Pharmacology & Pharmacy
Vol.4 No.2(2013), Article ID:30395,7 pages DOI:10.4236/pp.2013.42035
Down Regulation of MyD88 in Macrophages Treated with Liposomes Composed of Phosphatidylserine
![]()
School of Pharmacy Tokyo University of Pharmacy and Life Sciences, Tokyo, Japan.
Email: *aramaki@ps.toyaku.ac.jp
Copyright © 2013 Yuka Takasugi et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received January 17th, 2013; revised February 28th, 2013; accepted April 8th, 2013
Keywords: Phosphatidylserine; Liposome; MyD88; TGF-β; Macrophage
ABSTRACT
We have recently demonstrated that liposomes composed of phosphatidylserine (PS-liposomes) suppressed nitric oxide and inflammatory cytokine productions following LPS stimulation in macrophages. In this study, we examined the effect of PS-liposomes on expressions of TLR-4 and MyD88, which are essential for the signal transduction in LPS stimulation. Expression of MyD88 was suppressed when macrophages were treated with PS-liposomes, but not with liposomes of phosphatidylcholine. No change in TLR-4 expression was observed. MyD88 suppression was restored to the control levels when cells were pre-treated with anti-TGF-β antibody, suggesting that TGF-β plays an important role in down-regulation of MyD88 following PS-liposome treatment.
1. Introduction
Apoptosis has been recognized to be of major importance for embryonic development, tissue homeostasis, autoimmune disease, cancer progression, and the killing of cancer cells by anti-cancer drugs [1-4]. In the body, professional and non-professional phagocytes rapidly recognize and phagocytose apoptotic cells to prevent the release of noxious contents that could provoke inflammation and tissue damage [5-7]. Exposure of the anionic phospholipid, phosphatidylserine (PS), in the outer leaflet of the plasma membrane is one of the most striking and consistent changes on the surface of apoptotic cells [8,9]. The best known molecule involved in recognition by macrophages is PSas an “eat me signal” [9]. Furthermore, interaction between macrophages and apoptotic cells results in the secretion of cytokines such as TGF-β and IL-10, which are known as anti-inflammatory cytokines [5-7,10,11].
TGF-β is a multifunctional cytokine that regulates numerous physiological processes, including cell growth, differentiation, apoptosis, adhesion, and the synthesis of extracellular matrix proteins [12]. Engulfment of apoptotic cells is thought not only to remove them from the tissue but also to provide protection from local damage resulting from the release or discharge of pro-inflammatory contents, and the immune-suppressive effect was largely inhibited by TGF-β-neutralizing antibodies [13]. Liposomes composed of PS (PS-liposomes) can mimic the release of cytokines to apoptotic cells, and indeed the release of TGF-β1 from macrophages has been identified [14]. Hoffmann et al. have reported that PS-liposomes inhibit immune responses in vivo through a PS-specific receptor, and TGF-β plays a critical role in this inhibition [15].
In our series of studies, PS-liposomes inhibited the production of nitric oxide (NO) and tumor necrosis factor (TNF)-α fromthioglycollate-elicited mouse peritoneal macrophages stimulated with LPS, and TGF-β1 is one of the factors produced by PS-liposomes that suppresses the productions of NO and TNF-α in macrophages [16,17]. Furthermore, we demonstrated that the PI3K/Akt pathway and the downstream extracellular signal-regulated kinase (ERK), a MAP kinase, signaling pathway via PS-specific receptors are intimately involved in the production of TGF-β1 by macrophages treated with PS-liposomes [18]. However, it is not yet clear how TGF-β acts on macrophages and inhibits the secretion of NO or TNF-α from macrophages stimulated with LPS.
In this study, we focused on Toll-like receptor (TLR) 4 and MyD88. It is well known that the effect of LPS is mediated through a member of the highly conserved TLR family, TLR4, leading to the induction of numerous pro-inflammatory genes in most cell types, particularly macrophages [19,20]. Furthermore, TLR adaptor protein, MyD88 is essential for responses against a broad range of microbial components through TLRs [21]. Our data indicated down-regulation of MyD88 when the cells were treated with PS-liposomes, but no changes in TLR4 expression. This inhibitory effect of PS-liposomes was restored when the cells were treated with anti-TGF-β antibody, suggesting that inhibitory effects of PS-liposomes on macrophages result in the down-regulation of MyD88 expression through TGF-β production.
2. Materials and Methods
2.1. Materials
Phosphatidylserine (PS) was kindly provided from H. Holstein GmbH EtCo. KG (Tokyo, Japan) bovine brain and cholesterol (Chol) was obtained from Sigma Co., Ltd. (St. Louis, MO). Phosphatidylcholine (PC) from egg yolk was purchased from Nippon Oil and Fat Co. (Tokyo, Japan).
2.2. Preparation of Liposomes
Multilamellar liposomes composed of PS:PC:Cholesterol = 2:1:1 (by molar ratio, PS-liposomes) and PC:Cholesterol = 3:1 (by molar ratio, PC-liposomes) were prepared by vortexing and passed through a membrane filter (0.45 μm; Corning Glassworks, Corning, NY) before use. Liposomal size and Zeta potential were measured with a dynamic light-scattering spectrophotometer (DLS-7000, Otsuka Electronics, Tokyo) and Nicomp 380 ZLS (PSS Nicomp Particle Sizing System). The mean diameter and Zeta potential of SA-liposomes were 380 nm and −25 mV, respectively.
