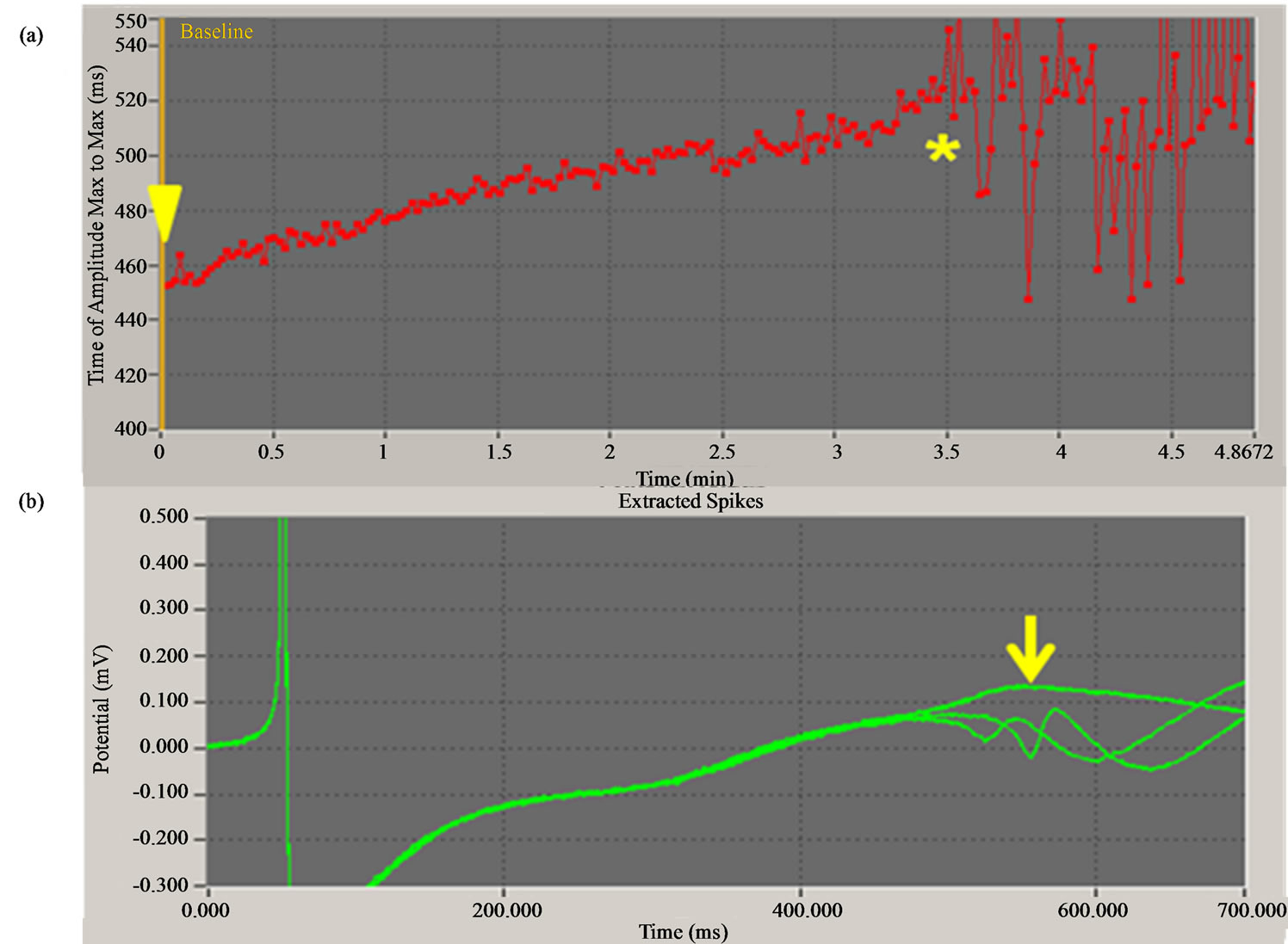
Figure 1. Compound X induces early afterdepolarization (EAD) of a human embryonic stem cell-derived cardiomyocyte cluster. (a) Changes in field potential duration (FPD, indicated by time of amplitude maximum to maximum) by compound X at 10 µM. Yellow arrowhead indicates the time at which compound X was added. FPD was prolonged with time, and EAD occurred abruptly 3.5 min after the addition (yellow asterisk). (b) Superimposed waveforms of field potential just before (indicated by yellow arrow) and after the occurrence of EAD.
