Health
Vol.6 No.10(2014), Article ID:44737,14 pages DOI:10.4236/health.2014.610123
Superoxide Dismutase: Therapeutic Targets in SOD Related Pathology
Filip Cristiana1*, Albu Elena2, Zamosteanu Nina1
1Department of Biochemistry, University of Medicine and Pharmacy “Gr. T. Popa”, Iasi, Romania
2Department Pharmacology, University of Medicine and Pharmacy “Gr. T. Popa”, Iasi, Romania
Email: *elenaalbu@yahoo.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 25 February 2014; revised 30 March 2014; accepted 7 April 2014
ABSTRACT
There are growing evidences on the role of adaptive mechanisms of all cell types in pathological processes: atherosclerosis, ischemic attack, bacterial infections, etc. All kinds of these processes involve as main mechanism oxidative stress. Aerobic organisms use oxygen in processes that accidentally or deliberately generate aggressive species for the biologic components in the form of radicals. Radicals were looked initially as “harmful” molecules and this is true for large quantities but in small or even moderate amounts these molecules prove to have a physiological role. Reactive species are highly reactive and as a consequence are short living species. Their impact is supposed to be limited in the proximity area of their formation. Instead recent evidences indicate their implications in cellular signaling suggesting that individual chemical properties of reactive species make a difference in their biological role. This paper presents superoxide, nitric oxide and peroxide radical generation under cellular changing conditions, the adapting behavior of the enzymes that synthesize and remove them as well as some therapeutic target in superoxide related pathology.
Keywords:Superoxide Anion, Nitric Oxide Radical, Superoxide Dismutase, Gene Therapy

1. Introduction
Reactive species are produced in living organisms in well establish purpose or by accidental events. These fast acting molecules are suited to produce quick responses (killing invading organisms, promoting vasodilatation) but they also can be damaging to the cells. Thus reactive species are double swords molecules depending on the generated amounts, time of action and the cellular changing conditions. Literature shows that enzymes are able to adapt their behavior in order to keep the “redox homeostasis”. Deficiencies occurring in enzymes belonging to the antioxidant defense alter the adapting capacity of this system. As a consequence different drugs are envisaged to reestablish or to mimic the activity of some antioxidant enzymes. Our paper will focus on free radicals generation, SOD types and activities, as well as SOD therapeutic utilization.
2. Reactive Species Generation
A radical (also known as free radical) is highly reactive chemical specie that possesses a single unpaired electron in outer orbitals and is able to independently exist. Radicals are involved in extracting electron from any neighbor molecule in order to complete their own orbitals. Small and fast moving molecules are very efficient in oxidative activity and oxygen and nitrogen are suited to generate reactive species.
Beside oxygen and nitrogen transitional metals also have single unpaired electrons in theirs outer orbitals. They don’t behave as free radicals (because in living organisms they are attached to proteins in most cases) but they are able to transfer electron and consequently to generate free radicals.
There are two main groups of free radicals: ROS or reactive species of oxygen, RNS or reactive nitrogen species. ROS and RNS can act together damaging cells and causing nitrosative stress. Therefore, these two species are often collectively referred to as ROS/RNS.
3. Superoxide Generation
The starting point in reactive oxygen species is superoxide radical generation. Briefly superoxide is generated from oxygen a molecule that has two impaired electron having the same spin in the outer orbital. Getting an electron from no matter what substrate, oxygen becomes a radical, thus very reactive, named superoxide.
The structure of oxygen and superoxide is shown in Figure 1.
Superoxide can generate in its turn other potent species that can be either radical or non-radical. The path for reactive species generation is shown in Figure 2.
Almost all type of cells and intracellular organelles may generate superoxide anion through two different ways: using the enzymatic complexes within any cell or by hazard events that involve radiation, xenobiotics.
The reaction of superoxide with non-radicals is spin forbidden. In biological systems, this means superoxide reacts with itself (dismutation) or with another biological radical such as nitric oxide (NO•) or it reacts with a transition-series metal. Several sources for superoxide radical are: 1) the mitochondrial electron transport chain (ETC), 2) cytosolic xanthine and xanthine oxidase (XO), 3) the group of nitric oxide synthetizes (NOS) 4) membrane-associated NADPH oxidase complex (NoX), 5) hemoglobin in erythrocyte and recent added 6) homocysteine.