2.3. Preparation of Macrophages
Mouse macrophage-like cell line, RAW264.7, was purchased from Riken Cell Bank (Ibaraki, Japan). C3H/HeN mice (male, 6 - 8 weeks old) were purchased from Japan SLC Inc. (Shizuoka, Japan). Animal use and relevant experimental procedures were approved by the Tokyo University of Pharmacy and Life Science Committee on the Care and Use of Laboratory Animals. C3H/HeN mice were injected intraperitoneally with 1.0 mL of 3% thioglycollate (Difco Laboratory, Detroit, MI). On day 4, murine peritoneal macrophages were prepared according to our previously described method [16].
2.4. FACS Analysis for TLR4 Expression
RAW264.7 cells and murine peritoneal macrophages (1 × 106 cells) were treated with PS-liposomes (125 μg of phospholipids/mL) at 37˚C for 24 hr. After treatment, the cells were washed with PBS, and then were treated withanti-TLR4 antibody (Imgenex, San Diego, CA) at 4˚C for 1 hr. After three washes with PBS, cells were re-suspended in FITC-labeled anti-rat IgG as secondary antibody (Imgenex, San Diego, CA) at 4˚C for 1 hr. The expression of TLR4 on the cell surface was evaluated by flow cytometry (FACSCalibur, Becton-Dickinson).
2.5. Western Blotting
RAW264.7 and murine peritoneal macrophages (1 × 106 cells/well) were treated with PS-liposomes (125 μg of phospholipids/mL) or anti-PS-receptor antibody (20 μg/mL) for given periods. Cells were then lysed as described previously [22]. For the determination of MyD88, cell lysates were separated by 12% SDS-PAGE, blotted on Immobilon P membranes (Nihon Millipore, Tokyo, Japan), and analyzed usinganti-MyD88 antibody and HRPconjugated-anti-rabbit IgG antibody (Imgenex, San Diego, CA, USA). Band intensity was analyzed with NIH Image in each case [14].
2.6. Induction of Apoptotic Cells
Jurkat T cellspurchased from Riken Cell Bank (Ibaraki, Japan) were exposed to UV irradiation at 254 nm for 5 min. The cells were cultured in RPMI 1640 with 10% fetal calf serum (Gemini Bio-Products) for 2 h at 37˚C in 5% CO2. Cell apoptosis was evaluated by determining the externalization of phosphatidylserine (PS) by flow cytometry (FACS Calibur, Becton Dickinson, San Jose, CA) using TACSTM Annexin V-FITC apoptosisdetection kits according to the manufacturer’s instructions (Biovision, Mountain View, CA), and the proportion of cells expressing PS at the surface was generally 60%.
2.7. Detection of TGF-β1
RAW264.7 cells (5 × 105 cells/well) were treated with PS-liposomes (125 μg of phospholipids/mL) for a specified period. TGF-β1 concentrations were determined by ELISA using pairs of purified capture and biotinylated detection antibodies recognizing murine TGF-β1 according to the manufacturer’s directions (BD Biosciences).
2.8. Statistical Analysis
Paired Student’s t test was used to compare paired groups. Aanalysis of variance (ANOVA) was used for multigroup analysis. Values of p < 0.05 were considered to indicate a significant difference.
3. Results and Discussion
In our previous study [14,16-18,22],we demonstrated that PS-liposomes inhibited the productions of nitric oxide
(NO) and inflammatory cytokine, TNF-α, frommurine peritoneal macrophages stimulated with LPS, and TGF-β plays an important role in this inhibitory effect. It is well known that the effect of LPS is mediated through a member of the highly conserved Toll-like receptor family, TLR4, leading to the induction of numerous pro-inflammatory genes in most cell types, particularly macrophages [20]. In TLR4 signaling, stimulation of TLR4 facilitates the activation of two pathways, MyD88-dependent and MyD88-independent pathways. The former involves the early phase of nuclear factor (NF)-κB activation leading to the production of inflammatory cytokines [20]. Furthermore, MyD88-deficient mice fail to produce TNF and IL-6 when exposed to LPS [23]. In this study, to clarify the mechanism of how TGF-β acts on macrophages and inhibits the secretion of NO or TNF-α from macrophages stimulated with LPS, we focused on TLR4 and MyD88 expressions.
3.1. Effects of PS-Liposomes on TLR4 Expression
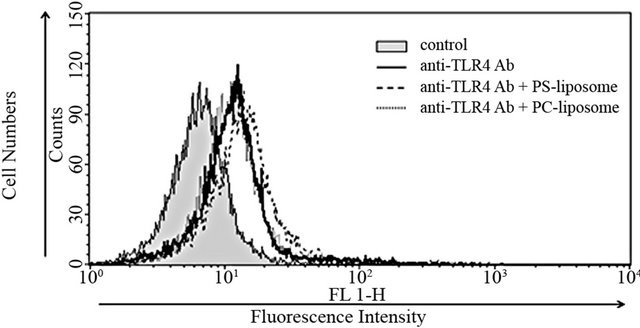
Effects of PS-liposomes on TLR4 expression were examined in murine macrophage cell line RAW264.7 cells and peritoneal macrophages. RAW264.7 cells were treated with liposomes for 24 h, and then the changes in TLR4 expression were estimated by flow cytometry. As shown in Figure 1(a), no changes were observed when RAW264.7 cells were treated with PS-liposomes and PC-liposomes, and the intensity of fluorescence was the same as that of the control. In the case of murine peritoneal macrophages, no changes in TLR4 expression were also observed when the cells were treated with PS-liposomes for 12 and 24 h (Figure 1(b)).
