Paper Menu >>
Journal Menu >>
 Vol.2, No.4, 286-290 (2010) Health doi:10.4236/health.2010.24041 Copyright © 2010 SciRes. Openly accessible at http://www.scirp.org/journal/HEALTH/ Coexistence of Duarte 1 and Duarte 2 variants of galactosemia with extrahepatic biliary atresia* Ramandeep Singh1, Ketan Kulkarni2, Gurjit Kaur3, Babu Ram Thapa2, Rajendra Prasad1 1Department of Biochemistry, Postgraduate Institute of Medical Education and Research, Chandigarh, India 2Division of Pediatric Gastroenterology, Postgraduate Institute of Medical Education and Research, Chandigarh, India 3Department of Physiology, Government Medical College and Hospital, Chandigarh, India; fateh1977@yahoo.com Received 3 December 2009; revised 11 January 2010; accepted 15 January 2010. ABSTRACT Galactosemia is an autosomal recessive disor- der caused by deficient or absent activities of one of the three enzymes involved in the ga- lactose metabolic pathway. The predominant form is classic type galactosemia caused by severe reduction or absence of the galactose- 1-phosphate uridyl transferase (GALT) enzyme. Coexistence of extrahepatic biliary atresia (EHBA) with Duarte 1 and 2 variants of galacto- semia has not been described earlier. Here we report a case of EHBA with concordant Duarte 1 and 2 variants of galactosemia in an infant with cholestasis. Genetic analysis of the index pa- tient for galactosemia revealed presence of Duarte 1/Duarte 2 variants of galactosemia with genotype N314D-L218L/N314D-G1105C-GI391A- G1323A-5’UTR-119delGTCA. Clinical evaluation of the patient showed the presence of EHBA. Henceforth, it may be hypothesized that EHBA may have a genetic basis with simultaneous involvement of the G ALT gene. Keywords: Duarte 1; Duarte 2; Extrahepatic Biliary Atresia; Galactosemia; Galatose-1-Phosphate Uridyl Transferase Ge ne; Los An gles 1. INTRODUCTION Galactosemia is an autosomal recessive disorder of ga- lactose metabolism caused by deficient or absent activi- ties of one of the three enzymes involved in the galac- tose metabolism. The deficiency of galactose-1-phos- phate uridyl transferase (GALT; MIM# 230400) is most common and presents as neonatal hepatitis and liver failure. In addition to the predominant classic form, there are two clinical variants: Duarte-1 and Duarte-2 [1]. Al- though coexistence of Duarte 1 and 2 variant of galacto- semia has been reported in literature, synchronous pres- ence of these 2 variants with extrahepatic biliary atresia (EHBA) has not been documented [1,2]. In this commu- nication, we report a case of EHBA coexisting with Duarte 1 and Duarte 2 variants of galactosemia detected at our center during evaluation of cholestatic infants for galactosemia. 