3.3. IC50 Values against the hERG Current
The IC50 values of cisapride and E-4031 against the hERG current were 13.9 ± 1.1 (n = 5) and 29.9 ± 1.0 nM (n = 7), respectively. The following IC50 values were calculated for the proprietary compounds: 0.30 ± 1.1 (n = 6), 10.6 ± 1.2 (n = 5), and 9.7 ± 1.1 μM (n = 5) for compounds X, Y, and Z, respectively (Table 3).
3.4. IC50 Values against the Cav1.2 Current
The IC50 value of verapamil toward the Cav1.2 current was calculated as 0.33 ± 0.09 μM (n = 3) as a control. The IC50 values of compounds X, Y, and Z were 27.2 ± 2.6 (n = 3), 2.8 ± 0.1 (n = 4), and 16.0 ± 0.9 μM (n = 3), respectively (Table 3).
3.5. Effects of Compounds X, Y, and Z on Electrocardiogram and Hemodynamics in Anesthetized Dogs
We examined the changes in plasma concentration of compound X at 10, 20, and 30 min, corresponding to the time points at the end of each 10-min infusion of 0.1, 0.3, and 1.0 mg/kg, respectively, in anesthetized dogs (Figure 2(a)). The concentration increased in a dosedependent manner. QTcF intervals were prolonged after the start of the 10-min infusion of 1 mg/kg: 11.9% and 20.5% prolongation (p < 0.001) was observed at 25 and 30 min, respectively (Figure 2(b)). At the 30-min point, the plasma concentration of compound X was 2.4 ± 0.7 μM. There was a strong positive correlation between QTcF prolongation and plasma concentration (slope = 6.905, p = 0.0004) (Figure 2(c)). Compound X did not seem to affect PQ interval, although there was a significant difference from the control at the 30-min point (p < 0.01) owing to a shortening trend in the control group (Figure 2(d)). There was no difference in the change in QRS interval between the compound Xand vehicletreated groups (Figure 2(e)). Heart rate (beats/minute) was significantly decreased by compound-X infusion at the 30- min point (p < 0.05) (Figure 2(f)). In the case of compound X, LVdP/dtmax was not determined.
Next, we investigated the changes in plasma concentrations of compounds Y and Z at 10, 20, and 30 min, corresponding to the time points at the end of each 10-min infusion of 1, 3, and 10 mg/kg, respectively (Figure 3(a)). With both compounds the QTcF interval seemed to be shortened slightly, although the changes were not significant (Figure 3(b)). With compound Y treatment, one dog showed ~10% shortening of the QTcF interval 10 min after the end of the 10-mg/kg infusion (at the 30-min point). Administration of compound Y or Z to dogs caused a dose-dependent PQ prolongation (Figure 3(c)); significant prolongation was observed at 25 (p < 0.01) and 30 (p < 0.001) min with compound Y treatment, and at 30 min (p < 0.05) with compound Z treatment. In contrast, the QRS interval was not affected by compound Y or Z (Figure 3(d)). Common and characteristic hemodynamic changes occurred in response to compounds Y and Z. Compound Y or Z infusion induced dose-dependent decreases in heart rate and LVdP/dtmax. There was a significant reduction in heart rate at 25 (p < 0.001) and 30 (p < 0.001) min with 10-mg/kg infusion of compound Y, and at 25 (p < 0.05) and 30 (p < 0.001) min with 10-mg/kg infusion of compound Z (Figure 3(e)); significant decreases in LVdP/dtmax were detected at 25 and 30 min with 10-mg/kg infusion of either compound Y or Z (p < 0.001 at all points; Figure 3(f)).