Das et al. reported that Leishmania infection induced tolerance in macrophages, leading to an inability to produce of inflammatory cytokines TNF-α and IL-1β in response to TLR stimulation with LPS. Overexpression of TGF-β, but not IL-10, resulted in suppressedLPS-induced production of TNF-α and down-regulation of TLR4 expression in Leishmania-infected macrophages [24]. Furthermore, Jones et al. reported that activin A, which is also named restrictin-P, could decrease the secretions of IL-1β and NO, as well as the mRNA expressions of IL-1β and iNOS, in mouse macrophage cell line RAW264.7 cells induced by LPS [25]. In addition, activin A could reduce the expressions of CD68 and CD14, as well as TLR4 on RAW264.7 cells. In our previous study [26], PS-liposomes inhibited the secretions of NO and inflammatory cytokines frommurine peritoneal macrophages stimulated with LPS, but there was no change in the expression of CD14 in peritoneal macrophages following PS-liposome treatment. PS-liposomes induced macrophages to exhibit tolerance towards LPS, leading to an inability to produceNO and inflammatory cytokines.
 (a)
(a)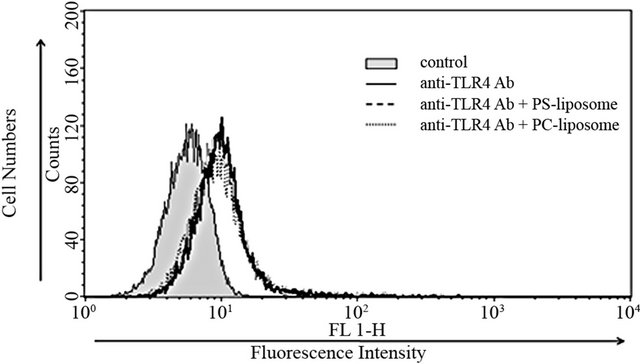 (b)
(b)
Figure 1. Effects of PS-liposomes on TLR4 expression in murine peritoneal macrophages (a) and RAW264.7 cells (b). Both cells (1 × 106 cells) were incubated with PS-liposomes (125 μg of phospholipids/mL) or PC-liposomes (125 μg of phospholipids/mL) for 24 h. After the incubation, cells were washed twice with PBS, and then incubated with anti-TLR4 antibody. After three washes with PBS, cells were incubated with FITC-labeled anti-rat IgG as second antibody, and then subjected to flow cytometry.
However, no change in TLR4 expression was observed in this study. The reason for the discrepancy in TLR4 expressionmay be attributable to the differences of cell type and stimuli, but the detailed mechanism is not yet understood.
3.2. PS-Liposomes Down-Regulate MyD88 Expression
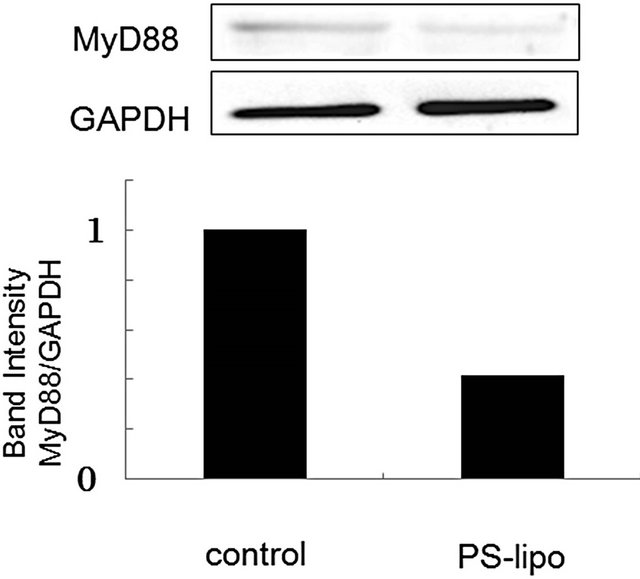
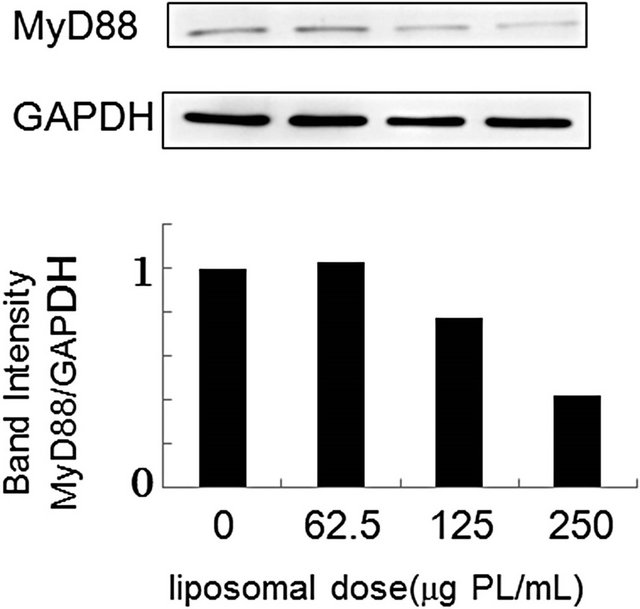