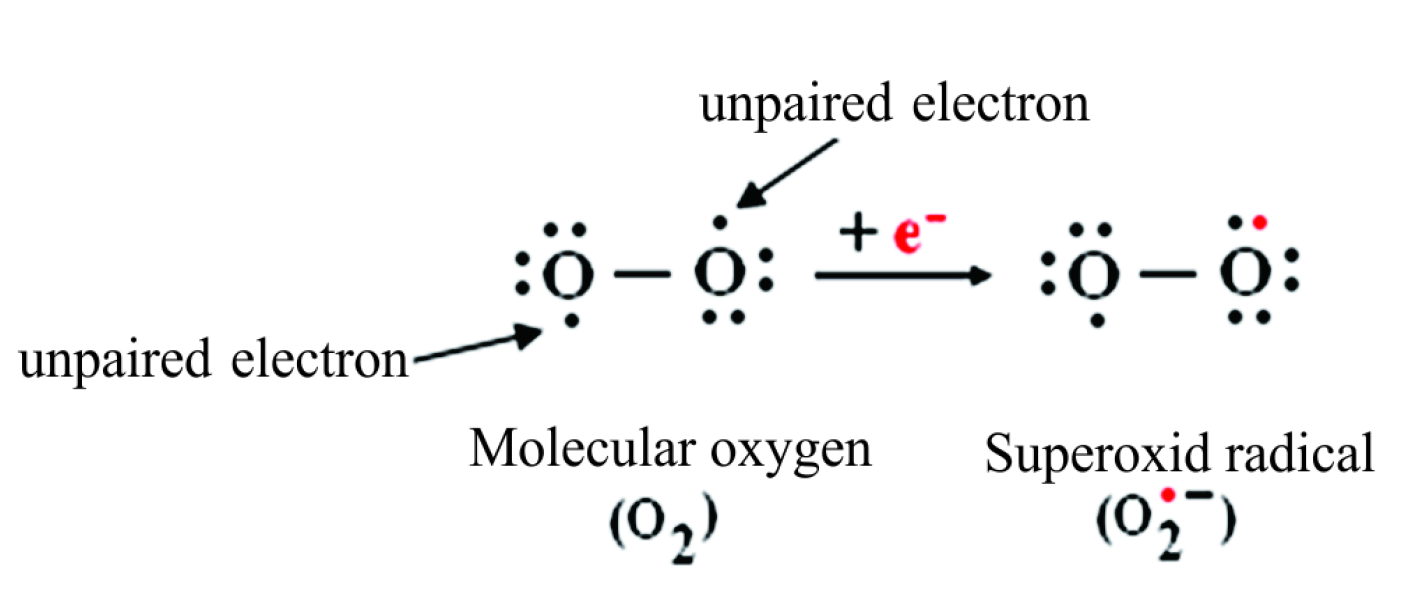
Figure 1. Chemical structure of oxygen and superoxide anion.
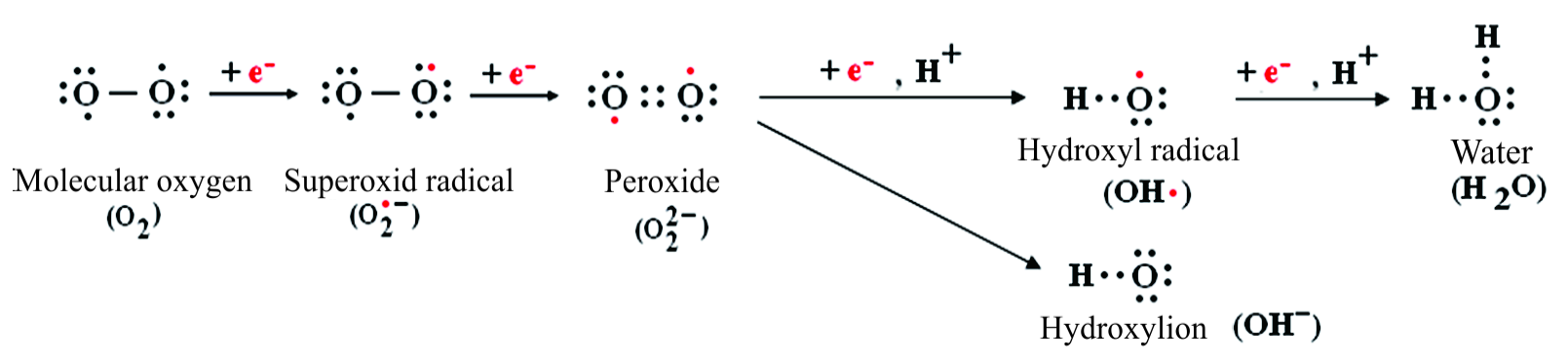
Figure 2. Reactive species oxidation from starting superoxide radical.
1) In mitochondria superoxide anions is mainly generated by complex I and III as a by-product. Superoxide is produced in mitochondria by slippage of an electron from the ETC to molecular oxygen during oxidative phosphorylation. In the process of electrons transport chain is considered that only 3% of total oxygen is consumed to generate superoxide radical [1] . Recent data present a method using a redox sensor that is able to catch the mitochondrial superoxide generation in complex I. The method provides new insights into the intimate relationship between mitochondrial energy production and ROS generation and signaling [2] .