4. Discussion
We evaluated STVFPDc values from Poincaré plots in the hES-CMC system, and we observed increases in STVFPDc in clusters treated with cisapride or E-4031, along with FPDc prolongation. It is well known clinically that cisapride is torsadogenic. Few clinical data about the concentration of E-4031 that causes TdP are available. This compound is, however, a potent hERG channel blocker, so it can be expected to prolong the action potential and cause TdP at low plasma concentrations [22]. Therefore, it is possible that STVFPDc in the hES-CMC system is an additional useful marker of TdP. Nalos et al. examined action potential duration (APD) and beat-to-beat variability of repolarization of hES-CMCs treated with moxifloxacin or E-4031 [23]. Moxifloxacin treatment of hES-CMCs gave slight but not significant APD prolongation without increases in STV, reflecting clinical observations. Nalos et al. considered that assays using
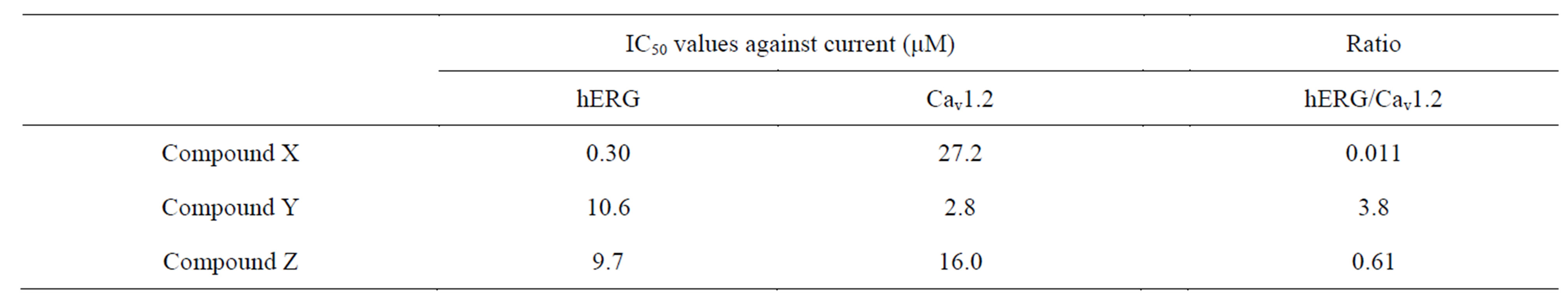
Table 3. Summary of IC50 values of compounds X, Y, and Z toward the hERG and Cav1.2 currents.

Figure 2. Changes in plasma concentration of compound (a), QTcF (b), PQ (d), QRS interval (e), and heart rate (f) in anesthetized dogs treated with compound X. Arrows indicate starting points of infusion of stepwise doses (0.1, 0.3, and 1.0 mg/kg body weight). Values are expressed as means ± S.E.M. Three dogs were used per group. *p < 0.05; **p < 0.01; ***p < 0.001 compared with the control vehicle-treated group. (c) is a constrained linear regression with the y-intercept to zero between the plasma concentration of compound X and changes in QTcF interval in anesthetized dogs treated with compound X; 95% confidence is indicated by dashed lines.

Figure 3. Changes in plasma concentration of compound (a), QTcF (b), PQ (c), QRS interval (d), heart rate (e), and LVdP/dtmax (f) in anesthetized dogs treated with compound Y or Z. Arrows indicate starting points of infusion of stepwise doses (1, 3, and 10 mg/kg body weight). Values are expressed as means ± S.E.M. Three dogs were used per group. *p < 0.05; **p < 0.01; ***p < 0.001 compared with the control vehicle-treated group.
hES-CMCs would have merits for further safety evaluation.
The class Ia antiarrhythmic drug quinidine causes serum or plasma concentration-dependent QT prolongation clinically. Quinidine at 5 μM prolongs APD at 90% repolarization (APD90) in guinea pig ventricles [24] and in canine Purkinje fibers [25]. Our concentrations of quinidine at 1 and 3 μM for FPDc prolongation agreed well with these data. Wu et al. reported significant increases in beat-to-beat variability of monophasic APD90 at 0.1 to 3 μM in rabbit left ventricles [26]. Our present study showed an increasing trend in STVFPDc in hES-CMCs treated with quinidine. Three of six clusters demonstrated >180% of the pre-value at 3 μM; this suggests that quinidine is torsadogenic.