Cellular responses to LPS are mediated by TLR4 [20]. Intracellular signaling pathways through TLR4 are divided into two categories: MyD88-dependent pathway and MyD88-independent pathway [20,21]. The MyD88- dependent pathway leads to early activation of the transcription factor NF-κB and the productions of proinflammatory cytokines and proteins, including TNF-α and NO synthase. On the other hand, the MyD88-independent pathway activates interferon-regulatory factor (IRF) -3 and involves the late phase of NF-κB activation, leading to the productions of IFN-β [20,21]. Since PS-liposomes inhibited the productions of cytokines and NO generation [16,17], we focused on the inhibitory effect of the liposomes on the MyD88-dependent pathway. We first investigated down-regulation of MyD88 expression in murine peritoneal macrophages and RAW264.7 cells by Western blotting, and band intensity was analyzed with NIH Image in each case. As shown in Figure 2, MyD88 expression in each cell treated with PS-liposomes was down-regulated to half of the level of the control. Inhibitory effects of PS-liposomes on MyD88 expression were observed in a dose-dependent manner (Figure 3). Furthermore, the inhibitory effect was observed when the cells were treated with PS-liposomes at least for 6 h, and no noteworthy time-dependent inhibitory effect was found in murine peritoneal macrophages (Figure 4). These findings revealed that PS-liposomes suppressed MyD88 expression in macrophages.
Exposure of PS in the outer leaflet of the plasma membrane is one of the most striking and consistent changes on the surface of apoptotic cells, and PS is known as an “eat me signal” involved in the recognition by macrophages [9]. We thus examined whether apoptotic cells could suppressed MyD88 expression in macrophages.
 (a)
(a)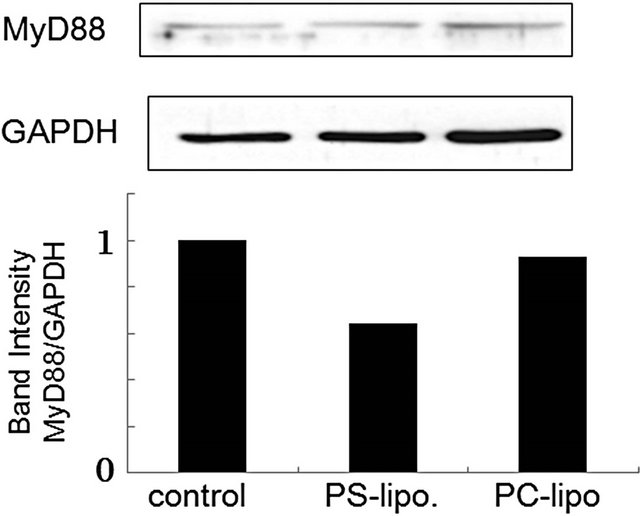 (b)
(b)
Figure 2. Effects of PS-liposomes on MyD88 expression in murine peritoneal macrophages (a) and RAW264.7 cells (b). Both cells (1 × 106 cells) were incubated with PS-liposomes (125 μg of phospholipids/mL) or PC-liposomes (125 μg of phospholipids/mL) for 18 h. MyD88 expression was evaluated by Western blotting. Cell lysates were separated by 12% SDS-PAGE, blotted on Immobilon P membranes, and analyzed using ananti-MyD88 antibody and HRP-conjugated anti-rabbit IgG antibody. Band intensity was analyzed with NIH Image in each case.
 (a)
(a)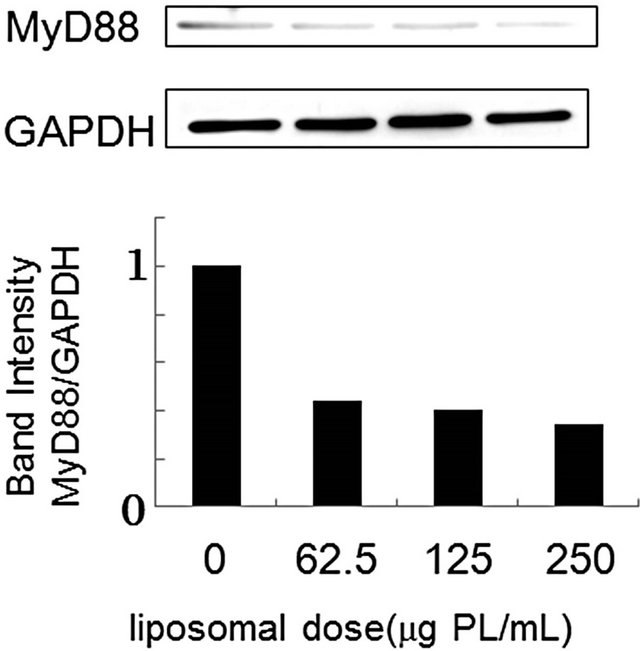 (b)
(b)
Figure 3. Effect of liposomal dose on MyD88 expression in murine peritoneal macrophages (a) and RAW264.7 cells (b). Both cells (1 × 106 cells) were incubated with various doses of PS-liposomes for 18 h. MyD88 expression was evaluated by Western blotting according to the METHODS described above.