2) Xanthine oxidase (XO) is a ubiquitous enzyme involved in a variety of physiological and pathophysiological processes. It plays a critical role in purine catabolism producing uric acid and hydrogen peroxide thereby contributing to other possible reactive species generation (Figure 2). In this process xanthine oxidase may also generate 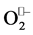 [3] . The ability of XO to generate
[3] . The ability of XO to generate  has been studied in the context of ischemia—reperfusion injury and heart failure [4] . In addition to that xanthine oxidase can also generate another radical NO• under hypoxic condition. J. O. Lunderg proposes the mechanism of NO• production by xanthine oxidase in ischemia [5] . XO can use as substrate either oxygen, in normal or hyperoxia, and nitrate in hypoxia. In hypoxia XO shifts from oxygen consumption to nitrite consumption. XO-catalyzed nitrite reduction to NO• is greatly enhanced in low oxygen tensions and in acidic conditions such as those seen during ischemia Figure 3.
has been studied in the context of ischemia—reperfusion injury and heart failure [4] . In addition to that xanthine oxidase can also generate another radical NO• under hypoxic condition. J. O. Lunderg proposes the mechanism of NO• production by xanthine oxidase in ischemia [5] . XO can use as substrate either oxygen, in normal or hyperoxia, and nitrate in hypoxia. In hypoxia XO shifts from oxygen consumption to nitrite consumption. XO-catalyzed nitrite reduction to NO• is greatly enhanced in low oxygen tensions and in acidic conditions such as those seen during ischemia Figure 3.
3) Nitric oxide synthases are a family of enzymes catalyzing the production of nitric oxide (NO•) from L-arginine. In human organism there are three enzymes isoforms: nNOS (neuronal NOS or NOS1), iNOS (inducible NOS or NOS2) and eNOS (endothelial NOS or NOS3). The isoform NOS1 is involved in cell communication, NOS3 in vasodilatation and both are constitutive enzymes. NOS 2 is inducible enzyme mainly involved in immune response [6] .
Recently it was found that endothelial NOS (eNOS) in “uncoupled situation” may generate 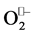 depending the availability of its substrates within cell (Figure 4) [7] . The endothelial nitric oxide synthase activity is regulated by a combination of mechanisms that allow eNOS to modulate its activity under physio-pathological condition [8] . eNOS contains 2 enzymatic domains, a flavin-containing reductase and a heme-containing oxygenase domain (Fe3+) connected by a regulatory calmodulin-binding domain. Binding of the Ca2+/calmodulin complex orients the other domains in such a position that NADPH-derived electrons generated on the reductase domain