The class Ib antiarrhythmic drug mexiletine shortens APD90 at 10 μM in guinea pig ventricles [24] and at 20 μM in canine Purkinje fibers [27]. However, mexiletine at 0.1 to 100 μM gave no significant FPDc shortening in our study. The class Ic antiarrhythmic drug flecainide, increases APD90 at 10 μM but decreases it at 30 μM in guinea pig right ventricular papillary muscles [28]. Additionally, Campbell reported that flecainide at 0.25 to 5 μM has minor effects on APD90 in guinea pig ventricles, although transitory prolongation was observed at 1 μM [24]. We observed no significant changes in FPDc or STVFPDc with flecainide from 10 nM to 10 μM. These results suggest that our preparations had little late sodium current [29] that was sensitive to sodium channel blockers.
We examined the correlation between FPDc in the hES-CMC system and QTcF interval in dogs by using our proprietary compounds, the channel involvement of which in regard to the QT interval has been determined only in terms of the hERG channel. The IC50 value of compound X toward the hERG current was 0.30 μM, and significant FPDc prolongation at 1 to 10 μM was observed in our in vitro assay using hES-CMCs. FPDc prolongation was thus explained by hERG channel blockage by compound X. In anesthetized dogs, significant prolongation of QTcF intervals was seen 10 min after the start of infusion of 1.0 mg/kg compound X, at which time the plasma concentration of this drug was 2.4 μM. This QTcF prolongation was correlated with the plasma concentration of compound X; the slope was 6.905 in the linear regression y = 6.905x, where x is the plasma concentration of compound X and y is the % change in the QTcF interval. When the plasma concentration of compound X was 1 and 3 μM, the QTcF interval change was estimated at +6.9% and +20.7%, respectively. Interestingly, the values calculated on the basis of our in vivo dog study were close to those from in the in vitro hESCMC system (12.1% and 21.0% FPDc prolongation at 1 and 3 μM, respectively). Therefore, with administration of compound X, the hES-CMC system displayed strong predictability toward QT prolongation in the in vivo dog studies. This good concordance may be related to the use of a measurement medium containing FBS in the hES-CMC system. Additionally, we found concentration-dependent increases in STVFPDc in hES-CMCs treated with compound X: at doses of 1, 3 and 10 μM, along with the FPDc prolongation, the STVFPDc was increased to 181%, 257%, and 259% of the pre-treatment value. These values seemed to be equivalent to those with E- 4031 and cisapride. The results of STVFPDc suggest that compound X is torsadogenic. Interestingly, compound X triggered typical EAD in one in five clusters.
Compounds Y and Z also had inhibitory activity against the hERG current at 10.6 and 9.7 μM, respectively, as IC50 values. Unlike with compound X, treatment with compound Y or Z at 10 to 30 μM gave a trend toward shortening of FPDc, rather than prolongation, in the hES-CMC system. In our in vivo study, neither compound Y nor Z induced QTcF prolongation in anesthetized dogs, although the plasma concentrations reached 38.0 μM (compound Y) and 29.0 μM (compound Z) at the end of infusion. Hence, we considered that with administration of compound Y or Z there was a similar trend between the FPDc changes in the hES-CMC system and the QTcF changes in the in vivo dog experiments. McAllister et al. found a strong positive linear relationship between plasma verapamil concentration and changes in PR intervals after i.v. drug administration to conscious dogs, although there were no changes in the QRS or QT interval [30]. Similarly, intravenous verapamil administration at 0.1 and 0.3 mg/kg induced significant prolongation (8.9% and 20.0%, respectively) of the PQ interval in anesthetized dogs, whereas no changes were seen in the QRS or QT interval [31]. In addition, dose-dependent decreases in heart rate and LVdP/dtmax were observed with verapamil (−12.3% and −23.1%, respectively, at 0.3 mg/kg) [31]. Furthermore, decreases in heart rate (−14%) and prolongation of the PQ interval (48%) have been reported after i.v. diltiazem infusion into conscious dogs [32]. Our results, together with the above-mentioned findings, suggest that compounds Y and Z block calcium channels. In our previous in vitro assessment using hES-CMCs, verapamil and diltiazem at 30 and 100 nM, respectively, produced >10% shortening of FPDc [8]. We therefore speculated that FPDc changes were determined by the potency relationships between hERG and Cav1.2 current inhibition by each compound. In relative terms, inhibitory activity against the hERG current was stronger than that against the Cav1.2 current in the case of compound X, and vice versa for compound Y (Table 3). Therefore, these relationships could result in FPDc prolongation with compound X treatment and FPD shortening with compound Y treatment. Compound Y possessed more potent inhibitory activity toward the Cav1.2 current than did compound Z, but their IC50 values toward the hERG current were similar. This may reflect the finding that compound Y shortened FPDc more clearly. Notably, the FPDc changes were reproduced in the canine in vivo experiments as slight shortening of the QTcF. Accordingly, our system is able to detect physiological QT changes caused by the interaction of the effects of a compound on plural channels; hERG assay alone cannot do this.
