Pharmacology & Pharmacy
Vol.3 No.2(2012), Article ID:18737,8 pages DOI:10.4236/pp.2012.32034
Synthesis and Screening of New 5-Substituted-1,3,4-oxadiazole-2-thioglycosides as Potent Anticancer Agents
![]()
1Green Chemistry Department, National Research Centre, Cairo, Egypt; 2Chemistry Department, Faculty of Science, Helwan University, Cairo, Egypt.
Email: gnawwar@yahoo.com
Received January 14th, 2012; revised February 26th, 2012; accepted March 8th, 2012
Keywords: 1,3,4-Oxadiazoles; Thioglycosides; Acyclic Sugars; Antitumor
ABSTRACT
A series of newly 1,3,4-oxadiazole-2-thioglycoside derivatives were synthesized. The key step of this protocol is the coupling between 5-herteroaryl-1,3,4-oxadiazole-2-thione and activated sugars (cyclic or acyclic sugar analogues) in the presence of basic medium. Among of the synthesized compounds, compounds 7, 10, 11 and 13 were screened for them in vitro anticancer activity against four human cancer cells. MCF-7 (Breast), HEPG2 (Liver), HCT116 (Colon) and HEP2 (Larynx) carcinoma cell lines with IC50 values ranging from 2.08 - 8.72 µg/well. Compounds 11 and 13 were highly specific and potent for four cell lines (MCF-7, HCT116, HEPG2 and HEP2).
1. Introduction
Among the various human diseases, cancer has proven to be one of the most intractable diseases to which humans are subjected, and as yet no practical and generally effective drugs or methods of control are available. According to the world health organization (WHO), cancer is an important health problem that claims the level of more than 7 million people world wide on an annual basis [1] Therefore, identification of novel potent, selective, and less toxic anticancer agents remains one of the most pressing health problems [1]. 1,3,4-oxadiazoles are an important class of heterocyclic compounds with wide range of biological activities such as anti-inflammatory [2-4], anticancer in various cancer cell lines [5-7], antiviral [8], antimicrobial [9], antineoplastic [10], fungicidal [11], inhibition of tyrosinase [12] and cathepsin k [13]. Carbohydrates are an important element in many natural antibiotics including bleomycin, neocarzinostatin chromophpre and other DNA-targeting drugs [14]. The role of the carbohydrate moiety includes enhancement of DNA sequence specificity and cell recognition [15]. Other properties augmented by carbohydrates are membrane permeability, water solubility, and chirality of chromophores or DNA damaging molecules. These characteristics suggest that introduction of carbohydrate moieties into synthetic drugs should generate hybrid molecules of considerable interest. Moreover, high levels of glucosylation are one of the many molecular changes that accompany malignant transformations. These changes are characteristic for cancer cells and can protect them from immune surveillance and chemotherapeutic agents, and enhance their metastatic capacity [16]. The carbohydrate moiety can be expected to play the role of drug carrier and improve the selectivity of compounds for cancerous cell lines. For example, vaccination using synthetic tumor associated antigens such as carbohydrate antigens, holds promise for generating a specific antitumor response by targeting the immune system to cancer cells [17].
According to the above mentioned facts, and in continuation of our interest in the synthesis of biologically and chemically valuable compounds [7,18,19], it seemed a good idea to prepare novel 1,3,4-oxadiazoles carrying thioglycosides in position 2 and various interesting heterocyclic moieties in position 5 and measure their anticancer activities in various cancer cell lines.
2. Results and Discussion
Chemistry
The synthesis of the target 1,3,4-oxadiazole-2-thione compounds containing various heterocyclic moieties at position 5 is depicted in Scheme 1. The selected hydrazide derivatives 2a, 2b were prepared according to literature [20]. Hydrazide derivatives 2c and 2d were prepared by reacting the ester derivatives 1c and 1d [21]
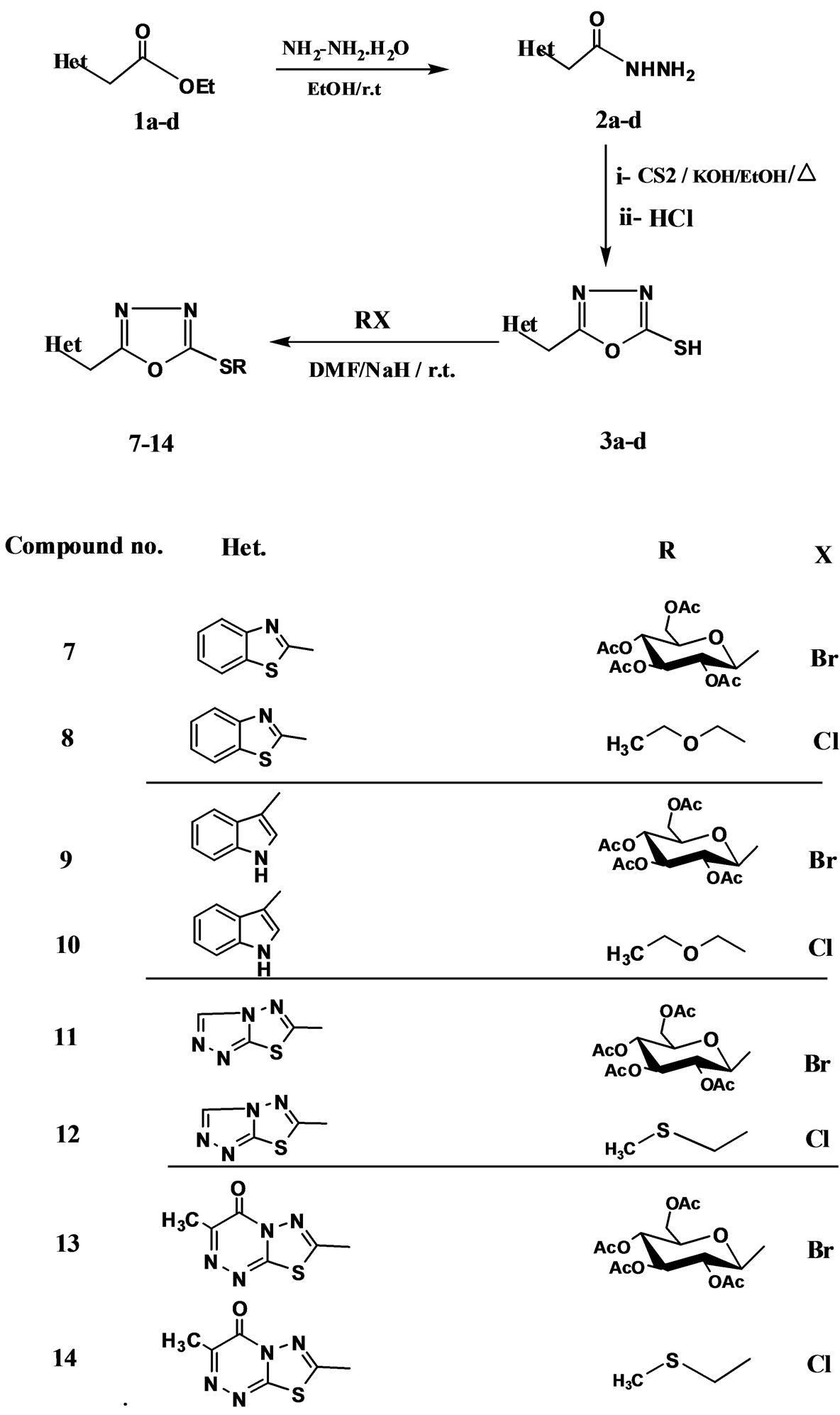
Scheme 1. Synthesis of 1,3,4-oxadiazole thioglycosides.