depending the availability of its substrates within cell (Figure 4) [7] . The endothelial nitric oxide synthase activity is regulated by a combination of mechanisms that allow eNOS to modulate its activity under physio-pathological condition [8] . eNOS contains 2 enzymatic domains, a flavin-containing reductase and a heme-containing oxygenase domain (Fe3+) connected by a regulatory calmodulin-binding domain. Binding of the Ca2+/calmodulin complex orients the other domains in such a position that NADPH-derived electrons generated on the reductase domain
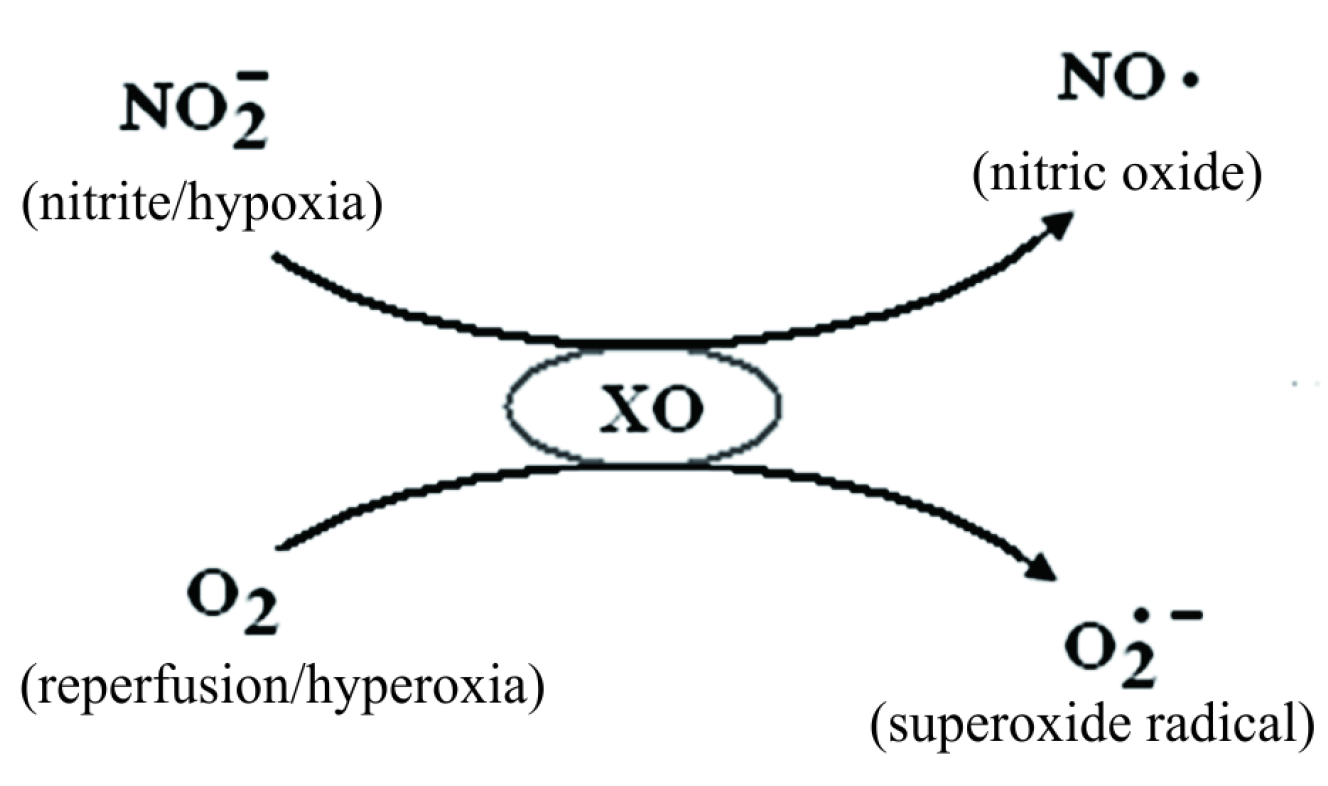
Figure 3. XO is able to generate either NO• or superoxide ( ) depending on the cellular changing conditions.
) depending on the cellular changing conditions.

Figure 4. Endothelial NOS differently behaves generating either NO• or  depending on the substrate availability.
depending on the substrate availability.
flow to the oxygenase domain [9] . The oxygenase domain of eNOS contains an iron ion (Fe3+) that binds oxygen on reduction Fe2+, and this complex finally causes the conversion of L-arginine to NO• and L-citrulline. This sequence of events properly rules if the cofactor tetrahidrobiopterine (BH4) “provides the connection” between the two domains. Deficiency of arginine or BH4 causes the reductase uncoupling from oxygenase. At the oxygenase domain intermediate Fe2+-O2 complex dissociates to form superoxide and the original Fe3+ group of the eNOS [10] . In this particular situation eNOS function in “uncoupled” way, here from the name “uncoupled eNOS”. Thus eNOS releases  instead of NO•.
instead of NO•.
4) The NADPH oxidase is a membrane-bound enzyme complex. It is made up of six subunits: one of them has GTP-ase activity while the others five have oxidase activity. One of these oxidase subunits is gp91-PHox (recently renamed NOX2) [11] [12] . The subunits enzyme complex can be found in the plasma membrane as well as in the membranes of phagosomes used by neutrophil white blood cells to engulf microorganisms.
Over the last decade, many studies have shown that the major source of ROS in the vascular wall is nonphagocytic NAD(P)H oxidase, which utilizes NADH/NADPH as the electron donor to reduce molecular oxygen and to produce . Activation of this enzyme requires the assembly of both cytosolic and membrane bound subunits to form a functional enzyme complex. In the vasculature the NAD(P)H oxidase complex is at least partly pre-assembled, as a significant proportion of NAD(P)H oxidase subunits are co-localized intracellularly in endothelial cells [4] .
. Activation of this enzyme requires the assembly of both cytosolic and membrane bound subunits to form a functional enzyme complex. In the vasculature the NAD(P)H oxidase complex is at least partly pre-assembled, as a significant proportion of NAD(P)H oxidase subunits are co-localized intracellularly in endothelial cells [4] .