The results obtained with our reference compounds, including hERG channel blockers, indicated that the hES-CMC system presents two parameters for the prediction of TdP: one is FPDc, corresponding to the QT interval, and the other is STVFPDc, corresponding to the beat-to-beat variability of repolarization. These findings suggest that combined detection of FPDc and STVFPDc can increase the predictability of ventricular fibrillation risk. In addition, we demonstrated that the hES-CMC system is a physiologically relevant multichannel detection system that reflects in vivo observations in evaluating proprietary compounds as drug candidates.
Acknowledgements
We are grateful to Ms. Atsuko Ojima for her help with examining the hES-CMC system.
REFERENCES
- H. R. Lu, E. Vlaminckx, A. N. Hermans, J. Rohrbacher, K. Van Ammel, R. Towart, M. Pugsley and D. J. Gallacher, “Predicting Drug-Induced Changes in QT Interval and Arrhythmias: QT-Shortening Drugs Point to Gaps in the ICHS7B Guidelines,” British Journal of Pharmacology, Vol. 154, No. 7, 2008, pp. 1427-1438. http://dx.doi.org/10.1038/bjp.2008.191
- P. L. Hedley, P. Jørgensen, S. Schlamowitz, R. Wangari, J. Moolman-Smook, P. A. Brink, J. K. Kanters, V. A. Corfield and M. Christiansen, “The Genetic Basis of Long QT and Short QT Syndromes: A Mutation Update,” Human Mutation, Vol. 30, No. 11, 2009, pp. 1486-1511. http://dx.doi.org/10.1002/humu.21106
- R. L. Martin, J. S. McDermott, H. J. Salmen, J. Palmatier, B. F. Cox and G. A. Gintant, “The Utility of hERG and Repolarization Assays in Evaluating Delayed Cardiac Repolarization: Influence of Multi-Channel Block,” Journal of Cardiovascular Pharmacology, Vol. 43, No. 3, 2004, pp. 369-379.
