 Open Journal of Pediatrics, 2011, 1, 79-86 doi:10.4236/ojped.2011.14019 Published Online December 2011 (http://www.SciRP.org/journal/ojped/ OJPed ) Published Online December 2011 in SciRes. http://www.scirp.org/journal/OJPed Mechanism of origin in two cases of chimerism Antonella Minelli1,2*, Andrea Guala3,4, Albert o Groppo1,2, Gabriella Restagno5, Roberto Lala6, Silvia Einaudi6, Mariaelena Repici6, Emilio Merlini7, Luca Sbaiz5, Valentina Asnaghi 5, Ana Graciela Lopez8, Paola Angellotti3, Silvia Cristina9, Cesare Danesino1,2 1Dipartimento di Medicina Molecolare, Sezione di Biologia Generale e Genetica Medica, Università degli Studi di Pavia, Pavia, Italy; 2Istituto di Ricerca e Cura a Carattere Scientifico IRCCS Policlinico San Matteo, Pavia, Italy; 3Struttura Complessa Pediatria, Ospedale Castelli, ASL VCO, Verbania, Italy; 4Master in Malattie Rare, Università degli Studi di Torino , Torino , Italy; 5Dipartimento di Diagnostica, Diagnosi e Consulenza Genetica, Ospedale Regina Margherita—S. Anna, Torino, Italy; 6Struttura Complessa Endocrinologia e Diabetologia, Ospedale Regina Margherita—S. Anna, Torino, Italy; 7Struttura Complessa Urologia, Ospedale Regina Margherita—S. Anna, Torino, Italy; 8Struttura Complessa Pediatria, Ospedale Civile, ASL NO, Borgomanero, Italy; 9Struttura Complessa Anatomia Patologica, Ospedale Civile, ASL NO, Borgomanero, Italy. Email: *antonella.minelli@unipv.it Received 3 October 2011; revised 16 November 2011; accepted 21 November 2011. ABSTRACT Chimerism is defined as the presence in a subject of more than one stable and genetically distinct cell line; cases reported so far include both patients with am- biguous genitalia and healthy subjects. The biological mechanisms, which may give origin to chimeras, are complex, and can be understood by analyzing DNA samples of the patients and their parents using mo- lecular techniques. The objective of this study is to identify the mechanism of origin for the 2 cases we report. The first patient is a phenotipically normal girl with normal (external and internal) genitalia; the second patient had ambiguous genitalia and under- went surgery. DNA was purified from blood samples and, limited to Patient 1, from a sample of biliary cyst. Short tandem repeat polymorphisms were ana- lyzed in order to identify the relative parental con- tribution to the patients. Molecular analyses carried out on the first patient are not fully informative be- cause of two possible explanations (i.e. parthenoge- netic and andrognetic chimera), while in the second case the presence of four alleles at some markers al- lowed us to identify a tetragametic chimera origin- nated from the fusion of two distinct embryos. Stud- ies carried on one single tissue may not always be conclusive as they do not allow the precise identifica- tion of the mechanism of origin. In these cases, stud- ies on more tissues are strongly suggested. Keywords: Chimerism; Androgenetic Chimera; Parthe- nogenetic Chimera; Tetragametic Chimera; Microsatel- lite Polymorphism Analysis 1. INTRODUCTION Chimerism is defined as the presence of more than one stable and genetically distinct cell line [1] originated independently from one another [2]. Thus chimerism is different from mosaicism, in which the cell lines have a common, single cell progenitor. This condition is rare in humans: Malan et al. in 2006 counted about 30 cases [3] and this number increased a little in these last years. When both female (46,XX) and male (46,XY) cell lines are present, the phenotype, ranging from normal male to normal female through various degree of am- biguous genitalia, is related to their distribution in the gonads. The ratio between the two cell lines in different tissues does not allow a precise prediction of the status of gonads or the phenotyp e of external genitalia [3]. The origin of the cell lines can be defined at a mo- lecular level using a number of polymorphic markers and comparing the genetic profile of the patient with those of the parents. Only few cases, especially in these past years, have been studied this way, allowing to pro- pose at least four different mechanisms of origin, namely tetragametic chimerism, parthenogenetic chimerism, chimera resulting from the fertilization of the second polar body and androgenetic chimerism. These mecha- nisms were reviewed by Malan in 2006 [3]; details are provided in the Discussion section of this paper. We report two cases of chimerism in which molecular analysis allowed us to suggest either parthenogenetic or  A. Minelli et al. / Open Journal of Pediatrics 1 (2011) 79-86 80 androgenetic origin in the first case and to identify a tetragametic mechanism of origin in the second. 