The general reaction catalyzed by phagocytic/non-phagocytic NADP oxidase is:
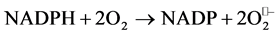
5) Molecular oxygen is carried from lung in order to be delivered to tissues and as a consequence it is found, for a short period of time, free, thus unbound. In this state it might be prone to generate reactive species. Oxygen binds to hemoglobin at the ferrous iron. The ferrous state (Fe2+) of iron is a condition for hemoglobin normal function. However a small percent of Fe2+ is slowly converted by O2 to ferric form (Fe3+) in resulting methemoglobin. An enzymatic system, methemoglobin reductase quickly restores Fe3+ to Fe2+ and reduces methemoglobin back to hemoglobin. Binding of oxygen to the iron in the hem is considered not to change the oxidation state of the metal. However oxygenated hem has some of the electronic characteristics of Fe3+-OO− peroxide anion [13] . Misra and Fridovich demonstrate that the Fe3+ O2− complex is able to generate superoxide radical [14] during the normal molecular oxygen transport to tissues through the hemoglobin auto-oxidation. Thus hemoglobin auto-oxidation causes superoxide formation within erythrocyte.
Other researchers confirm this finding showing that hemoglobin may undergo oxidative reaction in the oxygen releasing process. Balagopalakrishna [15] and coworkers demonstrate that at intermediate oxygen pressure, where hemoglobin partially releases molecular oxygen, the superoxide radical production increases. They show that superoxide radical is released in the hydrophobic hem pocket. The process in slow enough thus the formation of superoxide was followed for more than 15 min, and thus detected by low temperature electron paramagnetic resonance technique.
6) It was proved that homocysteine generates superoxide radicals thus promoting vasoconstriction. Lang et al. demonstrates [16] that the inhibitory effect of homocysteine on endothelium-dependent relaxation is caused by an increase of the intracellular levels of 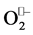 in the endothelial cell and provide a possible mechanism for the endothelial dysfunction associated with hyperhomocysteinemia.
in the endothelial cell and provide a possible mechanism for the endothelial dysfunction associated with hyperhomocysteinemia.
Intriguing new data put into a different light the superoxide anion. Pacher [17] shows that superoxide acts as a mild reducing agent under physiological conditions taking into account that its reduction potential is about −0.1 V at physiological oxygen concentrations.
The limited chemical reactivity of superoxide is supported by that fact that in biological environment its reaction with non-radicals is spin forbidden. As a consequence a more potent oxidant generated by superoxide was proposed in the form of hydroxyl radical non-enzymatic formation. Concentration of hydroxyl generation can’t be significantly high because of three reasonable doubts: first superoxide radical the major hydroxyl precursor (via hydrogen peroxide) is quickly removed by superoxide dismutase in normal cell, second generating hydroxyl Haber-Wiesse reaction (the reaction of iron with hydrogen peroxide) tends to have rather slow rate constants and third hydroxyl radical is so reactive that it will react with virtually every biological molecule within a very short diffusion distance. As a consequence the biological relevance of hydroxyl radical is limited and controversial in cellular toxicity [18] . As a conclusion another entity in the form of peroxynitrite was envisaged and added to reactive species starting from the above considerations.
4. NO Generation
A short inside in nitric oxide (NO•) generation is needed because it is linked to the other reactive species and to the activity of the enzymes operating with some of these radicals as also.
Nitrogen compounds found in the body comes from exogenous sources as nitrites/nitrates or from endogen production of nitric oxide. The group of nitrogen derivatives includes:
• NO• nitric oxide a natural free radical also named nitrogen monoxide is involved in vasodilatation in mammals; it is lipophilic and diffuses rapidly through membranes [19] .
• NO2− nitrogen dioxide or nitrite. In organism is found in its corresponding salts nitrites (from nitrous acid HNO2).
• NO3− nitrate (from nitric acid HNO3) also found in the body in corresponding salts.
Nitrogen derivatives convert into each other forward and backward continuously under shifting conditions within cells (Figure 5).
Nitric oxide structure is presented in Figure 6, is a colorless gas with low solubility in water.
Its hydrophobicity and, in consequence, its high diffusion rate in biological systems allows the molecule to reach the targets before degradation [20] . NO• is a messenger in many physiological processes: endothelial relaxation of the smooth muscle, inhibition of platelet aggregation, neurotransmission and cytotoxicity. NO• can be generated by NOS synthetizes group of enzymes in physiological conditions but also in particular cell conditions as hypoxia [5] by xanthine oxidase (Figure 3).
Reactions of nitric oxide differ between in vitro and in vivo systems. In vitro systems, the main degradation product of nitric oxide is NO2− (nitrite). In vivo nitric oxide can interact with other radicals or compounds. The main reactions of physiological relevance for nitric oxide are with: superoxide 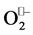 resulting ONOO− (peroxynitrite), himself resulting N2O3 (dinitrogen trioxide), hydroxyl resulting nitrites
resulting ONOO− (peroxynitrite), himself resulting N2O3 (dinitrogen trioxide), hydroxyl resulting nitrites 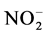 (from nitrous acid HNO2), and hemoglobin resulting methemoglobin and NO3− (nitrate) (Figure 7).