- Y. Asai, M. Tada, T. G. Otsuji and N. Nakatsuji, “Combination of Functional Cardiomyocytes Derived from Human Stem Cells and a Highly-Efficient Microelectrode Array System: An Ideal Hybrid Model Assay for Drug Development,” Current Stem Cell Research and Therapy, Vol. 5, No. 3, 2010, pp. 227-232. http://dx.doi.org/10.2174/157488810791824502
- M. K. B. Jonsson, G. Duker, C. Tropp, B. Andersson, P. Sartipy, M. A. Vos and T. A. B. van Veen, “Quantified Proarrhythmic Potential of Selected Human Embryonic Stem Cell-Derived Cardiomyocytes,” Stem Cell Research, Vol. 4, No. 3, 2010, pp. 189-200. http://dx.doi.org/10.1016/j.scr.2010.02.001
- M. Pekkanen-Mattila, H. Chapman, E. Kerkelä, R. Suuronen, H. Skottman, A. P. Koivisto and K. Aalto-Setälä, “Human Embryonic Stem Cell-Derived Cardiomyocytes: Demonstration of a Portion of Cardiac Cells with Fairly Mature Electrical Phenotype,” Experimental Biology and Medicine (Maywood), Vol. 235, No. 4, 2010, pp. 522- 530. http://dx.doi.org/10.1258/ebm.2010.009345
- S. Peng, A. E. Lacerda, G. E. Kirsch, A. M. Brown and A. Bruening-Wright, “The Action Potential and Comparative Pharmacology of Stem Cell-Derived Human Cardiomyocytes,” Journal of Pharmacological and Toxicological Methods, Vol. 61, No. 3, 2010, pp. 277-286. http://dx.doi.org/10.1016/j.vascn.2010.01.014
- K. Yamazaki, T. Hihara, T. Taniguchi, N. Kohmura, T. Yoshinaga, M. Ito and K. Sawada, “A Novel Method of Selecting Human Embryonic Stem Cell-Derived Cardiomyocyte Clusters for Assessment of Potential to Influence QT Intervals,” Toxicology in Vitro, Vol. 26, No. 2, 2012, pp. 335-342. http://dx.doi.org/10.1016/j.tiv.2011.12.005
- P. T. Nguyen, M. M. Scheinman and J. Seger, “Polymorphous Ventricular Tachycardia: Clinical Characterization, Therapy, and the QT Interval,” Circulation, Vol. 74, No. 2, 1986, pp. 340-349. http://dx.doi.org/10.1161/01.CIR.74.2.340
- S. H. Hohnloser, T. Klingenheben and B. N. Singh, “Amiodarone-Associated Proarrhythmic Effects. A Review with Special Reference to Torsade de Pointes Tachycardia,” Annals of Internal Medicine, Vol. 121, No. 7, 1994, pp. 529-535. http://dx.doi.org/10.7326/0003-4819-121-7-199410010-00009
- L. M. Hondeghem, L. Carlsson and G. Duker, “Instability and Triangulation of the Action Potential Predict Serious Proarrhythmia, but Action Potential Duration Prolongation Is Antiarrhythmic,” Circulation, Vol. 103, No. 15, 2001, pp. 2004-2013. http://dx.doi.org/10.1161/01.CIR.103.15.2004
- T. D. Gbadebo, R. W. Trimble, M. S. Khoo, J. Temple, D. M. Roden and M. E. Anderson, “Calmodulin Inhibitor W- 7 Unmasks a Novel Electrocardiographic Parameter That Predicts Initiation of Torsade de Pointes,” Circulation, Vol. 105, No. 6, 2002, pp. 770-774. http://dx.doi.org/10.1161/hc0602.103724
- S. Kääb, M. Hinterseer, M. Näbauer and G. Steinbeck, “Sotalol Testing Unmasks Altered Repolarization in Patients with Suspected Acquired Long-QT-Syndrome— A Case-Control Pilot Study Using i.v. Sotalol,” European Heart Journal, Vol. 24, No. 7, 2003, pp. 649-657. http://dx.doi.org/10.1016/S0195-668X(02)00806-0
- M. B. Thomsen, S. C. Verduyn, M. Stengl, J. D. Beekman, G. de Pater, J. van Opstal, P. G. Volders and M. A. Vos, “Increased Short-Term Variability of Repolarization Predicts d-Sotalol-Induced Torsades de Pointes in Dogs,” Circulation, Vol. 110, No. 16, 2004, pp. 2453-2459. http://dx.doi.org/10.1161/01.CIR.0000145162.64183.C8
- M. B. Thomsen, J. Matz, P. G. Volders and M. A. Vos, “Assessing the Proarrhythmic Potential of Drugs: Current Status of Models and Surrogate Parameters of Torsades de Pointes Arrhythmias,” Pharmacology and Therapeutics, Vol. 112, No. 1, 2006, pp. 150-170. http://dx.doi.org/10.1016/j.pharmthera.2005.04.009
- C. Antzelevitch, “Heterogeneity and Cardiac Arrhythmias: An Overview,” Heart Rhythm, Vol. 4, No. 7, 2007, pp. 964-972. http://dx.doi.org/10.1016/j.hrthm.2007.03.036