2. MATERIAL AND METHODS 2.1. Patients A cohort of 215 infants (age range: 5 days - 10.5 months), admitted in Pediatric Gastroenterology ward of Postgraduate Institute of Medical Education and Re- search (PGIMER) over a period of 30 months (from January, 2007 to June, 2009) with cholestasis were evaluated for galactosemia. This study was approved by Institute’s ethical committee. 2.2. Red Cell (Galactose-1-Phosphate Uridyl Transferase) GALT Assay GALT activity was measured using a Perkin-Elmer neo- natal GALT kit (Perkin Elmer Wallac Victor 2D fluoro- meter, Finland) which measured the uridylphosphoglu- cose (UDPG) consumption on samples of dried blood eluted from the filter. 2.3. DNA Is olation and Polymerase Chain Reaction (PCR) Genomic DNA was isolated from whole blood by me- thod of Daly et al. [3]. Polymerase Chain Reaction (PCR) was performed in order to amplify the required se- quences by using specific primers mentioned in Table 1 [4,5]. 2.4. Restriction Fragment Length Polymorphism (RFLP) Analysis A rapid PCR-based DNA analysis was performed to confirm or identify the sequence variations that create or abolish the given natural and amplification-created re- *This study was supported by Department of Biotechnology, Ministry of Science and Technology, Government of India. 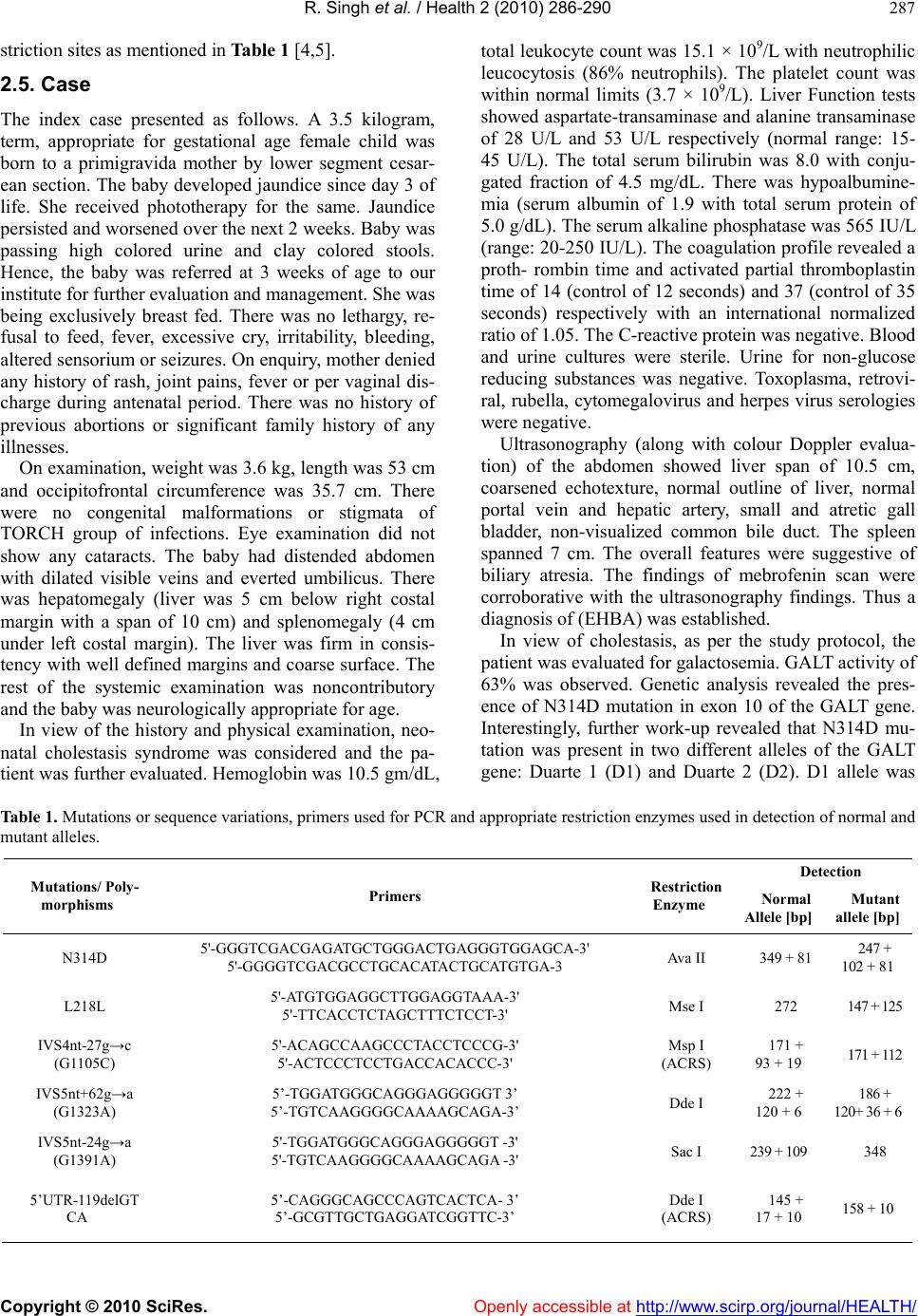 R. Singh et al. / Health 2 (2010) 286-290 Copyright © 2010 SciRes. Openly accessible at http://www.scirp.org/journal/HEALTH/ 287 striction sites as mentioned in Table 1 [4,5]. 2.5. Case The index case presented as follows. A 3.5 kilogram, term, appropriate for gestational age female child was born to a primigravida mother by lower segment cesar- ean section. The baby developed jaundice since day 3 of life. She received phototherapy for the same. Jaundice persisted and worsened over the next 2 weeks. Baby was passing high colored urine and clay colored stools. Hence, the baby was referred at 3 weeks of age to our institute for further evaluation and management. She was being exclusively breast fed. There was no lethargy, re- fusal to feed, fever, excessive cry, irritability, bleeding, altered sensorium or seizures. On enquiry, mother denied any history of rash, joint pains, fever or per vaginal dis- charge during antenatal period. There was no history of previous abortions or significant family history of any illnesses. On examination, weight