2. CASES REPORT 2.1. Patient 1 Patient 1 was born at 37 weeks’ gestation after an un- eventful pregnancy, to healthy, unrelated Caucasian par- ents. Birth weight was 2600 g, length was 45 cm, and head circumference was 43 cm. Parental karyotype is normal. Chorionic villus sampling was performed upon pa- rental request, and th e results showed two cell lines. One cell line with a 46,XX karyotype was seen in short term cultures, while a 46,XY cell line was observed in long term cultures. Control amniocentesis showed 46,XX in 21 clones from 3 independent cultures and 46,XY from 4 colonies from a single culture. Ultrasound examination consistently revealed a fetus with no malformations and with normal female external genitalia. At birth, physical examination revealed a fully normal child with no malformations or dysmorphic features, and normal female external genitalia; abdominal ultrasound examination in the first week of life demonstrated the presence of normal uterus and ovaries. The placenta showed morphological alterations sug- gestive of “chimerism”: it weighed 1730 g, and upon sectioning, a marginal area of vesicle formation consis- tent with molar changes was observed. At microscopic examination, the placental parenchyma showed mainly third trimester chorionic villi with widespread small ar- tery lesions in both secondary and tertiary stem villi. There were marked villous hydrops with central cistern formation consistent with complete hydatidiform mole. The mixed population of morphologically normal cho- rionic villi and villi with the typical chan ges of complete hydatidiform mole was highly suggestive of placental/ fetal mosaicism [4,5]. These data, in addition to the re- sults observed antenatally, were an indication to perform cytogenetic analysis of the newborn, which showed a 46,XX[31]/46,XY[19] karyotype. A similar ratio of the two cell lines was observed during follow up. Normal development was recorded during follow up. At the age of 2, a slightly enlarged liver was found, and an ultrasound examination demonstrated a cystic forma- tion of 7 cm which was diagnosed as a “congenital bi- liary cyst”, which was surgically removed. 2.2. Patient 2 Patient 2, Caucasian, was admitted to the hospital be- cause of a sex determination disorder. Male sex was at- tributed at birth, but external genitalia, ambiguous and classified as Prader type III, were represented by the presence of a penoclitoris, perineal hypospadia, uro- genital sinus, bifid scro tum and bilateral cryptorchidism. At birth, chromosomal analysis on peripheral blood lymphocytes revealed two cell lines, 46,XX[49]/46,XY [15]. A second karyotype analysis few months later con- firmed the presence of the two cell lines: 46,XX[67]/ 46,XY[33], while cultured fibroblasts from a skin biopsy showed only 46,XX[16]. Both parents showed normal karyotype. These results led to a diagnosis of 46,XX/ 46,XY chimer ism. QF-PCR analysis with chromosome specific probes (13, 18, 21, X and Y) revealed the presence of DNA be- longing to two different cell lines, one female and one male. The X-chromosome signal proved to be stronger than the Y-chromosome signal. Endocrine evaluation showed LH: 4.08 mU/ml; es- tradiol: 8 pg/ml; testosterone: 1.08 ng/ml; all these val- ues as well as 17OHP, androstenedione and DHEAS were within normal range. FSH (9.45 mU/ml) was out o f range, and it was similar to what is observ ed in females. Transabdominal ultrasound showed a uterus behind the bladder and two gonadal structures, while contrast X-rays demonstrated a straight female-type urethra, va- gina