with hydrazine hydrate in absolute ethanol to yield 2c and 2d in good yields. To obtain the desired 1,3,4-oxadiazole derivatives 3a-d, compounds 2a-d were allowed to react with carbon disulphide in the presence of ethanolic potassium hydroxide followed by acidification with dilute hydrochloric acid to give the 1,3,4-oxadiazoles derivatives 3a [7,20], 3b-d in good yields. The structures of the oxadiazoles 3a-d were established and confirmed for the reaction products on the basis of their elemental analysis and spectral data (MS, IR, 1HNMR, 13C NMR (cf. experimental).
To achieve the S-glycosides, the aglycone 3a-d was converted to sodium salt's via reaction with NaH in dry DMF followed by addition of the activated cyclic sugars (2,3,4,6-tetra-O-acetyl-α-D-glucopyranosyl bromide 4, to give the corresponding thioglycoside derivatives 7, 9, 11, and 13 in a good yields, respectively.
The structures of thioglycosides 7, 9, 11 and 13 were established and confirmed for the reaction products on the basis of their elemental analysis and spectral data (MS, IR, 1HNMR, 13C NMR). Thus, their 1HNMR spectrum showed the anomeric proton as a doublet at a range δ = 5.65 - 5.84 ppm with a spin spin coupling constant (J1’,2’ > 9 Hz) corresponding to a trans orientation of H-1’ and H-2’ protons indicating the β-configuration. On the other hand, the formation of S-glycosides and not the corresponding N-glycosides were proved using 13CNMR spectroscopy which revealed the absence of a signal at δ 178 ppm for the thione carbon and the appearance of a signal at δ = 164 corresponding to the C-2 carbon, whose chemical shift is the same as that of the corresponding S-methyl derivative [22-24]. Moreover, acyclic thioglycosides analogues 8, 10, 12 and 14 were also achieved upon treatment of 3a-d with the activated acyclic sugar analogues, chloromethyl methylthioether (5), or chloromethyl ethyl ether (6) following the above mentioned reaction condition for preparing 7, 9, 11 and 13 where the same results (S-acyclicthioglycosides analogous) were obtained in fairly good yields (cf. experimental) and not N-acyclicglycosides.
3. Pharmacology
3.1. Materials and Methods
Potential cytotoxicity effect of the newly synthesized compounds and Tamoxifen and 5-Flurouracil a reference drugs in four concentrations, were evaluated in the National Institute of Cancer, Cairo Egypt by SRB assay [25]. Cells were plated in 96-multiwell plate (104 cells/well) for 24 h before treatment with the compounds to allow attachment of cell to the wall of plate. Different concentration of each compound under test (0, 1, 2.5, 5 and 10 µg/well) were added to the cell monolayer triplicate wells were prepared for each individual dose. Monolayer cells were incubated with the compound(s) for 48 h at 37˚C and in atmosphere of 5% CO2. After 48 h, cells were fixed, washed and stained with sulfo-rhodamine B strain.
Excess stain was washed with acetic acid and attached stain was recovered with tris EDTA buffer. Coor intensity was measured in an ELISA reader. Finally, the relation between surviving fraction and drug conc. is plotted to get the survival curve of each tumor cell line after the specified compound.
3.2. Anticancer Screening Studies
In order to study structure—activity relationship (SAR), four of the newly synthesized compounds were screened in vitro for their anticancer potential against four human cancer cell lines one dose assay has been done for compounds 7, 10, 11 and 13. The 4-cell lines used in the present investigation are MCF-7 (Breast), HEPG2 (Liver), HCT116 (Colon) and HEP2 (Larynx) carcinoma cell lines and. IC50 was calculated with regard to saline control group and potency was calculated with regard to percentage of change of tamoxifen or 5-flurouracil and tested compounds, the cell viability results of compounds 7, 10, 11, 13 are summarized in Table 1. The obtained results showing that the four tested compounds have moderate to excellent antitumor activity towards the four cell lines with IC50 values ranging from 2.12 - 4.07 (µg/well) for Breast cell line (MCF-7), 2.28 - 8.72 (µg/well) for Liver cell line (HEPG2), 3.21 - 3.71 (µg/well) for colon cell line (HCT116) and 2.08 - 3.76 (µg/well) for larynx cell line (HEP2) as depicted in (Figures 1-5).
Compared with both standard drugs, compound 11, 13 were highly specific and potent for four cell lines (MCF- 7, HEPG2, HCT116 and HEP2). Its seam that these activities attributed to the presence of skeleton which are very close to Labradorin 1 and Labradorin 2 [naturally occurring 5-(3’-indolylooxazoles] which were found to be cytotoxic against NCl-H460 (lung-NSC) human cancer cell line [26].
4. Toxicity Studies
4.1. Animals and Treatment
All the compounds tested for their anticancer activity 7, 10, 11 and 13 showed very good or moderate anticancer
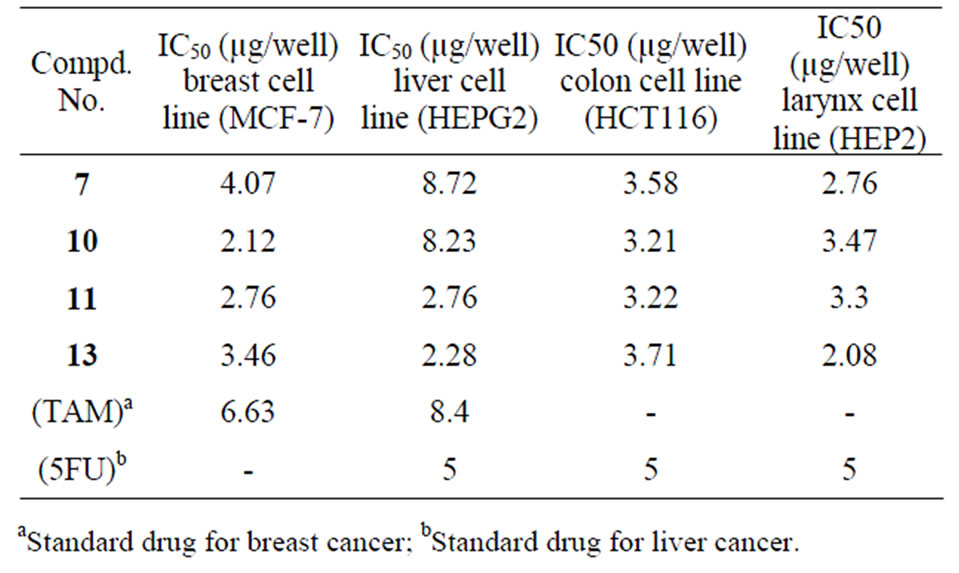
Table 1. Cytotoxicity of the synthesized candidates on breast (MCF-7), liver (HEPG2), colon (HCT116) and larynx (HEP2) cancer cell lines.