was 3.6 kg, length was 53 cm and occipitofrontal circumference was 35.7 cm. There were no congenital malformations or stigmata of TORCH group of infections. Eye examination did not show any cataracts. The baby had distended abdomen with dilated visible veins and everted umbilicus. There was hepatomegaly (liver was 5 cm below right costal margin with a span of 10 cm) and splenomegaly (4 cm under left costal margin). The liver was firm in consis- tency with well defined margins and coarse surface. The rest of the systemic examination was noncontributory and the baby was neurologically appropriate for age. In view of the history and physical examination, neo- natal cholestasis syndrome was considered and the pa- tient was further evaluated. Hemoglobin was 10.5 gm/dL, total leukocyte count was 15.1 × 109/L with neutrophilic leucocytosis (86% neutrophils). The platelet count was within normal limits (3.7 × 109/L). Liver Function tests showed aspartate-transaminase and alanine transaminase of 28 U/L and 53 U/L respectively (normal range: 15- 45 U/L). The total serum bilirubin was 8.0 with conju- gated fraction of 4.5 mg/dL. There was hypoalbumine- mia (serum albumin of 1.9 with total serum protein of 5.0 g/dL). The serum alkaline phosphatase was 565 IU/L (range: 20-250 IU/L). The coagulation profile revealed a proth- rombin time and activated partial thromboplastin time of 14 (control of 12 seconds) and 37 (control of 35 seconds) respectively with an international normalized ratio of 1.05. The C-reactive protein was negative. Blood and urine cultures were sterile. Urine for non-glucose reducing substances was negative. Toxoplasma, retrovi- ral, rubella, cytomegalovirus and herpes virus serologies were negative. Ultrasonography (along with colour Doppler evalua- tion) of the abdomen showed liver span of 10.5 cm, coarsened echotexture, normal outline of liver, normal portal vein and hepatic artery, small and atretic gall bladder, non-visualized common bile duct. The spleen spanned 7 cm. The overall features were suggestive of biliary atresia. The findings of mebrofenin scan were corroborative with the ultrasonography findings. Thus a diagnosis of (EHBA) was established. In view of cholestasis, as per the study protocol, the patient was evaluated for galactosemia. GALT activity of 63% was observed. Genetic analysis revealed the pres- ence of N314D mutation in exon 10 of the GALT gene. Interestingly, further work-up revealed that N314D mu- tation was present in two different alleles of the GALT gene: Duarte 1 (D1) and