and uterus. Contrast medium reached the tubae bi- laterally. Laparoscopy revealed on the left side an uterus as well as a female gonad and tuba, with internal inguinal ring. On the right side, the internal inguinal ring was open and a testicular structure was present in the abdo- men. Detection of a morphologically normal uterus, to- gether with a tuba and a female gonad led to decide to change the anatomical sex of the baby from male to fe- male. This decision was discussed with the baby’s par- ents and psychological support was provided. The baby underwent female genitoplasty with clitoral reduction. The right gonad was removed together with a cystic formation associated to it. Histopathology of the gonadal tissue revealed the presence of ovarian parenchyma and testicular tissue, in particular seminiferous tubules with interstitial fibrosis and Leydig cells. Structures similar to rete testis, epidi- dymis and vascular plexus were also detected. The find- ing was compatible with a diagnosis of ovotestis. The cystic structure was characterised by osteocartilaginous, thyroideal and mature connective tissue areas. Karyo- typing was performed on a sample of the cystic tissue and resulted 46,XX. Histological diagnosis was of a mature cystic teratoma. Biopsy was not performed on the left gonad because it presented with a macroscopically normal aspect, and in order to preserve the supposed ovarian tissue. C opyright © 2011 SciRes. OJPed  A. Minelli et al. / Open Journal of Pediatrics 1 (2011) 79-86 81 3. MATERIALS AND METHODS 3.1. DNA Purification After informed consent, blood samples from the two patients and their parents were obtained. Limited to Pa- tient 1, DNA was also purified from a fragment of bil- iary cyst surgically removed. DNA was purified by rou- tine methods using GenElute™ Blood Genomic DNA Kit (Sigma-Aldrich®). 3.2. STRPs Analysis The short tandem repeat polymorphisms (STRPs) to be analysed were chosen on several different chromosomes, on the basis of their heterozygosity (see Table 1 and Ta bl e 2 for a complete list of markers for patient 1 and patient 2 respectively). Genotyping of STRPs was performed using ABI PRISM™ multicolor fluorescent dye technology, based on labeling DNA fragments with different color fluores- cent dyes by PCR amplification. PCR conditions were developed in our lab. The PCR products are displayed as electropherograms showing fluorescence intensity as a function of fragment size or migration time. Peak Scan- ner SoftwareTM v1.0 by Applied Biosystems provides peak detection (areas and heights) relative to alleles am- plified as DNA fragments. When parents shared an allele of the same size, we calculated the ratio between peak heights and compared it with the ratio of cell lines as demonstrated by cytoge- netic analysis. This method allowed to assess the origin of that allele (maternal, paternal or both of them) and infer a single or double, maternal and/or paternal contri- bution. 4. RESULTS We analyzed several STRPs localized on different auto- somes, in addition to the X chromosome (see Table 1 and Table 2 for a complete list of markers for patient 1 and patient 2 respectively). Informative and partially informative markers have been found. We defined a marker as informative when it allowed us to prove a double paternal and/or maternal contribution. A marker was considered partially infor- mative when it showed three alleles and the origin of the extra allele could not be inferred because its origin could be maternal and/or paternal. Table 1 and Ta ble 3 show results for patient 1; Table 2 and Table 4 illustrate findings for patient 2. Figure 1 shows examples of informative markers for both patient 1 and 2. 