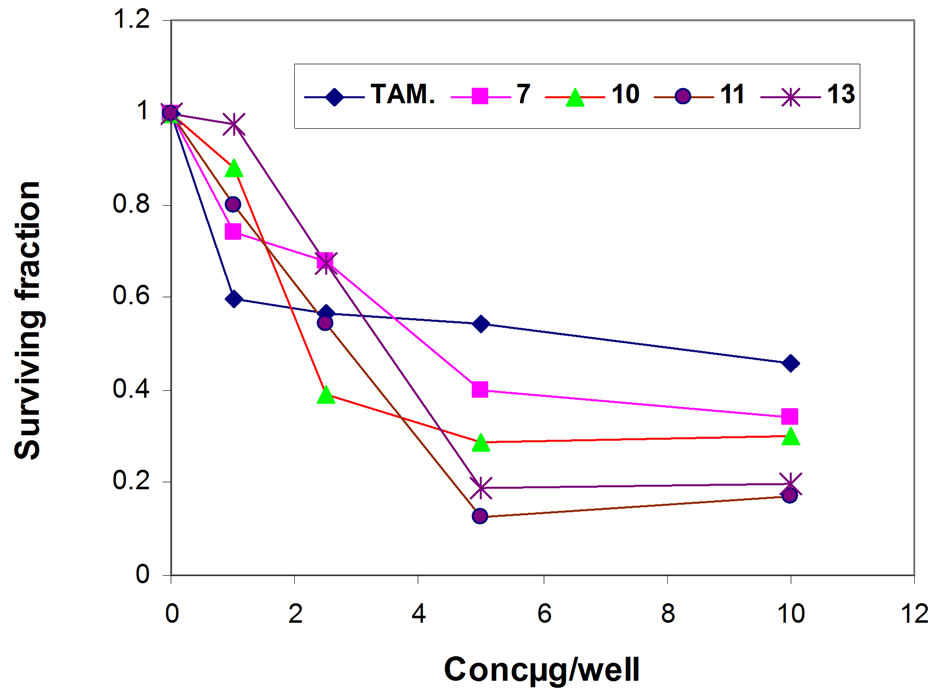
Figure 1. Representative graph showing survival of MCF-7 cell grown compared to TAM in the presence of increasing concentrations of compounds 7, 10, 11 and 13.
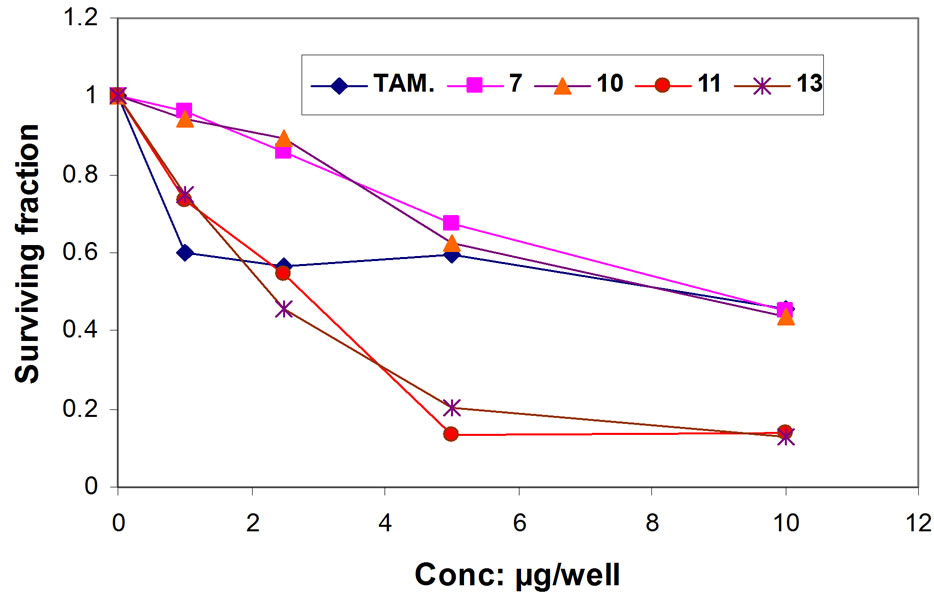
Figure 2. Representative graph showing survival of HEPG- 2 cell grown compared to TAM in the presence of increasing concentrations of compounds 7, 10, 11 and 13.
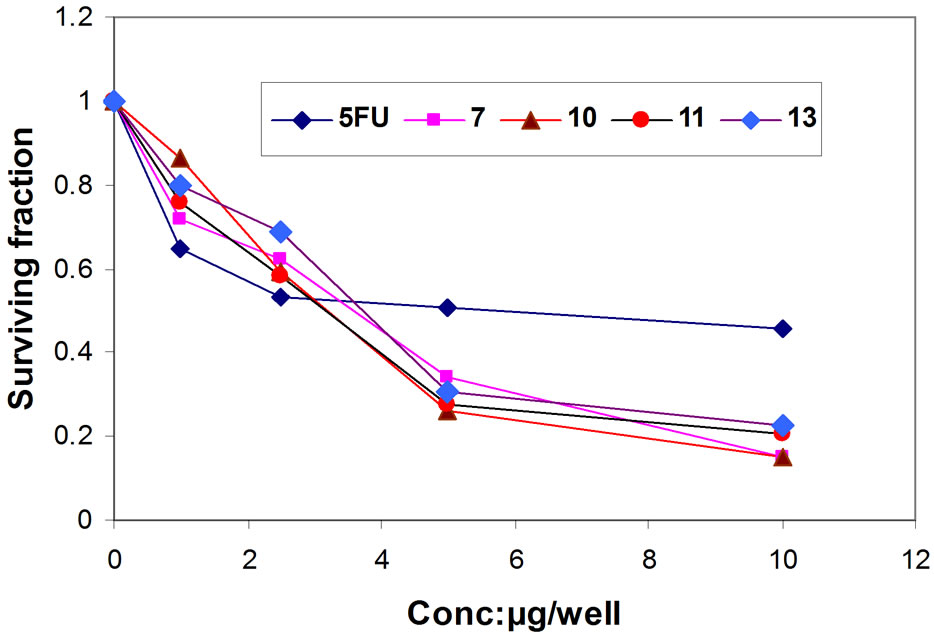
Figure 3. Representative graph showing survival of HCT- 116cell grown compared to 5FU in the presence of increasing concentrations of compounds 7, 10, 11 and 13.