Duarte 2 (D2). D1 allele was Table 1. Mutations or sequence variations, primers used for PCR and appropriate restriction enzymes used in detection of normal and mutant alleles. Detection Mutations/ Poly- morphisms Primers Restriction Enzyme Normal Allele [bp] Mutant allele [bp] N314D 5'-GGGTCGACGAGATGCTGGGACTGAGGGTGGAGCA-3' 5'-GGGGTCGACGCCTGCACATACTGCATGTGA-3 Ava II 349 + 81 247 + 102 + 81 L218L 5'-ATGTGGAGGCTTGGAGGTAAA-3' 5'-TTCACCTCTAGCTTTCTCCT-3' Mse I 272 147 + 125 IVS4nt-27g→c (G1105C) 5'-ACAGCCAAGCCCTACCTCCCG-3' 5'-ACTCCCTCCTGACCACACCC-3' Msp I (ACRS) 171 + 93 + 19 171 + 112 IVS5nt+62g→a (G1323A) 5’-TGGATGGGCAGGGAGGGGGT 3’ 5’-TGTCAAGGGGCAAAAGCAGA-3’ Dde I 222 + 120 + 6 186 + 120+ 36 + 6 IVS5nt-24g→a (G1391A) 5'-TGGATGGGCAGGGAGGGGGT -3' 5'-TGTCAAGGGGCAAAAGCAGA -3' Sac I 239 + 109 348 5’UTR-119delGT CA 5’-CAGGGCAGCCCAGTCACTCA- 3’ 5’-GCGTTGCTGAGGATCGGTTC-3’ Dde I (ACRS) 145 + 17 + 10 158 + 10  R. Singh et al. / Health 2 (2010) 286-290 Copyright © 2010 SciRes. Openly accessible at http://www.scirp.org/journal/HEALTH/ 288 found to carry a silent mutation L218L in exon 7 in ad- dition to the N314D in exon 10 of GALT gene (Figure 1). In contrast, D2 allele was found to co-exist with 3 different polymorphisms: IVS4nt-27g→c (G1105C), IVS5nt+62g→a (G1323A) and IVS5nt-24g→a (G1- 391A) and promoter region deletion of 4 nucleotides (5’UTR-119del GTCA) (Figure 2). The patient was managed with appropriate supportive and nutritional care along with cessation of breast feed- ing whilst soya-milk formula was initiated. Subsequently Kasai’s porto-enterostomy was performed at 3 months of age. Per operatively, the liver was greenish, hypertro- phied; biliary channels were fibrotic although right he- patic duct was visible. Per-operative cholangiogram con- firmed the diagnosis of EHBA. The patient was discharged on appropriate dietary ad- vice and multivitamin supplementation. At 4 months of follow-up, the infant is passing normal colored stools and is anicteric and gaining weight, although at a slower pace. In view of established biliary cirrhosis the family has been advised regular medical follow-up and has been counseled regarding the possible need of liver trans- plantation in future. 