4.1. Patient 1 Eight autosomal markers are informative, since they Table 1. List of the STRPs studied for patient 1, showing size (in bp) and which alleles were observed in the patient and in her parents. Informative STRPs are shown in bold print (I: informative marker; N/I: not informative marker). STRP Father Mother Patient Notes D1S1609 178/194174/182 178/182/194 Id D1S3723 182 190/194 182/190 N/I D2S1361 170/186 165/186 N/Ia D2S2739 291/317 291/301 N/Ia D3S4555 205/213205/209 205/213 N/Ia D3S2406 330/346326 326/330/346 I D4S3355 136 136/144 136/144 N/Ia D4S2426 260 248/264 248/260 N/I D5S1470 170/186 166/186 N/Ia D5S815 286/290 286/294 N/Ia D7S1808 255/273258/261 255/258/273 Id D7S1805 198/216198/216 216 N/I D7S1820 251/255263 251/255/263 I D7S796 176/184180/188 176/180/184 Id D7S1830 221 221/225 221/225 N/Ia D8S1130 133/137137/146 133/137/146 Ic D8S586 244/252240/248 244/248 N/Id D10S189 185 183/185 183/185 N/Ia D10S1779 266/268264/268 264/266/268 Ic D10S547 237/239239/247 239/247 N/Ia D10S570 285/295291/295 291/295 N/Ia D12S390 139/154148 139/148 N/I D12S1586 157/169165/167 157/165 N/I 15DUP10 398 395 395/398 N/I 15DUP12 238 234 234/238 N/I D15S822 284 /288260/264 260/284/288 I D15S643 195/213217 195/213/217 I COMPLEX 264/268264/268 264/268 N/I EVI20 191/193191 191/193 Ib D20S604 137/141137/141 137 N/I D20S1151 251/279243/247 243/279 N/Id DXS9908 222 224/230 222/224 I GATA172D05 106 114/126 106/126 Id aThe two alleles show peaks of similar size; bSTRP demonstrating double paternal contribution; cOnly partially informative marker; dData consistent with those found analyzing the biliary cyst. C opyright © 2011 SciRes. OJPed  A. Minelli et al. / Open Journal of Pediatrics 1 (2011) 79-86 82 Table 2. List of the STRPs studied for patient 2, showing size (in bp) and which alleles were observed in the patient and in her parents. Informative STRPs are shown in bold print. (I: informative marker; N/I: not informative marker). STRP Father MotherPatient Notes D1S3723 186/190 145/186 145/186 Ib D1S1609 190/202 178/186 178/186/190 I D2S1361 175/183 179/183 175/179/183 Ic D3S2406 330 326/342 326/330/342 I D5S1470 182/190 186/194 182/186/190/194I D5S815 256/290 286/290 256/286/290 Ic D7S1805 213/217 198/221 198/213/217/221I D7S796 188 176/191 176/188/191 I D8S1130 132/150 132/141 132 N/I D10S1779 265 274/276 265/274/276 I D10S570 288 294 288/294 N/I D12S390 148/157 152/157 148/152/157 Ic D15S643 209/213 209 209/213 N/Ia D20S1151 246/274 246/250 246/250/274 Ic DXS9908 224 228 224/228 I GATA172D05 130 114/122114/122/130 I aThe two alleles show peaks of similar size; bSTRP demonstrating double maternal contribution; cOnly partially informative marker. Table 3. Peak heights (in relative fluorescent units) and their ratios for STRPs showing two different alleles, and the parents sharing one of them for patient 1. Allele 1 Allele 2 STRP Size (b p) Height Size (bp) Height Ratio D2S1361 165 8876 186 8578 1.03 D2S2793 291 6720 301 6437 1.04 D3S4555 205 6418 213 3632 1.76 D4S3355 136 3049 144 2810 1.09 D5S1470 166 2555 186 2658 1.04 D5S815 286 4461 294 4166 1.07 D7S1830 221 268 225 241 1.11 D10S189 183 570 185 703 1.23 D10S547 239 215 247 185 1.16 D10S570 291 977 295 630 1.55 EVI20 191 1794 193 661 2.71 Tabl e 4. Peak heights (in relative fluorescent units) and their ratios for STRPs showing two different alleles, and parents sharing one of them for patient 2. Allele 1 Allele 2 STRP Size (bp)Height Size (bp) HeightRatio D1S3723145 767 186 1494 1.95 D15S643209 157 213 134 1.17 show double paternal and single maternal contribution. As regards EVI20, it has been considered an informative marker because the ratio between peak heights is con- sistent with the ratio of cytogenetic analysis (see Table 3). Markers D8S1130 and D10S1779 reveal three alleles and are reckoned partially informative, because the ori- gin of the extra allele cannot be assessed. X-linked markers are informative, since they show double paternal (considering the Y chromosome) and single maternal contribution. The same results were observed when analyzing DNA extracted from biliary cyst, tested only for D1S1609, D7S1808, D7S796, D8S586, D20S1151 and GATA 172D05 markers. The 10 informative markers clearly reveal double pa- ternal contribution, while, up to now, double maternal contribution has never been proven. 