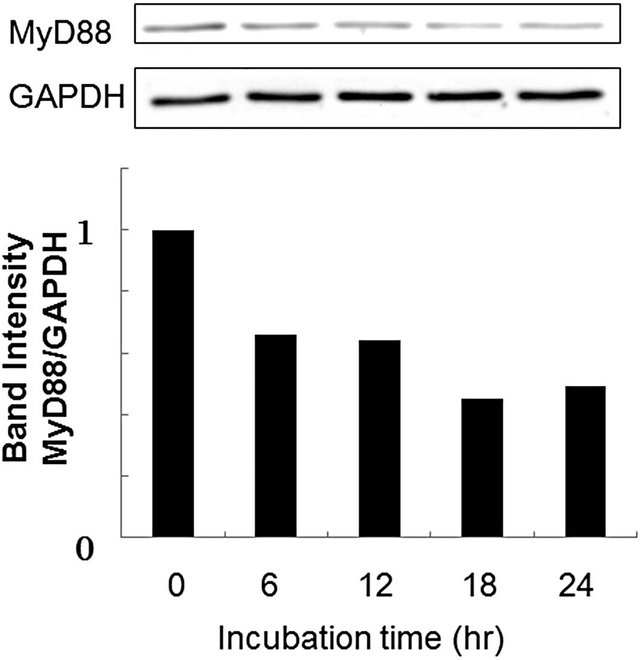
Figure 4. Effects of incubation time on MyD88 expression in murine peritoneal macrophages. Cells (1 × 106 cells) were incubated with various doses of PS-liposomes for 18 h. MyD88 expression was evaluated by Western blotting according to the METHODS described above.
Mouse peritoneal macrophages were treated with apoptotic Jurkat T cells, which were obtained by UV irradiation at 254 nm for 5 min, and MyD88 expression was clearly suppressed (Figure 5). Recently, several papers have indicated that apoptotic cells participate in immune modulations in antigen-presenting cells, macrophages and plasmacytoid dendritic cells, and immune responses such as cytokine production, inflammation response, and
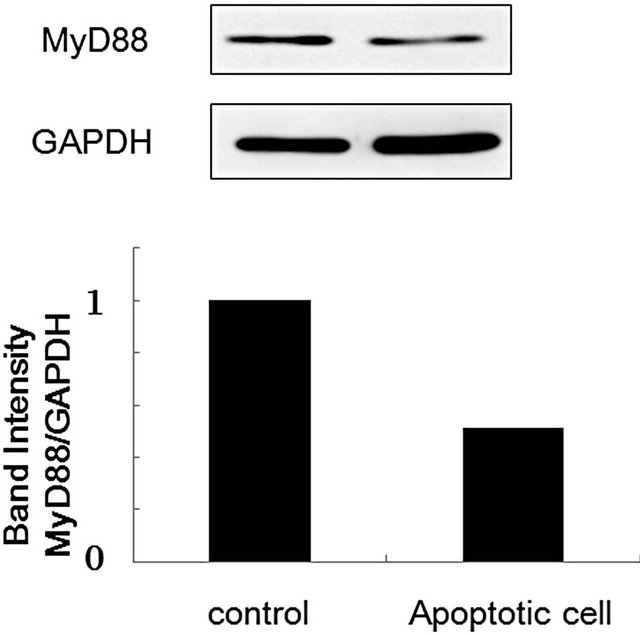
Figure 5. Effect of apoptotic cells on MyD88 expression in murine peritoneal macrophages. Murine peritoneal cells (1 × 106 cells) were incubated with apoptotic Jurkat T cells (2 × 105 cells) for 18 h. After the incubation, macrophage cell lysate was prepared, and MyD88 expression was evaluated by Western blotting according to the METHODS described above.
T cell differentiation were changed [27-30]. In these phenomena, apoptotic cells could not directly interact with macrophages or dendritic cells, and soluble factors including TGF-β are required in their immunomodulatoryfunctions.