3. DISCUSSION The report presents a peculiar case of coexistence of 2 variants of galactosemia together (Duarte 1 and Duarte 2 together) with EHBA in a solitary case amongst a cohort of 215 patients with neonatal cholestasis. The patient was found to carry the L218L transition along with N314D on one allele which is known as Duarte1 (D1) or Los Angeles variant (LA) of galacto- semia [6,7]. This variant is known to be associated with the increased GALT activity [1]. A molecular mechanism involving an increased rate of translation through codon bias has been proposed to be the possible mechanism [8]. In addition, three different transitions were also found: IVS4nt –27 g→c (G1105C) in intron 4 and IVS5nt –24 g→a (G1391A) and IVS5nt +62 g→a (G1323A) in intron 5, all in a heterozygous state. Besides, a deletion of 4 nu- cleotides (5’UTR-119delGTCA) was detected in the promoter region of the GALT gene. This type of geno- type, that is: N314D-G1105C-GI391A-G1323A-5’UTR- 119delGTCA is known to constitute another galacto- semia variant called Duarte 2 (D2) variant which is as- sociated with the decrease GALT activity [6,7]. Investi- gators have suggested that these genetic alterations might be regulatory mutations which may be responsible for the decrease in GALT activity in D2 variants [7]. So the genotype of the patient was N314D-L218L/N314D- G1105C-GI391A-G1323A-5’UTR-119delGTCA or D1/ D2. This unique genotype causing coexistence of D1 and D2 variant galactosemia along with EHBA has not been earlier reported in literature. The coexistence of EHBA alongwith galactosemia made us ponder that although EHBA is not believed to be an inherited disease; genetic factors have been re- A. N314D mutation by Ava II Lane 1: 100 bp DNA ladder Lane 2: Wild type Lane 3: Homozygous mutant C. Sequence of GALT gene showing: (a) Normal sequence (b) N314D mutation B. L218L mutation by Mse I Lane 1: Wild type Lane 2: Heterozygous mutant Lane 3: 50 bp DNA ladder D. Sequence of GALT gene showing: (a) Normal sequence (b) L218L mutatio Figure 1. Restriction digestion analysis and electrophoregram of N314D and L218L mutation. 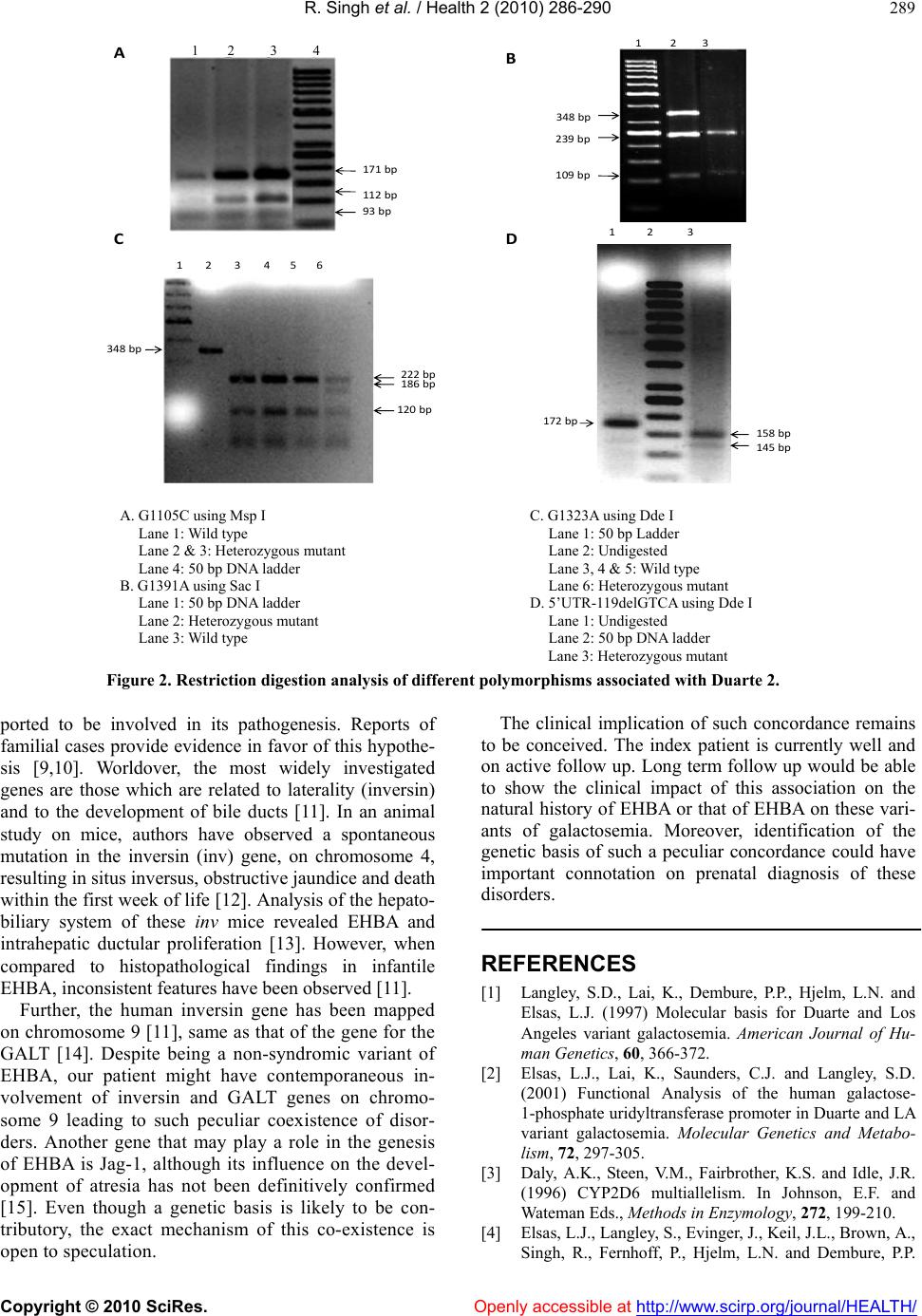 R. Singh et al. / Health 2 (2010) 286-290 Copyright © 2010 SciRes. Openly accessible at http://www.scirp.org/journal/HEALTH/ 289 172bp 158bp 145bp 123 171bp 112bp 93bp 123456 222bp 186bp 120bp 123 348bp 239bp 109bp A DC B 348bp 1 2 3 4 A. G1105C using Msp I Lane 1: Wild type Lane 2 & 3: Heterozygous mutant Lane 4: 50 bp DNA ladder B. G1391A using Sac I Lane 1: 50 bp DNA ladder Lane 2: Heterozygous mutant Lane 3: Wild type C. G1323A using Dde I Lane 1: 50 bp Ladder Lane 2: Undigested Lane 3, 4 & 5: Wild type Lane 6: Heterozygous mutant D. 5’UTR-119delGTCA using Dde I Lane 1: Undigested Lane 2: 50 bp DNA ladder Lane 3: Heterozygous mutant Figure 2. Restriction digestion analysis of different polymorphisms associated