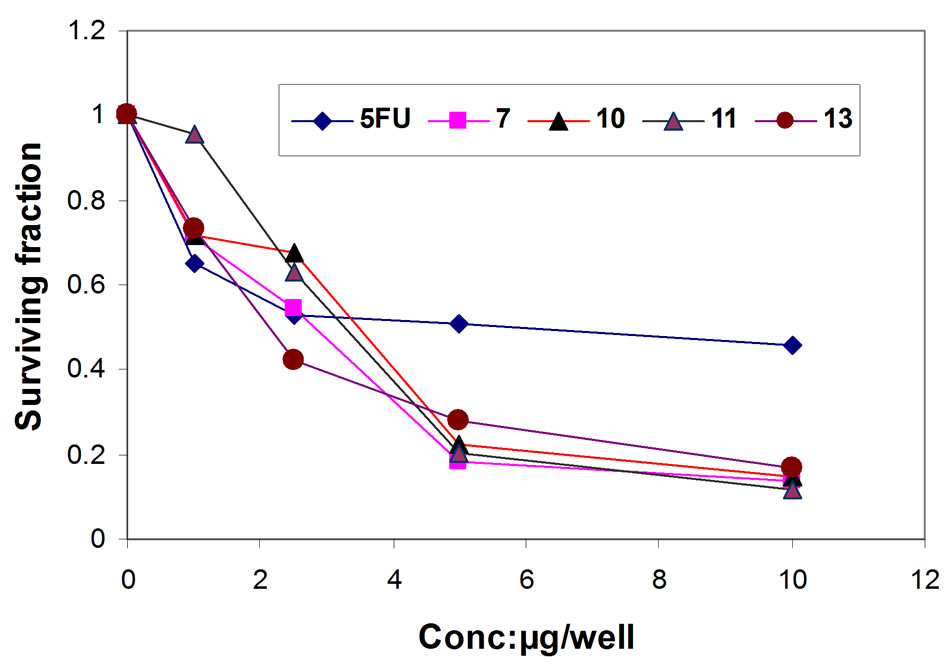
Figure 4. Representative graph showing survival of HEP-2 cell grown compared to 5FU in the presence of increasing concentrations of compounds 7, 10, 11 and 13.
activity in terms of growth inhibitory effect on cancer cell lines. Hence, it could be a potential drug candidate for cancer treatment. Hence this particular compounds were analyzed for its acute toxicity (lethal dose) which is
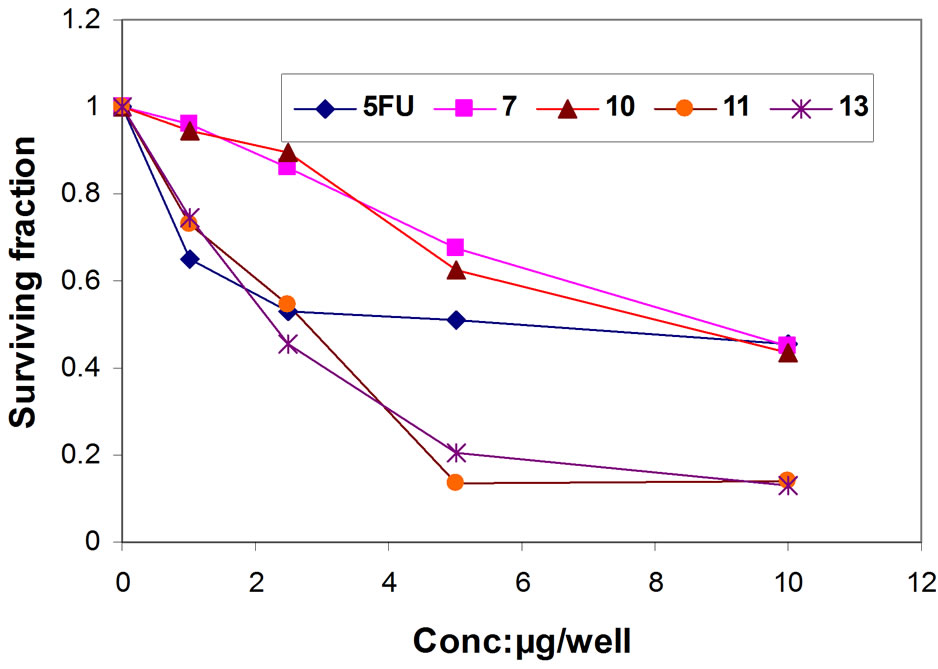
Figure 5. Representative graph showing survival of HEPG- 2 cell grown compared to 5FU in the presence of increasing concentrations of compounds 7, 10, 11 and 13.
one of the basic requirements in fixing the therapeutic dose in drug development. Male albino rats of the Wistar strain (Rattus norvegicus) weighing 110 - 115 g were obtained from Animal Breeding House of the National Research Centre (NRC), Dokki, Cairo, Egypt. Animals were housed in clean plastic cages in the laboratory animal room (23˚C ± 2˚C) on the standard pellet diet and tap water ad-libitum, a minimum relative humidity of 40% and a 12 h dark/light cycle. Rats were allowed to acclimate to laboratory conditions for 7 days prior to dosing. All compounds were dissolved in DMSO and administered by gavages at a fixed volume of 0.5 ml/rats. Animals were randomly divided into five groups, eight rats of each. Four groups were used for single dose treatments of compounds 7, 10, 11, and 13 at 600 mg/kg body weight, and one served as control (0.5 ml DMSO/rat). Then mortality of treated-rats was recorded after 24 h. The experimental protocols and procedures were approved by the Local Ethics Committee at the National Research Centre (NRC), Dokki, Cairo, Egypt, and confirmed with the Guide for the Care and Use of Laboratory Animals (NRC, 1999).
4.2. Acute Oral Toxicity
Our results in the present study showed that no mortality in rats traded with compound 13 while compounds 7, 10 and 11 caused 25%, 25% and 37.50% mortality at 500 mg/kg·b·wt, respectively. These results indicated that the lethal doses (LD50) of tested compounds are >600 mg/kg·b·wt and all compounds are consider a moderate toxicity based on oral LD50 values (no label, >600, <2000 mg/kg), which recommended by the Organization for Economic Co-operation and Development [24].
5. Conclusion
We have achieved the synthesis of 1,3,4-oxadiazole- 5-aryl derivatives having cyclic carbohydrate residues through S-glycosidic bond formation in position 2 or 1,3,4-oxadiazole-2-aryl derivatives having acyclic carbohydrate residues through S-glycosidic bond formation in position 5 an efficient manner. Pharmacological evaluation of compounds 7, 10, 11 and 13 against 4-cell lines MCF7 (breast), HEPG2 (liver), HCT116 (colon) and HEP2 (larynx) revealed them to possess high or moderate antitumor activities. Among the compounds tested for their anticancer activity 11, and 13 showed very good anticancer activity in terms of growth inhibitory effect on both cancer cell lines. Hence, it could be a potential drug candidate for cancer treatment. None of the tested compounds presented any toxicity in doses ranging from 50 to 600 mg·kg–1 of the animal body weight. The lethal doses (LD50) of tested compounds are >600 mg/kg·b·wt.