4.2. Patient 2 Two informative mark ers (D5S1470 and D7S1 805) on 2 chromosomes show 4 different alleles, proving a double maternal and double paternal contribution. In addition, other 5 autosomal markers are also infor- mative, since they show double maternal and single pa- ternal contribution. Though marker D1S3723 shows only two alleles, it has been considered an informative marker, because the ratio between peak heights is con- sistent with the ratio of cytogenetic analysis (see Table 4). Four markers are partially informative, since they show three different alleles and the origin of the extra allele cannot be assessed. Both X-linked markers are informative because they show double paternal contribution (considering the Y chromosome). Moreover, marker GATA172D05 shows double ma- ternal contribution. On the whole, these data are consistent with double paternal and double maternal contribu tions for this case. 5. DISCUSSION Since chimerism is characterized by the independent origins of the two cell lines, three or four alleles at a C opyright © 2011 SciRes. OJPed 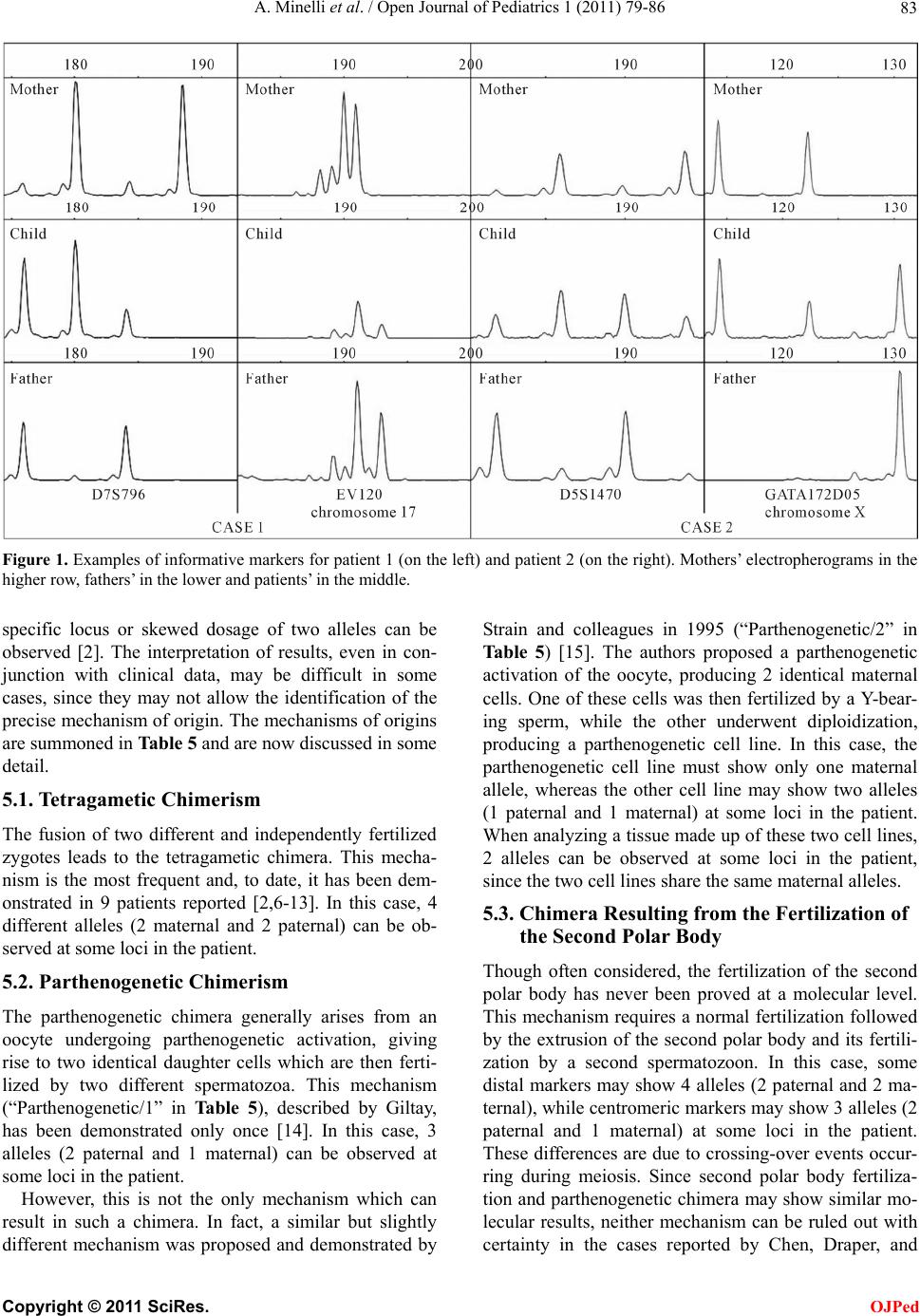 A. Minelli et al. / Open Journal of Pediatrics 1 (2011) 79-86 Copyright © 2011 SciRes. 83 Figure 1. Examples of informative markers for patient 1 (on the left) and patient 2 (on the right). Mothers’ electropherograms in the higher row, fathers’ in the lower and patients’ in the middle. specific locus or skewed dosage of two alleles can be observed [2]. The interpretation of results, even in con- junction with clinical data, may be difficult in some cases, since they may not allow the identification of the precise mechanism of origin. The mechanisms of origins are summoned in Table 5 and are now discussed in some detail. 5.1. Tetragametic Chimerism The fusion of two different and independently fertilized zygotes leads to the tetragametic chimera. This mecha- nism is the most frequent and, to date, it has been dem- onstrated in 9 patients reported [2,6-13]. In this case, 4 different alleles (2 maternal and 2 paternal) can be ob- served at some loci in the patient. OJPed 5.2. Parthenogenetic Chimerism The parthenogenetic chimera generally arises from an oocyte undergoing parthenogenetic activation, giving rise to two identical daughter cells which are then ferti- lized by two different spermatozoa. This mechanism (“Parthenogenetic/1” in Table 5), described by Giltay, has been demonstrated only once [14]. In this case, 3 alleles (2 paternal and 1 maternal) can be observed at some loci in the patient. However, this is not the only mechanism which can result in such a chimera. In fact, a similar but slightly different mechanism was proposed and demonstrated by Strain and colleagues in 1995 (“Parthenogenetic/2” in Table 5) [15]. The authors proposed a parthenogenetic activation of the oocyte, producing 2 identical maternal cells. One of these cells was then fertilized by a Y-bear- ing sperm, while the other underwent diploidization, producing a parthenogenetic cell line. In this case, the parthenogenetic cell line must show only one maternal allele, whereas the other cell line may show two alleles (1 paternal and 1 maternal) at some loci in the patient. When analyzing a tissue made up of these two cell lines, 2 alleles can be observed at some loci in the patient, since the two cell lines share the same maternal alleles. 5.3. Chimera Resulting from the Fertilization of the Second Polar Body Though often considered, the fertilization of the second polar body has never been proved at a molecular level. This mechanism requires a normal fertilization followed by the extrusion of the second polar body and its fertili- zation by a second spermatozoon. In this case, some distal markers may show 4 alleles (2 paternal and 2 ma- ternal), while centromeric markers may show 3 alleles (2 paternal and 1 maternal) at some loci in the patient. These differences are due to crossing-over events occur- ring during meiosis. Since second polar body fertiliza- tion and parthenogenetic chimera may show similar mo- lecular results, neither mechanism can be ruled out with certainty in the cases repoted by Chen, Draper, and r  A. Minelli et al. / Open Journal of Pediatrics 1 (2011) 79-86 84 Table 5. Mechanisms of origin of chimeras, possible results of STRP analysis and related references. In case of parthenogenetic and androgenetic chimerism, different