with Duarte 2. ported to be involved in its pathogenesis. Reports of familial cases provide evidence in favor of this hypothe- sis [9,10]. Worldover, the most widely investigated genes are those which are related to laterality (inversin) and to the development of bile ducts [11]. In an animal study on mice, authors have observed a spontaneous mutation in the inversin (inv) gene, on chromosome 4, resulting in situs inversus, obstructive jaundice and death within the first week of life [12]. Analysis of the hepato- biliary system of these inv mice revealed EHBA and intrahepatic ductular proliferation [13]. However, when compared to histopathological findings in infantile EHBA, inconsistent features have been observed [11]. Further, the human inversin gene has been mapped on chromosome 9 [11], same as that of the gene for the GALT [14]. Despite being a non-syndromic variant of EHBA, our patient might have contemporaneous in- volvement of inversin and GALT genes on chromo- some 9 leading to such peculiar coexistence of disor- ders. Another gene that may play a role in the genesis of EHBA is Jag-1, although its influence on the devel- opment of atresia has not been definitively confirmed [15]. Even though a genetic basis is likely to be con- tributory, the exact mechanism of this co-existence is open to speculation. The clinical implication of such concordance remains to be conceived. The index patient is currently well and on active follow up. Long term follow up would be able to show the clinical impact of this association on the natural history of EHBA or that of EHBA on these vari- ants of galactosemia. Moreover, identification of the genetic basis of such a peculiar concordance could have important connotation on prenatal diagnosis of these disorders. REFERENCES [1] Langley, S.D., Lai, K., Dembure, P.P., Hjelm, L.N. and Elsas, L.J. (1997) Molecular basis for Duarte and Los Angeles variant galactosemia. American Journal of Hu- man Genetics, 60, 366-372. [2] Elsas, L.J., Lai, K., Saunders, C.J. and Langley, S.D. (2001) Functional Analysis of the human galactose- 1-phosphate uridyltransferase promoter in Duarte and LA variant galactosemia. Molecular Genetics and Metabo- lism, 72, 297-305. [3] Daly, A.K., Steen, V.M., Fairbrother, K.S. and Idle, J.R. (1996) CYP2D6 multiallelism. In Johnson, E.F. and Wateman Eds., Methods in Enzymology, 272, 199-210. [4] Elsas, L.J., Langley, S., Evinger, J., Keil, J.L., Brown, A., Singh, R., Fernhoff, P., Hjelm, L.N. and Dembure, P.P.  