3.3. Involvement of TGF-β in MyD88 Down-Regulation
TGF-β is implicated as an important regulator of almost all major cell behaviors and activities, such as proliferation, adhesion, motility, apoptosis, and differentiation [12]. Which of these are affected and how they are regulated in response to TGF-β depend entirely on the cell type and the cell state in which the TGF-β signals are received. Many researchers reported that phagocytes can engage PS exposed on apoptotic cells through multiple distinct receptors, such as Tim-4, Stabilin-2, MER, BAI1, and integrin αvβ3 [9,25,31]. Uptake of apoptotic cellsthroughthese receptors leads macrophages to produce TGF-β [9,27]. Therefore, we evaluated the production of TGF-β in RAW264.7 cells following PS-treatment. As shown in Figure 6, secretion of TGF-β began at 3 h and reached a plateau at 9 h after the treatment of the cells with PS-liposomes. Our previous report indicated that the secretion of TGF-β was also observed in murine peritoneal macrophages [14]. To clarify whether TGF-β is involved in the MyD88 suppression, the effect of antiTGF-β antibody on the MyD88 expression was evaluated by ELISA. As shown in Figure 7, the band intensity suppressed by PS-liposomes was restored to the control level when RAW264.7 cells were pre-treated with anti-TGF-β antibody. These findings suggested that TGF-β, which is produced by PS-liposome treatment, was involved in the suppression of MyD88 expression in macrophages. TGF- β plays a fundamental role in regulation of cell growth,
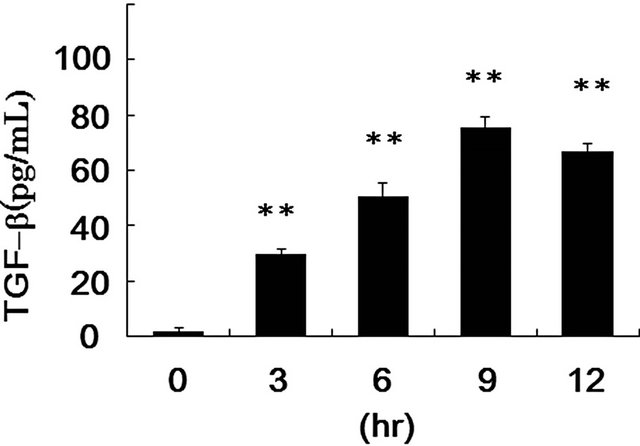
*: p < 0.05 significantly different from control (incubation time 0 h).
Figure 6. TGF-β production from RAW264.7 cells after stimulation with PS-liposomes. RAW264.7 cells (5 × 105 cells) were treated with PS-liposomes (125 μg of phospholipids/mL) for the indicated times. Supernatant was collected and TGF-β protein concentrations were evaluated by ELISA as described in METHODS. The values are mean + SD of triplicate results from three independent experiments.
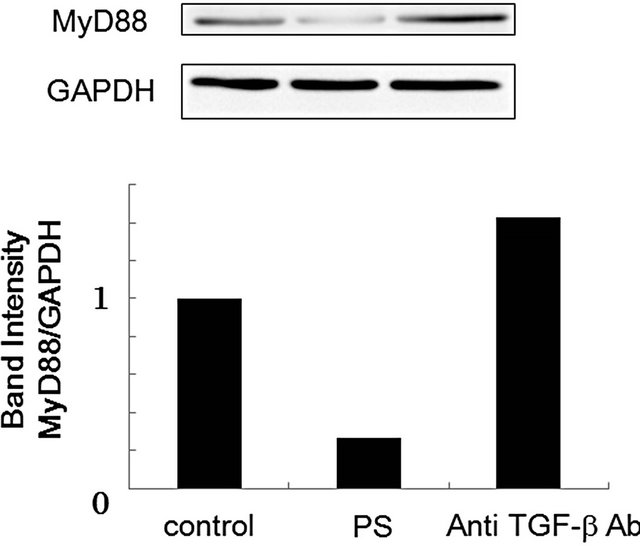
Figure 7. Effect of anti-TGF-β antibody on MyD88 expression in RAW264.7 cells. RAW264.7 cells (1 × 106 cells) were pre-treated with anti-TGF-β antibody for 2 h, and then incubated with PS-liposomes (125 μg of phospholipids/mL) for 18 h. Cell lysate was prepared and subjected to Western blotting as mentioned above.
and intense interest was focused on understanding the molecular details of the signaling pathway. Recently, Naiki et al. reported that TGF-β facilitates ubiquitination and proteasomal degradation of MyD88 and thereby attenuates MyD88-dependent signaling by decreasing cellular levels of MyD88 protein [32]. Ubiquitination and proteasomal degradation of MyD88 might be involved in the down-regulation induced by PS-liposomes. TLRs and MyD88 are central to the host response to bacterial infections such as Mycobacterium tuberculosis. In host defense against M. tuberculosis infection, lethal inflammation was observed in TGF-β-null mice, and uncontrolled inflammation may result directly from lack of inhibition of TLR signaling and sustained induction of pro-inflammatory gene targets [32]. PS-liposomes, which have the ability to suppress the production of inflammatory cytokines, may be a candidate for anti-inflammation treatment.
In conclusion, we examined the effect of PS-liposomes on expressions of TLR-4 and MyD88, which are essential for the signal transduction in LPS stimulation. MyD88 expression was suppressed when macrophages were treated with PS-liposomes; however, no change in TLR-4 expression was observed. MyD88 suppression was restored to the control levels when cells were pre-treated with anti-TGF-β antibody, suggesting that TGF-β plays an important role in the down-regulation of MyD88 following PS-liposome treatment.
REFERENCES
- R. E. Ellis, J. Y. Yuan and H. R. Horvitz, “Mechanisms and Functions of Cell Death,” Annual Reviews of Cell Biology, Vol. 7, 1991, pp. 663-698.