- M. Hinterseer, M. B. Thomsen, B.-M. Beckmann, A. Pfeufer, R. Schimpf, H.-E. Wichmann, G. Steinbeck, M. A. Vos and S. Kaab, “Beat-to-Beat Variability of QT Intervals Is Increased in Patients with Drug-Induced LongQT Syndrome: A Case Control Pilot Study,” European Heart Journal, Vol. 29, No. 2, 2008, pp. 185-190. http://dx.doi.org/10.1093/eurheartj/ehm586
- N. Abi-Gerges, J. P. Valentin and C. E. Pollard, “Dog Left Ventricular Midmyocardial Myocytes for Assessment of Drug-Induced Delayed Repolarization: ShortTerm Variability and Proarrhythmic Potential,” British Journal of Pharmacology, Vol. 159, No. 1, 2010, pp. 77- 92. http://dx.doi.org/10.1111/j.1476-5381.2009.00338.x
- T. Taniguchi, M. Uesugi, T. Arai, T. Yoshinaga, N. Miyamoto and K. Sawada, “Chronic Probucol Treatment Decreases the Slow Component of the Delayed-Rectifier Potassium Current in CHO Cells Transfected with KCNQ1 and KCNE1: A Novel Mechanism of QT Prolongation,” Journal of Cardiovascular Pharmacology, Vol. 59, No. 4, 2012, pp. 377-386. http://dx.doi.org/10.1097/FJC.0b013e318245e0c5
- R. L. Martin, J. T. Limberis, X. Liu, K. Houseman, Z. Su, W. Niforatos, B. F. Cox and G. A. Gintant, “Optimization of a Cav1.2 Cell Line for Use on QPatch and PatchXpress,” Biophysical Journal, Vol. 98, No. 3, 2010, p. 339a. http://dx.doi.org/10.1016/j.bpj.2009.12.1836
- J. Kutchinsky, S. Friis, M. Asmild, R. Taboryski, S. Pedersen, R. K. Vestergaard, R. B. Jacobsen, K. Krzywkowski, R. L. Schrøder, T. Ljungstrøm, N. Hélix, C. B. Sørensen, M. Bech and N. J. Willumsen, “Characterization of Potassium Channel Modulators with QPatch Automated Patch-Clamp Technology: System Characteristics and Performance,” Assay and Drug Development Technologies, Vol. 1, No. 5, 2003, pp. 685-693. http://dx.doi.org/10.1089/154065803770381048
- R. Webster, G. Allan, K. Anto-Awuakye, A. Harrison, T. Kidd, D. Leishman, J. Phipps and D. Walker, “Pharmacokinetic/Pharmacodynamic Assessment of the Effects of E4031, Cisapride, Terfenadine and Terodiline on Monophasic Action Potential Duration in Dog,” Xenobiotica, Vol. 31, No. 8-9, 2001, pp. 633-650. http://dx.doi.org/10.1080/00498250110054632
- L. Nalos, R. Varkevisser, M. K. B. Jonsson, M. J. C. Houtman, J. D. Beekman, R. van der Nagel, M. B. Thomsen, G. Duker, P. Sartipy, T. P. de Boer, M. Peschar, M. B. Rook, T. A. B. van Veen, M. A. G. van der Heyden and M. A. Vos, “Comparison of the IKr Blockers Moxifloxacin, Dofetilide and E-4031 in Five Screening Models of Pro-Arrhythmia Reveals Lack of Specificity of Isolated Cardiomyocytes,” British Journal of Pharmacology, Vol. 165, No. 2, 2012, pp. 467-478. http://dx.doi.org/10.1111/j.1476-5381.2011.01558.x
- T. J. Campbell, “Kinetics of Onset of Rate-Dependent Effects of Class I Antiarrhythmic Drugs Are Important in Determining Their Effects on Refractoriness in GuineaPig Ventricle, and Provide a Theoretical Basis for Their Subclassification,” Cardiovascular Research, Vol. 17, No. 6, 1983, pp. 344-352. http://dx.doi.org/10.1093/cvr/17.6.344
- K. R. Wyse, V. Ye and T. J. Campbell, “Action Potential Prolongation Exhibits Simple Dose-Dependence for Sotalol, but Reverse Dose-Dependence for Quinidine and Disopyramide: Implication for Proarrhythmia Due to Triggered Activity,” Journal of Cardiovascular Pharmacology, Vol. 21, No. 2, 1993, pp. 316-322. http://dx.doi.org/10.1097/00005344-199302000-00019
- L. Wu, D. Guo, H. Li, J. Hackett, G. X. Yan, Z. Jiao, C. Antzelevitch, J. C. Shryock and L. Belardinelli, “Role of Late Sodium Current in Modulating the Proarrhythmic and Antiarrhythmic Effects of Quinidine,” Heart Rhythm, Vol. 5, No. 12, 2008, pp. 1726-1734. http://dx.doi.org/10.1016/j.hrthm.2008.09.008
- V. Elharra, “Recovery from Use-Dependent Block of Vmax and Restitution of Action Potential Duration in Canine Cardiac Purkinje Fibers,” The Journal of Pharmacology and Experimental Therapeutics, Vol. 246, No. 1, 1988, pp. 235-242.