R. Singh et al. / Health 2 (2010) 286-290 Copyright © 2010 SciRes. Openly accessible at http://www.scirp.org/journal/HEALTH/ 290 (1995) 7Galactosemia: A strategy to identify new bio- chemical phenotypes and molecular genotypes. American Journal of Human Genetics, 56, 630-639. [5] Kozak, L., Francova, H., Fajkusov, L., Pijackova, A., Macku, J.M.S., Peskovova, K., Martincova, O., Krijt, J. and Bzduch, J. (2000) Mutation analysis of the GALT gene in Czech and Slovak galactosemia populations: Identification of six novel mutations, including a stop codon mutation (X380R). Human Mutation, 15(2), 206. [6] Greber, S., Guldberg, P., Scheibenreiter, S. and Strobl, W. (1995) Mutations in classical and Duarte 2 galactosemia. Pediatric Research, 38, 434 (Abstr.). [7] Podskarbi, T., Kohlmetz, T., Gathof, B.S., Kleinlein, B., Bieger, W.P., Greser, U. and Shin, Y.S. (1996) Molecular characterization of Duarte 1 and Duarte 2 variants of ga- lactose-1-phosphate uridyl transferase. Journal of Inher- ited Metabolic Disorders, 19, 638-644. [8] Kurland, C.G. (1991) Codon bias and gene expression. FEBS Letters, 285(2), 165-169. [9] Cunningham, M.L., Sybert, V.P., Opitz, J.M. and Rey- nolds, J.F. (1988) Idiopathic extrahepatic biliary atresia: Recurrence in sibs in two families. American Journal of Medical Genetics, 31, 421-426. [10] Danesino, C., Spadoni, E. and Buzzi, A. (1999) Familial biliary atresia. American Journal of Me dical Gene tic s, 85, 195. [11] Carvalho, E.D., Ivantes, C.A.P. and Bezerra, J.A. (2007) Extrahepatic biliary atresia: Current concepts and future directions. Jornal de Pediatria, 83(2), 105-120. [12] Yokoyama, T., Copeland, N.G., Jenkins, N.A., Mont- gomery, C.A., Elder, F.F. and Overbeek, P.A. (1993) Re- versal of left-right asymmetry: A situs inversus mutation. Science, 260, 679-682. [13] Mazziotti, M.V., Willis, L.K., Heuckeroth, R.O., LaRegi- na, M.C., Swanson, P.E., Overbeek, P.A. and Perlmutter, D.H. (1999) Anomalous development of the hepatobil- iary system in the Inv mouse. Hepatology, 30, 372-378. [14] Shih, L.Y., Suslak, L., Rosin, I., Searle, B.M. and Despo- sito, F. (1984) Gene dosage studies supporting localiza- tion of the structures genes for galactose-1-phosphate uridyl transferase (GALT) to band 913 of chromosome 9. American Journal of Medical Genetics, 19, 539-543. [15] Kohsaka, T., Yuan, Z.R., Guo, S.X., Tagawa, M., Naka- mura, A., Nakano, M., Kawasasaki, H., Inomata, Y., Ta- naka, K. and Miyauchi, J. (2002) The significance of human jagged 1 mutations detected in severe cases of extrahepatic biliary atresia. Hepatology, 36, 904-912. |