6. Experimental Protocols
6.1. Apparatus
All melting points were determined on an Electrothermal 9100 digital melting point apparatus. NMR and mass spectra were recorded on: 1H, 13C NMR spectra were determined using Jeol JMS-AX 500 MHz, Jeol GLM EX 270 MHz FT NMR spectrophotometer, DMSO-d6, TMS as internal standard, chemical shift in δ (ppm). Mass spectra were recorded on Varian MAT 311 A at 70 ev. Elemental analysis (in accord with the calculated values) were carried out in the microanalytical unit, Faculty of Science, Cairo University. Precoated silica gel 60 F254 plats with a layer thickness 0.25 nm from Merck were used for thin layer chromatography. Yields are not optimized.
6.2. General Method for the Synthesis of Hydrazide Derivatives 2c and 2d
To a stirred, cooled solution of 1c or 1d (10 mmole) in 10 ml absolute ethanol, hydrazine hydrate (25 mmole) was added drop wisely within 30 min. and the stirring was continued for another 1 hr. the yellow crystals which was formed during stirring, filtered, washed with ethanol and crystallized from methanol to give.
6.2.1. 2-([1,2,4]Triazolo[3,4-b][1,3,4]thiadiazol-6-yl)- acetohydrazide (2c)
Yellow crystals (methanol); yield (76%); m.p. 185˚C; IR (KBr, cm–1) υ 3450, 3245 (NH2), 3133 (NH), 3028 (CH), 1666 (CO), 1632 (C=N); 1HNMR (DMSO-d6): δ 4.02 (s, 2H, CH2), 4.39 (brs., 2H, NH2), 9.50 (s, 1H, triazole CH), 9.55 (brs., 1H, NH). 13C NMR: δ 36.12 (CH2), 136.68 (triazole C-5), 162.83 (triazoleC-3), 163.98 (thiazole C-2), 168.11 (C=O), ppm, MS: m/z (M+, 198, 40%). Anal. Calcd. For. C5H6N6OS (198.21): C, 30.30; H, 3.05; N, 42.30. Found: C, 30.35; H, 3.15; N, 42.40.
6.2.2. 2-(3-Methyl-4-oxo-4H-[1,3,4]thiadiazolo [2,3-c][1,2,4]Triazin-7-yl)acetohydrazide (2d)
Colorless crystals (methanol); yield (80%); m.p. 200˚C - 201˚C; IR(KBr, cm−1) υ 3392, 3300 (NH2), 3190 (NH), 2918 (CH), 1695, 1670 (2CO); 1H NMR (DMSO-d6): δ 2.35 (s, 3H, CH3), 4.48 (s, 2H, CH2), 4.79 (brs., 2H, NH2), 9.61 (s, 1H, NH); 13CNMR: δ 18.97 (CH3), 37.9 1(CH2), 155.81 (thiadiazole-C-2), 158.42 (triazine C-6), 164.52 (triazine C-3), 168.42 (triazine C-5), 170.54 (C=O). Anal. Calcd. For. C7H8N6O2S (240.24): C, 35.10; H, 3.25; N, 34.90. Found: C, 35.00; H, 3.36; N, 34.98.
6.3. General Method for the Synthesis of 1,3,4-Oxadiazole Derivatives 3b-d
To a solution of compounds 2a-d (10 mmol) in ethanolic potassium hydroxide (20 mL), carbon disulfide (2 mL) was dropped in over period of half hour at room temperature (25˚C). After an additional half hour of stirring, the reaction mixture was refluxed until the evaluation of H2S gas stop, a precipitate was formed and collected by filtration. The salt formed was dissolved in water, then acidify with HCl the precipitate thus formed was filtered off and crystallized from methanol to give:
6.3.1. 5-((1H-Indol-3yl)methyl)-1,3,5-oxadiazole-2- (3H)-thione (3b)
Yellow solid (methanol); yield (70%); m.p. 240˚C; IR (KBr, cm−1) υ 3220, 3135 (2NH), 2988 (CH), 1607 (C=N), 1320 (C=S); 1HNMR (DMSO-d6): δ 4.11 (s, 2H, CH2), 7.25 - 7.40 (m, 5H, 4 Ar-H, NH), 8.20 (s, 1H, indole H-2), 11.12 (brs., 1H, indoleNH); 13CNMR: δ 35.34 (CH2), 106.25 (indoleC-3), 112.21, 118.61, 119.45, 121.98 (4C, Ar-C), 125.00 (indole C-2), 128.85 (indole C-4), 136.67 (indole C-5), 164.05 (C=N oxadiazole) 178.30 (C=S). Anal. Calcd. For. C11H9 N3 O S (231.27): C, 57.13; H, 3.92; N, 18.17. Found: C, 57.25; H, 3.90; N, 18.10.
6.3.2. 5-([1,2,4]Triazolo[3,4-b][1,3,4]thiadiazol-6- ylmethyl)-1,3,4-oxadiazole-2(3H)-thione (3c)
Yellow solid (methanol); yield (72%); m.p. 170˚C; IR (KBr, cm−1) υ 3440 (NH), 3030 (CH), 1618 (C=N), 1320 (C=S); 1HNMR (DMSO-d6): δ 4.05 (s, 2H, CH2), 7.11 (s, 1H, NH), 9.45 (s, 1H, triazole H-5); 13CNMR: δ 35.78 (CH2), 138.65 (triazole C-5), 156.25 (thiadiazole C-2), 158.21 (oxadiazole C=N), 168.43 (triazole C-3), 178.35 (C=S). Anal. Calcd. For. C6 H 4N6OS2 (240.27): C, 29.99; H, 1.68; N, 34.98. Found: C, 29.75; H, 1.60; N, 34.92.
6.3.3. 3-Methyl-7-((5-thioxo-4,5-dihydro-1,3,4- oxadiazol-2-yl)methyl)-4H-[1,3,4]thiadiazolo [2,3-c][1,2,4]triazin-4-one(3d)
Yellow solid, yield (methanol); (79%), m.p. 178˚C; IR (KBr, cm−1) υ 3020, 2980 (CH), 1685 (CO), 1610 (C=N), 1325 (CS); 1H NMR (DMSO-d6): δ 2.60 (s, 3H, CH3), 4.20 (s, 2H, CH2), 7.56 (s, 1H, NH). 13C NMR: δ 18.65 (CH3), 37.53 (CH2), 154.63 (thiazole C-2), 154.68 (oxadiazole C-5), 158.23 (triazine C-6), 163.24 (triazine C-3), 166.13 (C=O), 178.33 (C=S). Anal. Calcd. For., C8H6N6O2S2 (282.30): C, 34.04; H, 2.14; N, 29.77. Found: C, 34.14; H, 2.10; N, 29.60.
6.4. General Method for the Synthesis of Thioglycosides 7-14
To a solution of compounds 3a-d (10 mmol) in dry DMF (20 ml), NaH (15 mmol) was added portion wise through 15 min. and the solution stirred at room temperature for another 30 min. Then, a solution of 2,3,4,6-tetra-O-acetyl-α-D-glucopyronosyl bromide (4), or chloromethyl ethyl ether (5), or chloromethyl methylthioether (6) in DMF (10 mL) was dropped within 30 min. and the reaction mixture was stirred at room temperature until completion (TLC, 10 - 12 h). After completion, the reaction mixture was poured on H2O and acidified with diluted acetic acid . the aqueous phase was extracted with ethyl acetate (3 × 20 mL), the combined organic phase was washed with water dried over anhydrous sodium sulphate. Removal of solvent gave a residue, which was purified by column chromatography using an appropriate solvent system to give compounds 7-14 respectively.