authors suggested different mechanisms resulting in such chimeras. Mechanism of origin Paternal contribution Maternal contribution Max number of alleles that may be observed in the patient References Tetragametic 2 2 4 2, 6 - 13 Parthenogenetic/1 2 1 3 14 Parthenogenetic/2 1 1 2 15 Fertilization of the second polar body2 2/3a 3/4a 16 - 18 Androgenetic/1 1 1 2 19 - 20 Androgenetic/2 2 1 3 21 Androgenetic/3 2 1 3 22 Androgenetic/4 2 1 3 22 aDue to crossing-over events during meiosis, centromeric markers may show 3 maternal alleles, whereas distal markers may show only 2, thus altering the maximum number of alleles in a tissu e made up o f 2 cell lines. Mosebach [16-18] . 5.4. Androgenetic Chimera After a normal event of fertilization of an oocyte and a X spermatozoon, endoreplication of the paternal pronu- cleous takes place; the following cell cleavage leads to a diploid (with maternal and paternal pronuclei) and a haploid (with only a paternal pronucleus) cells. A second event of endoreplication of the paternal genome in the haploid cell brings to the androgenetic cell line. This mechanism has been demonstrated twice [19,20]. In this case, the androgenetic cell line must show only one pa- ternal allele, whereas the other cell line may show two different alleles (1 paternal and 1 maternal) at some loci in the patient. When analyzing a tissue made up of these two cell lines, 2 alleles can be observed at some loci in the patient, since the two cell lin es share the same pater- nal alleles (“Androgene tic/1” in Table 5). A related mechanism was proposed by Surti in 2005 [21]: it requires the fusion of a normal zygote (fertilized by a Y-carrying spermatozoon) and an empty oocyte fertilized by a X-carrying spermatozoon and undergone to endomitosis. This second mechanism has been dem- onstrated only once. In this case, the androgenetic cell line must show only one paternal allele, while the other cell line may show two different alleles (1 paternal and 1 maternal) at some loci in the patient. When analyzing a tissue made up of these two cell lines, 3 alleles can be found at some loci in the patient, since the two cell line originated from two different spermatozoa (“Androge- netic/2” in Table 5). We would like to point out that Surti described the placenta as cystic when reporting the clinical history, and that chimerism was confined to the placenta [21]. Furthermore, other 2 mechanisms, each involving a tri-pronuclear (3PN) zygote, were proposed by Robinson in 2007 [22]. The authors suggested that, after the fer- tilization of an oocyte with two normal spermatozoa (leading to a 3PN zygote), fully diploid two-celled em- bryos can occur when only one of the three haploid ge- nomes replicates and segregates at the end of the one- cell stage. In this case, the androgenetic cell line may show 2 alleles (both paternal) at some loci in the patient, whereas the other cell line may show 2 alleles (1 pater- nal and 1 maternal) at some loci. When analyzing a tis- sue made up of these two cell lines, 3 alleles may be found at some loci in the patient, because 2 spermatozoa are involved (“Androgenetic/3” in Table 5). Alternatively, such embryos can also arise when a 3PN zygote undergoes cell division without genome replication, leading to a diploid and a haploid cell, and consequent replication of the haploid genome. In this case, the androgenetic cell line must show only one pa- ternal allele, while the other cell line may show two dif- ferent alleles (1 paternal and 1 maternal) at some loci in the patient. When analyzing a tissue made up of these two cell lines, 3 alleles can be found at some loci in the patient, because 2 spermatozoa are involved (“Androge- netic/4” in Table 5). It is worth noting that in cases of androgenetic chi- merism, complete hydatidiform moles, placental mes- enchimal dysplasia, cystic placenta, hemangiomas and liver cysts are often found during pregnancy [22]. 