(from nitrous acid HNO2), and hemoglobin resulting methemoglobin and NO3− (nitrate) (Figure 7).
NO• reacts in a fast way with superoxide, generating toxic peroxynitrite (ONOO−) a powerful nitrating agent. Peroxynitrite reacts in its turn with amino acids in proteins resulting nitrosothiols and nitrosamines, these products being considered an evidence for an intense oxidative stress. This type of stress is known as nitrosative stress. Nitrosylated proteins were found in many pathologic and signaling processes as well.
Intriguing new data reveal adaptive behavior in superoxide generation for mitochondria, eNOS and XO enzymes.

Figure 5. Nitrogen derivatives.
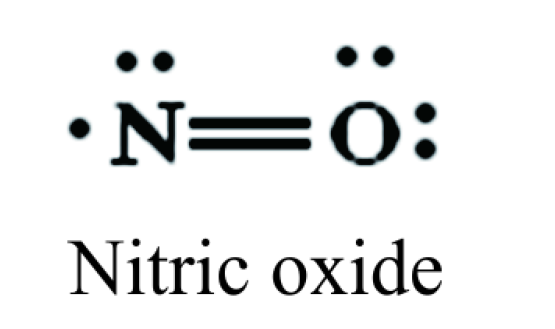
Figure 6. Nitric oxide chemical structure.

Figure 7. In vivo nitric oxide chemical reactions.
The main site of reactive oxygen species production is well known to be complex I and III in electron transport in mitochondria, in well oxygenated state, as electron from the ETC slip to molecular oxygen during oxidative phosphorylation. But recent studies demonstrate that endothelium cells surprisingly produce ROS under hypoxia also. The paradoxically increase in ROS production under low oxygenation is still not fully understood but it is considered that reactive oxygen species released during hypoxia act as signaling agents that trigger erythropoietin, vascular endothelial growth factor and glycolytic enzymes. Systemically, these responses enhance the delivery of O2 to cells and facilitate the production of glycolytic ATP instead of mitochondrion production. Induction of these genes is mediated by “specialized” hypoxia inducible factor 1 (HIF-1) [21] . As a conclusion when mitochondrion “senses” hypoxia it releases ROS as signaling molecules that activate diverse functional responses, including activation of gene expression that promote cell survival [2] . Wei and Dirksen indicates that the production of low to moderate levels of ROS/RNS and the frequency of production dictated by the particular cellular context and metabolic state is critical for the proper regulation of many essential cellular processes including gene expression and signal transduction [22] .
Stroes et al. demonstrate [23] [24] an intriguing activity for eNOS only, the simultaneous generation of both NO• and superoxide, even in the presence of BH4 and L-arginine, under physiological conditions, thereby not in “uncoupled state”. The consequence is the production of peroxynitrite by eNOS (Figure 7) a highly reactive molecule. Peroxynitrite anion (ONOO−) is a reactive species of increasingly recognized biological relevance that contributes to oxidative tissue damage. Recent research indicates [25] that peroxynitrite is able to cross the erythrocyte membrane and also other cells membrane by two different mechanisms: in the anionic form through the anion exchange channel, and in the protonated form by passive diffusion and thus influencing cell adaptive response.
These findings come into contradiction with previous presented data that sustained 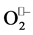 generation is possible only in uncoupled eNOS state [10] .
generation is possible only in uncoupled eNOS state [10] .
The balance between substrate supply (arginine, tetrahidrobiopterine (BH4), nitrite/nitrate) and tissue oxygenation may determine whether the net effects of the combined activity of the above mentioned enzymes are beneficial or harmful in a particular situation. Thus, the dominating species generated could be NO•, oxygen radicals, or their reaction products.
5. Superoxide Dismutase
Superoxide dismutase (SOD) represents a group of enzymes that use as cofactor copper and zinc, or manganese, iron, or nickel ions. There are three major families of superoxide dismutase, depending on the metal cofactor: Cu/Zn (which binds both copper and zinc), Fe and Mn types (which bind either iron or manganese), and the Ni type, which binds nickel (only in prokaryotes). SOD1 is located in the cytoplasm, SOD2 in the mitochondria, and SOD3 is extracellular. The first is a dimer (consists of two units), whereas the others are tetramers (four subunits). SOD1 and SOD3 contain copper and zinc, whereas SOD2, the mitochondrial enzyme, has manganese in its reactive site.
5.1. Superoxide Dismutase Activity
Superoxide dismutase remove  by catalyzing its dismutation, one
by catalyzing its dismutation, one 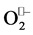 is being oxidized to O2 and another is reduced to H2O2 (Figure 8).
is being oxidized to O2 and another is reduced to H2O2 (Figure 8).