6.4.1. 2-(2,3,4,6-Tetra-O-acetyl-β-Dglucopyranosyl thio)-5-(benzo[d]thiazol- 2-ylmethyl)-1,3,4-oxadiazole (7)
Colorless crystals (ethanol); yield (72%); m.p. 210˚C; 1H NMR (DMSO-d6): δ 1.90 - 2.01 (4s, 12H, 4x OAc), 2.97 (s, 2H, CH2), 3.96 (dd, 1H, J = 2.2, 12.2 Hz, 6’H), 4.02 - 4.14 (m,5’-H ,6”H), 5. 18 (t, 1H, J3’,4’ = 9.95 Hz, 4’-H), 5.35, (t, J2’,3’ = 9.55Hz, 2’-H), 5.55 (t, 1H, J =9.19 Hz, 3’-H), 5.77 (d, 1H, J1’,2’ = 9.55 Hz, 1’-H), 7.3 - 7.81 (m, 5H, Ar-H); 13C NMR: δ 22.66 (CH3), 30.94 (CH2), 61.21 (C-4’) ,65.71 (C-6’), 67.30 (C-3’), 73.02 (C-2’)), 76.86 (C-5’), 81.87 (C-1’), 121.75, 121.94, 124.92, 125.50 (benzothiazole C-4, C-7, C-6, C-5), 135.82 (benzothiazole C-9),153.42 (benzothiazole C-8), 166.41 (oxadiazole C-5), 167.32 (benzothiazole C-2), 168.52 (oxadiazole C-2) 169.21 - 170.10 (4C=O). Anal. Calcd. For., C24H 25N 3O10S2 (579.60): C, 49.73; H, 4.35; N, 7.25. Found: C, 49.65; H, 4.25; N, 7.10.
6.4.2. 2-(Benzo[d]thiazol-2-ylmethyl)-5- (ethoxymethylthio)-1,3,4-oxadiazole (8)
Colorless crystals(ethanol); yield (60%); m.p. 80˚C; 1H NMR (DMSO-d6): δ 1.30 (t, 3H, CH3), 3.65 (q, 2H CH2), 3.92 (s, 2H, CH2), 5.45 (s, 2H, CH2), 7.55 - 7.70 (m, 2H, Ar-H), 8.10 - 8.15 (m, 2H, Ar-H); 13C NMR : δ 14.60 (CH3), 34.32 (CH2), 66.82 (CH2), 75.71 (CH2), 121.6, 121.8, 124.7, 125.6, 135.5, 151.98 (6C Ar-C), 165.4 (benzothiazole C-2), 166.5 (oxadiazole C-2), 166.70 (oxadiazole C-5). Anal. Calcd. For. C13H13N3O2S2 (307.39): C, 50.79; H, 4.26; N, 13.67. Found: C, 50.60; H, 4.20; N, 13.50.
6.4.3. 2-(2,3,4,6-Tetra-O-acetyl-β-Dglucopyranosylthio)-5-((1H-indol-3-yl)methyl)- 1,3,4-oxadiazole (9)
Colorless crystals (ethanol); yield (69 %); m.p. 190˚C; IR (KBr, cm−1) υ 3397 (NH), 3020, 2928 (CH), 1734 (CO). 1H NMR (500MH CDCl3): δ 2.00 - 2.07 (m, 12H, 4x OAc), 3.96 (m, 2H, H-6’,H-6”), 4.16 (S, 2H, CH2) 4.18 (m, 1H, H-5’), 5.22 (t,1H, H -4’), 5.35 (t, 1H, H-2’), 5.59 (t, 1H, H-3’) ,5.84 (d, 1H, J1,2 = 9.15, H-1’), 7.25 -7.36 (m, 4H, Ar-H) 7.56 (s, 1H , indole H-2), 8.2 (s, IH, NH); 13C NMR: δ 20.48 - 22.68 (CH3), 27.08 (CH2), 61.40 (C-6’), 67.48 (C-4’), 69.14 (C-2’), 73.14 (C-3’), 74.74 (C-5’), 82.69 (C-1’), 105.93, 111.40, 118.8, 120.08, 122.74, 123.477, 126.56, 136.02 (8C, indole), 166.21 (oxadiazoleC-5), 166.5 (oxadiazole C-2), 169.4 - 170.4 (4C=O). Anal. Calcd. For. C25H27N3O10S (561.56): C, 53.47; H, 4.85; N, 7.48. Found: C, 53.35; H, 4.70; N, 7.35.
6.4.4 2-((1H-Indol-3-yl) methyl)-5- ((ethoxymethyl)thio)-1,3,4-oxadiazole (10)
Colorless crystals (ethanol); yield (70%); m.p. 120˚C; IR (KBr, cm−1) υ 3338 (NH), 3060 (CH-aromatic), 2920 (CH-aliphatic); 1HNMR (DMSO-d6): δ 1.67 (t, 3H, CH3), 3.70 (q, 2H, CH2), 4.22 (s, 2H, CH2), 5.64 (s, 2H, CH2), 6.98, 7.33 - 7.86 (m, 5H, Ph-H), 7.92 (s, 1H, indole H-2), 11.04 (brs., 1H, NH); 13C NMR: δ 15.12 (CH3), 22.31 (CH2), 67.12 (CH2), 76.11 (CH2), 106.28 (indole C-3), 112.19 - 121.92 (4C,Ar-C), 125.01 (indole C-2), 127.12 (indole C-8), 136.71 (indole C-9), 162.84 (oxadiazole C-2), 163.98 (oxadiazole C-5). Anal. Calcd. For. C14H15N3O2S (289.35): C, 58.11; H, 5.23; N, 14.52. Found: C, 58.20; H, 5.15; N, 14.45.
6.4.5. 2-(2,3,4,6-Tetra-O-aceyl-β-Dglucopyranosylthio)-5- ([1,2,4]triazolo[3,4-b][1,3,4]thiadiazol-6- ylmethyl)-1,3,4-oxadiazole (11)
Colorless crystals (ethanol); yield (65%); m.p. 170˚C; 1HNMR (CDCl3): δ 2.00 - 2.11 (m, 12H, 4x OAc), 3.92 (s, 2H, CH2), 4.11 - 4.20 (m, 2H, H-6’,6”), 4.34 (m, 1H, H-5’), 5.20 (t, 1H, H -4’), 5.35 (t, 1H, H-2’), 5.58 (t, 1H, H-3’), 5.83 (d , 1H, J1,2 = 9.35, H-1’), 8.45 (triazolo H-5); 13CNMR: δ 20.87 - 21.03 (4CH3), 31.29(CH2), 62.19 (C-6’), 68.37 (C-4’), 68.56 (C-2’), 69.90 (C-3’), 73.02 (C-5’), 82.69 (C-1’), 142.59 (triazoleC-5), 162.86 (thiadiazoleC-2), 166.29 (oxadiazoleC-5), 169.80 (triazole C-3), 170.20 - 170.53 (4C=O). MS: m/z (M+, 570, 14%). Anal. Calcd. For. C20H22N6O10S2 (570.55): C, 42.10; H, 3.89; N, 14.73. Found: C, 42.23; H, 3.77; N, 14.68.