5.5. Patient 1 Results showed 3 alleles at some loci, two of them have a paternal origin, while the remaining is maternal. Hence they are consistent with both parthenogenetic and an- drogenetic hypothesis. Considering both molecular ana- lysis and the clinical description of the placenta, we con- sidered the androgenetic hypo thesis very likely. We then studied other tissues in order to identify an androgenetic C opyright © 2011 SciRes. OJPed  A. Minelli et al. / Open Journal of Pediatrics 1 (2011) 79-86 85 cell line alone which could support this hypothesis, but we were not able to find it. Based on these data, we cannot rule out either mechanism. 5.6. Patient 2 Results are consistent only with tetragametic chimerism, since it is the only hypothesis which can explain the 4 alleles we found. 6. CONCLUSIONS A routine chromosome analysis on a single tissue may not identify all cases of chimerism (in fact, in case 2 fibroblasts only show a 46,XX karyotype). In addition, even when two cell lines are observed in one single tis- sue, the interpretation of resu lts may not lead to the clear identification of the mechanism which gave rise to the chimera. It is therefore strongly sugg ested to study more than one tissue in patients with ambiguous genitalia, in order to rule out the possibility of chimerism or mosaic- ism and to identify clearly the mechanism of origin. In every case of ambiguous genitalia, sistematic sur- velliance is certanly needed to check the oncologic risk of the dysgenetic gonad. It is currently still a matter of debate whether ultrasound examination alone can be used as a reliable method for diagnosing the presence of structural abnormalities in the gonads, or biopsy should be performed in all cases. Biochemical markers of neo- plasia ought to be, of course, included in follow up [23-25]. Specific counseling issues arise when chimerism is diagnosed prenatally [12]. In fact it is not possible to predict what the phenotype will be at term, since it may range from completely normal to the presence of am- biguous genitalia. Chimerism is an interesting biological problem, in which the genotype-phenotype correlation is still far from being defined. Moreover, given that chimeras can be phenotypically normal male or female, and since the number of cases studied is, up to now, limited, it is rea- sonable to assume that chimeras are under-diagnosed and less rare than previously believed. Accurate clinical examination and extended genetic investigations will provide new insights into the biological questions still pending. 7. ACKNOWLEDGEMENTS Dr. Andrea Guala achieved a Master in Rare Diseases at Università degli Stud i di Torino, Italy. REFERENCES [1] Koenigsberg, R. (Ed.) (1994) Churchill’s Medical Dic- tionary. Centro Scientifico Editore, 355. [2] Ramsay, M., Pfaffenzeller, W., Kotze, E., Bhengu, L. and Essop, F. (2009) Chimerism in black southern african pa- tients with true hermaphroditism 46,XX/47XY,+21 and 46,XX/46,XY. Annals of the New York Academy of Sci- ences, 1151, 68-76. d oi:10.1111/j.1749-6632.2008.03570.x [3] Malan, V., Vekemans, M. and Turleau, C. (2006) Chi- mera and other fertilization errors. Clinical Genetics, 70, 363-373. d oi:10.1111/j.1399-0004.2006.00689.x [4] Makrydimas, G., Sebire, N., Thornton, S., Zagorianakou, N. and Lolis, D. (2002) Complete hydatidiform mole and normal live birth: A novel case of confined placental mosaicism: Case re port. Human Reproduction, 17, 2459- 2463. doi:10.1093/humrep/17.9.2459 [5] Golubovsky, M.D. (2003) Postzygotic diploidization of triploids as a source of un usual cases of mosaicism, chimerism and twinning. Human Reproduction, 18, 236- 242. doi:10.1093/humrep/deg060 [6] Green, A., Barton, D., Je nks, P., Pearson, J. and Yates, J. (1994) Chimaerism shown by cytogenetics and DNA polymorphism analysis. Journal of Medical Genetics, 31, 816-817. doi:10.1136/jmg.31.10.816 [7] Uehara, S., Nata, M., Nagae, M., Sagisaka, K. and Oka- mura, K. (1995) Molecular biologic analyses of tet- ragametic chimerism in a true hermaphrodite with 46,XX/46,XY. Fertility and Sterilility, 63, 189-192. [8] Bonthron D.T., Strain L. and Dean J.C.S. (1997) Amal- gamation of in vitro fertilized embryos, resulting in birth of a true hermaphrodite chimera. American Journal of Human Genetics, Supplement to Volume 61, A147. [9] Strain, L., Dean, J.C.S., Hamilton, M.P. R. and