In other words one radical loses its electron and the other gains an extra-electron, in “unequal and disproportioned” way. The disproportion is accomplished by Fe-, Mnand Cu, Zn-superoxide dismutases (SODs) in two steps, which are both first-order with respect to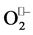 . The dismutation of superoxide
. The dismutation of superoxide  by SOD is very efficient having the largest kcat/KM (an approximation of catalytic efficiency) of any known enzyme (~7 × 109 M−1·s−1) [8] . SOD catalyst activity is limited only by the frequency of collision with superoxide. That means the reaction rate is limited only by the diffusion of superoxid radical. Diffusion limitation becomes canceled in radicals over production.
by SOD is very efficient having the largest kcat/KM (an approximation of catalytic efficiency) of any known enzyme (~7 × 109 M−1·s−1) [8] . SOD catalyst activity is limited only by the frequency of collision with superoxide. That means the reaction rate is limited only by the diffusion of superoxid radical. Diffusion limitation becomes canceled in radicals over production.

Figure 8. Superoxide dismutase activity.
The mechanism of reaction in presented in well-argued reference [26] which explains how this group of enzymes can at neutral pH to overcome the electrostatic repulsion between two anions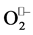 . The dismutation reaction takes place in two steps in a Ping-Pong mechanism [27] . In the first step the ion metal is reduced by a first
. The dismutation reaction takes place in two steps in a Ping-Pong mechanism [27] . In the first step the ion metal is reduced by a first 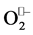 which loses its electron and is converted to molecular oxygen. In this first step the net positive charge of ion metal (Cu2+, Zn2+, Mn3+, Fe3+) facilitates the
which loses its electron and is converted to molecular oxygen. In this first step the net positive charge of ion metal (Cu2+, Zn2+, Mn3+, Fe3+) facilitates the 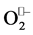 binding (at neutral pH, HO2• ↔
binding (at neutral pH, HO2• ↔ 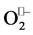 + H+ has a pK of 4.5), thus this first step requires only substrate binding and electron transfer, and so it is very favorable. In the second step the electron taken from
+ H+ has a pK of 4.5), thus this first step requires only substrate binding and electron transfer, and so it is very favorable. In the second step the electron taken from 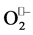 must be “mutated” (in order to restore the initial oxidation state of metal ion) on a second
must be “mutated” (in order to restore the initial oxidation state of metal ion) on a second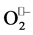 . By supplying even only one of the protons required for formation of H2O2, the enzyme can greatly promote and accelerate reduction of the second
. By supplying even only one of the protons required for formation of H2O2, the enzyme can greatly promote and accelerate reduction of the second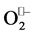 . The proton aid is necessary (for
. The proton aid is necessary (for 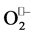 binding/reduction) because the reduced metal ion itself is less positively charged (Cu+, Zn2+, Mn2+, Fe2+) and less disposed to coordinate another
binding/reduction) because the reduced metal ion itself is less positively charged (Cu+, Zn2+, Mn2+, Fe2+) and less disposed to coordinate another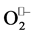 . Thus, the favorable first half-reaction is coupled to uptake of one proton, which in turn can facilitate second
. Thus, the favorable first half-reaction is coupled to uptake of one proton, which in turn can facilitate second 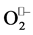 binding and subsequent reduction. Proton uptake coupled to metal ion reduction was demonstrated in the basic paper of Bull and Fee [28] and in more recent studies [29] [30] .
binding and subsequent reduction. Proton uptake coupled to metal ion reduction was demonstrated in the basic paper of Bull and Fee [28] and in more recent studies [29] [30] .
The dismutation of superoxide radical is accompanied by H2O2 generation a non-radical product. Hydrogen peroxide (H2O2) is lipid soluble and as a consequence it can diffuse through lipid membranes. It has a longer biological life span than 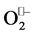 thus being able of activating different signaling pathways [4] . The products of superoxide dismutation, i.e. hydrogen peroxide and oxygen, may also play direct signaling roles in the intracellular milieu as well [31] .
thus being able of activating different signaling pathways [4] . The products of superoxide dismutation, i.e. hydrogen peroxide and oxygen, may also play direct signaling roles in the intracellular milieu as well [31] .
Recent data consider that another physiological “task” of SOD activity is the inhibition of oxidative inactivation of nitric oxide, and preventing also peroxynitrite formation (Figure 9) [32] and dysfunction of mitochondrion and endothelium [33] .