- C. B. Thompson, “Apoptosis in the Pathogenesis and Treatment of Disease,” Science, Vol. 267, No. 5203, 1995, pp. 1456-1462. doi:10.1126/science.7878464
- S. S. Bacus, A. V. Gudkov, M. Lowe, L. Lyass, Y. Yung, A. P. Komarov, K. Keyomarsi, Y. Yarden and R. Seger, “Taxol-Induced Apoptosis Depends on MAP Kinase Pathways (ERK and p38) and Is Independent of p53,” Oncogene, Vol. 20, No. 2, 2001, pp. 147-155. doi:10.1038/sj.onc.1204062
- Y. Fujiwara, K. Kawada, D. Takano, S. Tanimura, K. Ozaki and M. Kohno, “Inhibition of the PI3 Kinase/Akt Pathway Enhances Doxorubicin-Induced Apoptotic Cell Death in Tumor Cells in a p53-Dependent Manner,” Biochemical Biophysical Research Communications, Vol. 340, No. 2, 2006, pp. 560-566. doi:10.1016/j.bbrc.2005.12.039
- P. M. Henson and D. A. Hume, “Apoptotic Cell Removal in Development and Tissue Homeostasis,” Trends Immunology, Vol. 27, No. 5, 2006, pp. 244-250. doi:10.1016/j.it.2006.03.005
- V. A. Fadok, D. L. Bratton and P. M. Henson, “Phagocyte Receptors for Apoptotic Cells: Recognition, Uptake, and Consequences,” Journal of Clinical Investigation, Vol. 108, No. 7, 2001, pp. 57-962.
- C. N. Serhan and J. Savill, “Resolution of Inflammation: The Beginning Programs the End,” Nature Immunology, Vol. 6, No. 12, 2005, pp. 1191-1197. doi:10.1038/ni1276
- V. A. Fadok, D. L. Bratton, S. C. Frasch, M. L. Warner and P. M. Henson, “The Role of Phosphatidylserine in Recognition of Apoptotic Cells by Phagocytes,” Cell Deathand Differentiation, Vol. 5, No. 7, 1998, pp. 551- 562. doi:10.1038/sj.cdd.4400404
- J. Savilland C. Gregory, “Apoptotic PS to Phagocyte TIM-4: Eat Me,” Immunity, Vol. 27, No. 6, 2007, pp. 830-832. doi:10.1016/j.immuni.2007.12.002
- V. A. Fadok, D. L. Bratton, A. Konowal P. W. Freed, J. Y. Westcott and P. M. Henson, “Macrophages that Have Ingested Apoptotic Cells in Vitro Inhibit Proinflammatory Cytokine Production through Autocrine/Paracrine Mechanisms Involving TGF-β, PGE2, and PAF,” Journal of Clinical Investigation, Vol. 101, No. 4, 1998, pp. 890- 898. doi:10.1172/JCI1112
- V. A. Fadok, D. L. Bratton, D. M. Rose, A. Pearson, R. A. Ezekewitz and P. M. Henson, “Phosphatidylserine-Specific Clearance of Apoptotic Cells,” Nature, Vol. 405, No. 6782, 2000, pp. 85-90. doi:10.1038/35011084
- S. H. Schilling, “A Multipotential Cytokine,” In: B. Hjelmel, J. N. Rich and X. F. Wang, Eds., TGF-Family, Cold Spring Harbor, The TGF-Laboratory Press, New York, 2008, pp. 45-78.
- R. W. Vandivier, V. A. Fadok, P. R. Hoffmann, D. L. Bratton, C. Penvari, K. K. Brown, J. D. Brain, F. J. Accurso and P. M. Henson, “Elastase-Mediated Phosphatidylserine Receptor Cleavage Impairs Apoptotic Cell Clearance in Cystic Fibrosis and Bronchiectasis,” Journal of Clinical Investigation, Vol. 109, No. 5, 2002, pp. 661- 670.
- R. Matsuno, Y. Aramak and S. Tsuchiya, “Involvement of TGF-β in Inhibitory Effects of Negatively Charged Liposomes on Nitric Oxide Production by Macrophages Stimulated with LPS,” Biochemical Biophysical Research Communications, Vol. 281, No. 3, 2001, pp. 614-620. doi:10.1006/bbrc.2001.4419
- P. R. Hoffmann, J. A. Kench, A. Vondracek, E. Kruk, D. L. Daleke, M. Jordan, P. Marrack P. M. Henson and V. A. Fadok, “Interaction between Phosphatidylserine and the Phosphatidylserine Receptor Inhibits Immune Responses in Vivo,” Journal of Immunology, Vol. 174, No. 3, 2005, pp. 1393-1404.
- Y. Aramaki, F. Nitta, R. Matsuno, Y. Morimura and S. Tsuchiya, “Inhibitory Effects of Negatively Charged Liposomes on Nitric Oxide Production from Macrophages Stimulated by LPS,” Biochemical Biophysical Research Communications, Vol. 220, No. 1, 1996, pp. 1-6. doi:10.1006/bbrc.1996.0346
- M. Otsuka, K. Goto, S. Tsuchiya and Y. Aramaki, “Phosphatidylserine-Specific Receptor Contributes to TGF-β Production in Macrophages through a MAP Kinase, ERK,” Biological Pharmaceutical Bulletin, Vol. 28, No. 9, 2005, pp. 1707-1710. doi:10.1248/bpb.28.1707
- M. Otsuka, Y. Negishi and Y. Aramaki, “Involvement of Phosphatidylinositol-3-Kinase and ERK Pathways in the Production of TGF-β1 by Macrophages Treated with Liposomes Composed of Phosphatidylserine,” FEBS Letters, Vol.581, No. 3, pp. 25-330.