- U. Borchard and M. Boisten, “Effects of Flecainide on Action Potentials and Alternating Current-Induced Arrhythmia in Mammalian Myocardium,” Journal of Cardiovascular Pharmacology, Vol. 4, No. 2, 1982, pp. 205- 212. http://dx.doi.org/10.1097/00005344-198203000-00007
- A. Zaza, L. Belardinelli and J. C. Shryock, “Pathophysiology and Pharmacology of the Cardiac ‘Late Sodium Current’,” Pharmacology and Therapeutics, Vol. 119, No. 3, 2008, pp. 326-339. http://dx.doi.org/10.1016/j.pharmthera.2008.06.001
- R. G. McAllister Jr., D. W. Bourne and L. W. Dittert, “The Pharmacology of Verapamil. I. Elimination Kinetics in Dogs and Correlation of Plasma Levels with Effect on the Eletrocardiogram,” The Journal of Pharmacology and Experimental Therapeutics, Vol. 202, No. 1, 1977, pp. 38-44.
- S. Wakabayashi, S. Mochizuki, A. Tomiyama and S. Shibata, “Effects of KT-362, a New Calcium Release Blocker, on Vascular Selectivity and Hemodynamic Actions in Anesthetized Dogs,” The Japanese Journal of Pharmacology, Vol. 54, No. 1, 1990, pp. 23-32. http://dx.doi.org/10.1254/jjp.54.23
- M. Manabe, S. Motomura and K. Hashimoto, “Interaction between Diltiazem and Halothane or Enflurane in the Canine Blood-Perfused Papillary Muscle and Sinoatrial Node Preparations Cross-Circulated by Chronically Instrumented Conscious Donor Dog,” Journal of Anesthesia, Vol. 2, No. 1, 1988, pp. 50-62. http://dx.doi.org/10.1007/s0054080020050
Abbreviations
APD: action potential duration;
APD90: action potential duration at 90% repolarization;
CHO: Chinese hamster ovary;
DMEM: Dulbecco’s modified Eagle’s medium;
DMSO: dimethyl sulfoxide;
E-4031: N-[4-[1-[2-(6-methylpyridin-2-yl)ethyl]piperidine- 4-carbonyl]phenyl]methanesulfonamide dihydrochloride dihydrate;
EAD: early afterdepolarization;
EGTA: ethylene glycol-bis(2-aminoethylether)-N,N,N’,N’- tetraacetic acid;
FBS: fetal bovine serum;
FPD, field potential duration;
FPDc: corrected FPD;
HEPES: 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid;
hERG: human ether-à-go-go-related gene;
hES-CMC: human embryonic stem cell-derived cardiomyocyte cluster;
ISI: inter-spike interval;
LVdP/dtmax: maximal upstroke velocity of the left ventricular pressure;
QTc: corrected QT;
QTcF: QT corrected by Fridericia’s formula;
STV: short-term variability;
TdP: torsades de pointes.
NOTES
*Corresponding author.