6.4.6. 2-([1,2,4]Triazolo[3,4-b][1,3,4]thiadiazol- 6-ylmethyl)-5-(methylthiomethylthio)-1,3,4- oxadiazole (12)
Colorless crystals (ethanol); yield (80%), mp 90˚C - 91˚C; 1H NMR (DMSO-d6): δ 2.20 (s, 3H, CH3), 3.95 (s, 2H, CH2), 4.11 (s, 2H, CH2), 9.35 (s, 1H, triazolo H-5). 13CNMR:, δ 14.5 (CH3), 30.10 (CH2), 42.29 (CH2), 138.54 (triazole C-5), 162.22 (thiadiazole C-2), 168.23 (oxadiazole C-2), 169.14 (triazole C-3), 169.52 (oxadiazole C-5). MS: m/z (M+ = 300, 45%). Anal. Calcd. For. C8H8N6OS3 (300.38): C, 31.99; H, 2.68; N, 27.98. Found: C, 31.85; H, 2.52; N, 27.90.
6.4.7. 2-(2,3,4,6-Tetra-O-aceyl-β-Dglucopyranosylthio)-(3-methyl-4-oxo-4H-[1,3,4] thiadiazolo[2,3-c][1,2,4]triazin-7-yl)methyl)- 1,3,4-oxadiazole (13)
Colorless crystals (ethanol); yield (75%), mp 230˚C; 1H NMR (DMSO-d6): δ 2.05 - 2.07 (4S, 12H, 4x OAc), 2.31 (s, 3H, CH3), 4.01 (s, 2H, CH2), 4.16 - 4.18 (m, 2H, 26’-H), 4.36 (m, 1H, 5’ H), 5.22 (t, 1H, 4’-H), 5.35 (t, 2’-H), 5.59 (t, 1H, 3’-H), 5.83 (d, 1H, J1’,2’ = 9.15 Hz, 1’-H). 13C NMR: δ 18.11 (CH3), 21.52 (CH3), 32.34 (CH2), 62.21 (C-4’), 65.79 (C-6’), 67.30 (C-3’), 70.60 (C-2’)), 75.20 (C-5’), 82.70 (C-1’), 146.51 (thiadiazole C-2), 157.43 (triazine C-6), 157.84 (oxadiazoleC-5), 162.55 (triazineC-3), 164.15 (oxadiazole C-2), 166.34 (triazine C-5), 169.16 - 170.82 (4C=O). Anal. Calcd. For C22H24N6O11S2 (612.59): C, 43.13; H, 3.95; N, 13.72. Found: C, 43.20; H, 3.90; N, 13.60.
6.4.8. 3-Methyl-7-((5-(((methylthio)methyl)thio)- 1,3,4-oxadiazol-2-yl)methyl)-4H-[1,3,4]- thiadiazolo[2,3-c][1,2,4]triazin-4-one (14)
Colorless crystals (ethanol); yield (80%); mp 110˚C; 1H NMR (DMSO-d6): δ 2.25 (s, 3H, CH3), 2.72 (s, 3H CH3), 4.20 (s, 2H, CH2), 4.88 (s, 2H,CH2), 13C NMR: δ 14.86 (CH3), 32.24 (CH2), 42.26 (CH2), 146.51 (thiadiazole C-2), 157.43 (triazine C-6), 157.84 (oxadiazole C-5), 163.42 (triazine C-3), 164.15 (oxadiazole C-5), 166.34 (triazineC-5) MS: m/z (M+ = 342, 74%). Anal. Calcd. For C10H10N6O2S3 (342.42): C, 35.08; H, 2.94; N, 24.54. Found: C, 35.18; H, 2.86; N, 24.48.
REFERENCES
- S. Eckhardt, “Recent Progress in the Development of Anti Cancer Agents,” Current Medicinal Chemistry, Vol. 2, No. 3, 2002, pp. 419-439.
- V. Jakubkiene, M. M. Burbuliene, C. Mekuskiene, E. Udrenaite, P. gaidelis and P. Vainilavicius, “Synthesis and Anti-Inflammatory Activity of 5-(6-Methyl-2-substituted 4-Pyrimidinyloxymethyl)-1,3,4-oxadiazole-2-thiones and Their 3-Morpholinomethyl Derivatives,” IL Farmaco, Vol. 58, No. 4, 2003, pp. 323-328. doi:10.1016/S0014-827X(02)00022-8
- E. Palaska, G. Sahin, P. Kelicen, N. T. Durlu and G. Altinok, “Synthesis and Anti-Inflammatory Activity of 1-Acylthiosemicarbazides, 1,3,4-Oxadiazoles, 1,3,4-Thiadiazoles and 1,2,4-Triazole-3-thiones,” IL Farmaco, Vol. 57, No. 2, 2002, pp. 101-107. doi:10.1016/S0014-827X(01)01176-4
- B. Jayashankar, K. M. L. Rai, N. Baskaran and H. S. Sathish, “Synthesis and Pharmacological Evaluation of 1,3,4-Oxadiazole Bearing Bis(heterocycle)derivatives as Anti-Inflammatory and Analgesic Agents,” European Journal of Medicinal Chemistry, Vol. 44, No. 10, 2009, pp. 3898-3902. doi:10.1016/j.ejmech.2009.04.006
- D. Kumar, S. Sundaree, E. O. Johnson and K. Shah, “An Efficient Synthesis and Biological Study of Novel Indolyl-1,3,4-oxadiazoles as Potent Anticancer Agents,” Bioorganic & Medicinal Chemistry Letters, Vol. 19, No. 15, 2009, pp. 4492-4494. doi:10.1016/j.bmcl.2009.03.172
- W. S. Li, S. V. More, C. H. Wang, Y. C. Jen, C. F. Yao, T. F. Wang, C. C. Hung and S. C. Jao, “Synthesis and Structure-Activity Relationships of Novel Furazan-3,4- diamide Analogs as Potent Anti-Cancer Agents,” Bioorganic & Medicinal Chemistry Letters, Vol. 20, No. 3, 2010, pp. 1148-1152. doi:10.1016/j.bmcl.2009.12.017
- M. A. Abu-Zaied, E. M. El-Telbani, G. H. Elgemeie and G. A. M. Nawwar, “Synthesis and in Vitro Anti-Tumor Activity of New Oxadiazole Thioglycosides,” European Journal of Medicinal Chemistry, Vol. 46, No. 1, 2011, pp. 229-235. doi:10.1016/j.ejmech.2010.11.008
- T. M. C. Tan, Y. Chen, K. H. Kong, J. Bai, Y. Li, S. G. Lim, T. H. Ang and Y. Lam, “Synthesis and the Biological Evaluation of 2-Benzenesulfonylalkyl-5-substitutedsulfanyl-[1,3,4]-oxadiazoles as Potential Anti-Hepatitis B Virus Agents,” Antiviral Research, Vol. 71, No. 1, 2006, pp. 7-14. doi:10.1016/j.antiviral.2006.02.007
- M. A. Ali and M. Shaharyar, “Oxadiazole Mannich Bases: Synthesis and Antimycobacterial Activity,” Bioorganic & Medicinal Chemistry Letters, Vol. 17, No. 12, 2007, pp. 3314-3316. doi:10.1016/j.bmcl.2007.04.004