- P. J. Godowski, “A Smooth Operator for LPS Responses,” Nature Immunology, Vol. 6, No. 6, 2005, pp. 544-546. doi:10.1038/ni0605-544
- S. Akira and K. Takeda “Toll-Like Receptor Signaling,” Nature Review Immunology, Vol. 4, No. 7, 2004, pp. 499-511. doi:10.1038/nri1391
- T. Kawai, O. Adachi, T. Ogawa, K. Takeda and S. Akira, “Unresponsiveness of MyD88-Deficient Mice to Endotoxin,” Immunity, Vol. 11, No. 1, 1999, pp. 115-122. doi:10.1016/S1074-7613(00)80086-2
- Y. Aramaki, R. Matsuno and S. Tsuchiya, “Involvement of p38 MAP Kinase in the Inhibitory Effects of Phosphatidylserine Liposomes on Nitric Oxide Production from Macrophages Stimulated with LPS,” Biochemical Biophysical Research Communications, Vol. 280, No. 4, 2001, pp. 982-987. doi:10.1006/bbrc.2000.4204
- K. Takeda, T. Kaisho and S. Akira, “Toll-Like Receptors,” Annual Review Immunology, Vol. 21, 2003, pp. 335-376. doi:10.1146/annurev.immunol.21.120601.141126
- S. Das, K. Pandey, A. Kumar, A. H. Sardar, B. Purkait, M. Kumar, S. Kumar, V. N. Ravidas, S. Roy, D. Singh and P. Das, “TGF-β1 Re-Programs TLR4 Signaling in L. donovani Infection: Enhancement of SHP-1 and UbiquitinEditing Enzyme A20,” Immunological Cell Biology, Vol. 90, No. 6, 2011, pp. 640-654. doi:10.1038/icb.2011.80
- K. L. Jones, A. Mansell, S. Patella, B. J. Scott, P. Hedger, D. M. Kretse and D. J. Phillips, “Activin A Is a Critical Component of the Inflammatory Response, and Its Binding Protein, Follistatin, Reduces Mortality in Endotoxemia,” Proceedings of the National Academy of Sciences in USA, Vol. 104, No. 41, 2007, pp. 16239-16244.
- R. Matsuno, Y. Aramaki, H. Arima, Y. Adachi, N. Ohno, T. Yadomae and S. Tsuchiya, “Contribution of CR3 to Nitric Oxide Production from Macrophages Stimulated with High-Dose of LPS,” Biochemical Biophysical Research Communications, Vol. 244, No. 1, 1998, pp. 115- 119. doi:10.1006/bbrc.1998.8231
- C. G. Freire-de-Lima, Y. Q. Xiao, S. J. Gardai, D. L. Bratton, W. P. Schiemann and P. M. Henson, “Apoptotic Cells, through Transforming Growth Factor-Beta, Coordinately Induce Anti-Inflammatory and Suppress Pro-Inflammatory Eicosanoid and No Synthesis in Murine Macrophages,” Journal of Biological Chemistry, Vol. 281, No. 50, 2006, pp. 38376-38384. doi:10.1074/jbc.M605146200
- S. Nagata, “Apoptosis and Autoimmune Diseases,” Annals New York Academy of Sciences, Vol. 1209, pp. 10-16. doi:10.1111/j.1749-6632.2010.05749.x
- F. Bonnefoy, S. Perruche,M. Couturier, A. Sedrati, Y. Sun, P. Tiberghien, B. Gaugler and P. Saas, “Plasmacytoid Dendritic Cells Play a Major Role in Apoptotic Leukocyte-Induced Immune Modulation,” Journal of Immunology, Vol. 186, No. 10, 2011, pp. 5696-5705. doi:10.4049/jimmunol.1001523
- K. Köröskényi, E. Duró, A. Pallai, Z. Sarang, D. Kloor, D. S. Ucker, S. Beceiro, A. Castrillo, A. Chawla, C. A. Ledent, L. Fésüs and Z. Szondy, “Involvement of Adenosine A2A Receptors in Engulfment-Dependent Apoptotic Cell Suppression Ofinflammation,” Journal of Immunology, Vol. 186, No. 122, 2011, pp. 7144-7155.
- L. M. Stuart and R. A. B. Ezekowitz, “Phagocytosis: Elegant Complexity,” Immunity, Vol. 22, No. 5, 2005, pp. 539-550. doi:10.1016/j.immuni.2005.05.002
- Y. Naiki, K. S.Michelsen, W. Zhang, S. Chen, T. M. Doherty and M. Arditi, “Transforming Growth Factor-Beta Differentially Inhibits MyD88-Dependent, but Not TRAMand TRIF-Dependent, Lipopolysaccharide-Induced TLR4 Signaling,” Journal of Biological Chemistry, Vol. 280, No. 7, 2004, pp. 5491-5495. doi:10.1074/jbc.C400503200
NOTES
*Corresponding author.