Bonthron, D.T. (1998) A true hermaphrodite chimera resulting from embryo amalgamation after in vitro fertilization. New England Journal of Medicine, 338, 166-169. doi:10.1056/NEJM199801153380305 [10] Yu, N., Kruskall, M.S., Yunis, J.J., Knoll, J.H.M. and Uhl, L. (2002) Disputed maternity leading to identification of tetragametic chimerism. New England Journal of Medi- cine, 346, 1545-1552.doi:10.1056/NEJMoa013452 [11] Drexler, C., Glock, B., Vadon, M., Staudacher, E. and Dauber, E.M. (2005) Tetragametic chimerism detected in a healthy woman with mixed field agglutination reactions in ABO blood grouping. Transfusion, 45, 698-703. d oi:10.1111/j.1537-2995.2005.04304.x [12] Malan, V., Gesny, R., Morichon-Delvallez, N., Aubry, M. and Benachi, A. (2007) Prenatal diagnosis and normal outcome of a 46,XX/46,XY chimera: A case report. Hu- man Reproduction, 22, 1037-1041. doi:10.1093/humrep/del480 [13] Ve rdiani, S., Bonsignore, A., Casarino, L., Ferrari, G. and Zia, S. (2009) An unusual observation of tetragametic chimerism: Forensic aspects. International journal of le- gal medicine, 123, 431-435. doi:10.1007/s00414-009-0332-0 [14] Giltay, J.C., Brunt, T., Beemer, F.A., Wit, J.M. and Ploos Van Amstel, H.K. (1998) Polymorphic detection of a parthenogenetic maternal and double paternal contribu- tion to a 46,XX/46,XY hermaphrodite. The American Journal of Human Genetics, 62, 937-940. doi:10.1086/301796 [15] Strain, L., Warner, J.P., Johnston, T. and Bonthron, D.T. (1995) A human parthenogenetic chimaera. Nature Gene- C opyright © 2011 SciRes. OJPed  A. Minelli et al. / Open Journal of Pediatrics 1 (2011) 79-86 Copyright © 2011 SciRes. 86 OJPed tics, 11, 164-169. doi:10.1038/ng1095-164 [16] Chen, C.P., Chern, S.R., Sheu, J.C., Lin, S.P. and Hsu, C.Y. (2005) Prenatal diagnosis, sonographic findings and molecular genetic analysis of a 46,XX/46,XY true her- maphrodite chimera. Prenatal diagnosis, 25, 502-506. doi:10.1002/pd.1181 [17] Draper, N.L., Conley, C., Smith, C. and Benson, K. (2008) Dispermic chimerism identified during HLA typ- ing for stem cell transplantation. Transfusion, 48, 1398- 1402. d oi:10.1111/j.1537-2995.2008.01711.x [18] Mosebach, M., Parkner, A., Jakubiczka, S., Wieacker, P. and Heim, M.U. (2006) Dispermic chimerism identified during blood group determination and HLA typing. Transfusion, 46, 1978-1981. d oi:10.1111/j.1537-2995.2006.01005.x [19] Kaiser-Rogers, K.A., Mcfadden, D.E., Livasy, C.A., Dansereau, J. and Jiang, R. (2006) Androgenetic/bipa- rental mosaicism causes placental mesenchymal dyspla- sia. Journal of Medical Genetics, 43, 187-192. doi:10.1136/jmg.2005.033571 [20] Giurgea, I., Sanlaville, D., Fournet, J. C., Sempoux, C. and Bellanne-Chantelot, C. (2006) Congenital hyperinsu- linism and mosaic abnormalities of the ploidy. Journal of Medical Genetics, 43, 248-254. doi:10.1136/jmg.2005.034116 [21] Surti, U., Hill, L. M., Dunn, J., Prosen, T. and Hoffner, L. (2005) Twin pregnancy with a chimeric androgenetic and biparental placenta in one twin displaying placental mesenchymal dysplasia phenotype. Prenatal Diagnosis, 25, 1048-1056. doi:10.1002/pd.1255 [22] Robinson, W.P., Lauzon, J.L., Innes, A.M., Lim, K. and Arsovska, S. (2007) Origin and outcome of pregnancies affected by androgenetic/biparental chimerism. Human Reproduction, 22, 1114-1122. doi:10.1093/humrep/del462 [23] Hughes, I.A., Nihoul-Fékété, C., Thomas, B. and Cohen- Kettenis, P. (2007) Consequences of the ESPE/LWPES guidelines for diagnosis and treatment of disorders of sex development. Best Practice & Research Clinical Endo- crinology & Metabolism, 21, 351-365. doi:10.1016/j.beem.2007.06.003 [24] Looijenga, L.H.J., Hersmus, R., Oosterhuis, J.W., Cools, M. and Drop, S.L.S. (2007) Tumor risk in disorders of sex development (DSD). Best Practice & Research Clinical Endocrinology & Metabolism, 21, 480-495. doi:10.1016/j.beem.2007.05.001 [25] Cools, M., Drop, S.L.S., Wolffenbuttel, K.P., Oosterhuis, J.W. and Looijenga, L.H.J. (2006) Germ cell tumors in the intersex gonad: Old paths, new directions, moving frontiers. Endocrine reviews, 27, 468-484. doi:10.1210/er.2006-0005
|