- A. S. Aboraia, H. M. Abdel-Rahman, N. M. Mahfouz and M. A. El-Gendy, “Novel 5-(2-Hydroxyphenyl)-3-substituted-2,3-dihydro-1,3,4-oxadiazole—2-Thione Derivatives: Promising Anticancer Agents,” Bioorganic & Medicinal Chemistry Letters, Vol. 14, 2006, pp. 1236-1246. doi:10.1016/j.bmc.2005.09.053
- Y. Li, J. Liu, H. Zhang, X. Yang and Z. Liu, “Stereoselective Synthesis and Fungicidal Activities of (E)-Alpha- (me-thoxyimino)-benzeneacetate Derivatives Containing 1,3,4-Oxadiazole Ring,” Bioorganic & Medicinal Chemistry Letters, Vol. 16, No. 8, 2006, pp. 2278-2282. doi:10.1016/j.bmcl.2006.01.026
- M. T. H. Khan, M. I. Choudhary, K. M. Khan, M. Rani and A. U. Rahman, “Structure-Activity Relationships of Tyrosinase Inhibitory Combinatorial Library of 2,5-Disubstituted-1,3,4-oxadiazole Analogues,” Bioorganic & Medicinal Chemistry, Vol. 13, No. 6, 2005, pp. 3385- 3395. doi:10.1016/j.bmc.2005.03.012
- J. T. Palmer, B. L. Hirschbein, H. Cheung, J. McCarter, J. W. Janc, Z. W. Yu and G. Wesolowski, “Keto-1,3,4- oxadiazoles as Cathepsin K Inhibitors,” Bioorganic & Medicinal Chemistry Letters, Vol. 16, No. 11, 2006, pp. 2909-2914. doi:10.1016/j.bmcl.2006.03.001
- G. Pratviel, J. Bernadou and B. Meunie, “Carbon-Hydrogen Bonds of DNA Sugar Units as Targets for Chemical Nudeases and Drugs,” Angewandte Chemie International Edition, Vol. 34, 1995, pp. 746-769. doi:10.1002/anie.199507461
- R. A. Dwek, “Glycobiology
 toward Understanding the Function of Sugars,” Chemical Reviews, Vol. 96, No. 2, 1996, pp. 683-720. doi:10.1021/cr940283b
toward Understanding the Function of Sugars,” Chemical Reviews, Vol. 96, No. 2, 1996, pp. 683-720. doi:10.1021/cr940283b - J. Kralora, J. Koivukorpi, Z. Kejik, P. PoucKova, E. Sievanen, E. Kolehmainen and V. Kral, “Porphyrin-Bile Acid Conjugates: From Saccharide Recognition in the Solution to the Selective Cancer Cellfluorescence Detection,” Organic & Biomolecular Chemistry, Vol. 6, No. 9, 2008, pp. 1548-1552. doi:10.1039/b717528k
- T. Minko, S. S. Dharap, R. I. Pakunlu and Y. wang, “Molecular Targeting of Drug Delivery Systems to Cancer,” Current Drug Targets, Vol. 5, No. 4, 2004, pp. 389- 406. doi:10.2174/1389450043345443
- H. N. Hafez, A. B. A. El-Gazzar and G. A. M. Nawwar, “Synthesis, Biological and Medicinal Significance of S-Glycosido-thieno[2,3-d]-pyrimidines as New Anti-Inflammatory and Analgesic Agents,” European Journal of Medicinal Chemistry, Vol. 45, No. 4, 2010, pp. 1485- 1493. doi:10.1016/j.ejmech.2009.12.056
- H. N. Hafez, A. B. El-GAzzar and G. A. M. Nawwar, “New Acyclic Nucleosides Analogues as Potential Analgesic, Anti-Inflammatory, Anti-Oxidant and Anti-Microbial Derived from Pyrimido[4,5-b]quinolines,” European Journal of Medicinal Chemistry, Vol. 44, No. 4, 2009, pp. 1427-1436. doi:10.1016/j.ejmech.2008.09.030
- E. S. M. Yakout, Y. A. Allam and G. A. M. Nawwar, “Simple Syntheses of Benzothiazol-2-Ylazoles,” Heteroatom Chemistry, Vol. 10, No. 3, 1999, pp. 177-182. doi:10.1002/(SICI)1098-1071(1999)10:3<177::AID-HC1>3.0.CO;2-D
- E. M. El-Telbani, R. H. Swellem and G. A. M. Nawwar, “Facile Synthesis of 6-Heteroaryl [1,2,4] Triazolo [3,4- b][1,3,4] Thiadiazoles and 7-Hetaryl[1,3,4] Triadiazolo [2,3-c][1,2,4]thiazines with Fungicidal Activity,” Russian Journal of Organic Chemistry, Vol. 43, No. 12, 2007, pp. 1815-1820. doi:10.1134/S1070428007120135
- A. M. Attia and G. H. Elgemeie, “A New Class of Dihy Dropyridine Thioglycosides via Piperidinium Salts,” Synthetic Communications, Vol. 33, No. 13, 2003, pp. 2243- 2255. doi:10.1081/SCC-120021503
- I. W. J. Still, N. Plavac, D. M. Mackinnon and M. S. Chauhan, “Carbon-13 Nuclear Magnetic Resonance Spectra of Organic Sulfur Compounds. Comparison of Chemical Shifts for Carbonyl and Thiocarbonyl Compounds in the Pyrone, Thiopyrone, and Pyridone Series,” Canadian Journal of Chemistry, Vol. 54, No. 2, 1976, pp. 280-289. doi:10.1139/v76-042
- L. Stefaniak, “14N and 13C NMR of Tautomeric Systems of Mercaptoand Amino-Pyridines,” Organic Magnetic Resonance, Vol. 12, No. 6, 1979, pp. 379-382. doi:10.1002/mrc.1270120611
- P. Skehan, R. Strong, D. Scadiaro, A. Monks, J. McMahon, D. Vistica, J. T. Warren, H. Bokesch, S. Kenney and M. R. Boyed, “New Colorimetric Cytotoxicity Assay for Anticancer-Drug Screening,” Journal of the National Cancer Institute, Vol. 82, No. 13, 1990, pp. 1107-1112. doi:10.1093/jnci/82.13.1107
- G. R. Pettit, J. C. Knight, D. L. Herald, R. Davenport, R. K. Pettit, B. E. Tucker and J. M. Schmidt, “Isolation of Labradorins 1 and 2 from Pseudomonas Syringae pv. Coronafaciens,” Journal of Natural Products, Vol. 65, No. 12, 2002, pp. 1793-1797. doi:10.1021/np020173x