5.2. Superoxide Dismutase Types
5.2.1. SOD 1
The intracellular copper, zinc dependent superoxide dismutase play an important role in protecting components within many compartments of eukaryotic cells against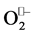 . Our team had studied the activity of intra-erythro-
. Our team had studied the activity of intra-erythro-
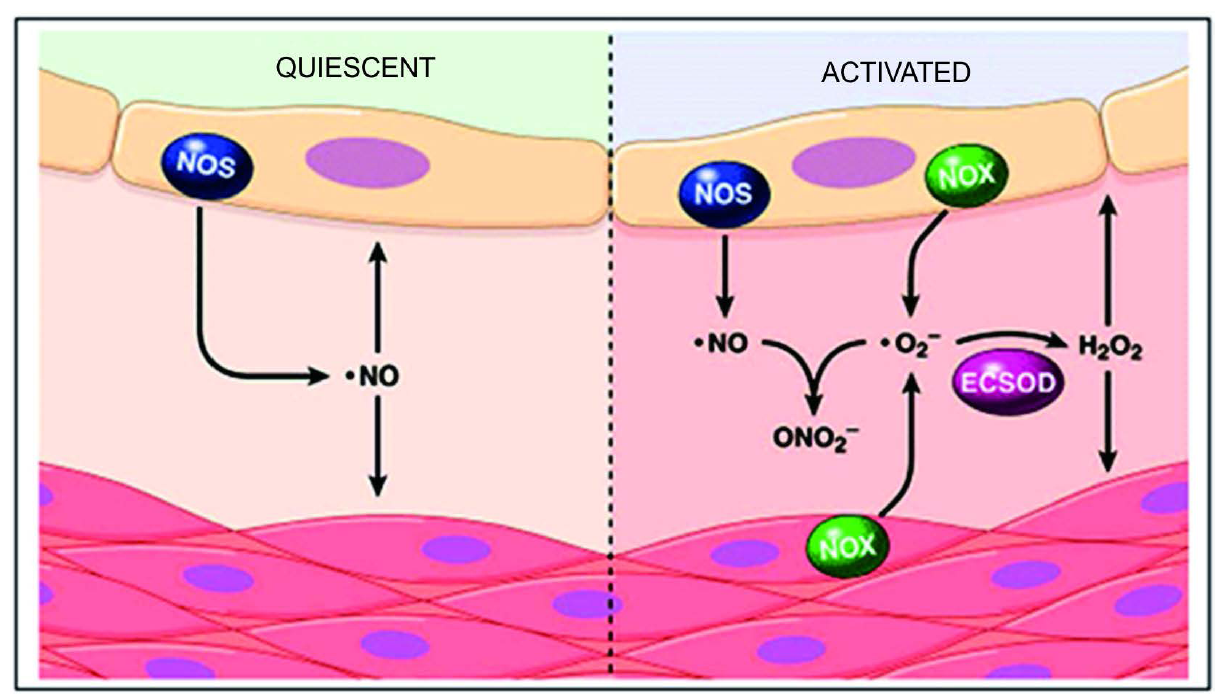
Figure 9. In quiescent state in the vasculature, NO• is generated predominantly by endothelial NO• synthase (NOS) in order to diffuses into endothelial and smooth muscle cells in order to sustain the vascular relaxation. In activated state in the vasculature, 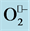 is generated in the extracellular space by endothelial and smooth muscle cell NADPH oxidase (NOX) complexes. The
is generated in the extracellular space by endothelial and smooth muscle cell NADPH oxidase (NOX) complexes. The 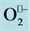 reacts with and inactivates NO•, forming peroxynitrite (ONO2). The action of extracellular superoxide dismutase (ECSOD), removes
reacts with and inactivates NO•, forming peroxynitrite (ONO2). The action of extracellular superoxide dismutase (ECSOD), removes 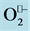 by converting into H2O2 thus preventing NO inactivation. Taken from [32] .
by converting into H2O2 thus preventing NO inactivation. Taken from [32] .
cyte SOD in three type of pathologic condition, experimentally induced in rats: diabetes (alloxan induced), hyperhomocysteinemia (induced by methionine loading) and chronic induced stress (by reversing normal daynight cycle). We have published data regarding superoxide dismutase activity in hyperhomocysteinemia experimentally induced in rats where we found a positive association of SOD activity with homocysteine levels, in red blood cells lysate [34] . In the induced diabetes and chronic stress SOD activity was found higher than control groups but lower when compared to SOD activity in hyperhomocysteine induced model (unpublished data). This data suggests a higher oxidative potential in hyperhomocystemia.
5.2.2. SOD2
In contrast to SOD1, the manganese containing SOD2 in mitochondria plays an essential role in oxidative stress protection. Complete loss of the enzyme results in neonatal lethality in mice [35] and is also critical for growth and viability of other eukaryotic organisms [36] .
Mn-SOD can be inactivated by oxidative and nitrosative stress as well. Nitration of a single tyrosine residue (Tyr-34) leads to complete enzyme inactivation [37] , with the possible consequence to favor peroxynitrite generation in mitochondrion. Nitration-induced Mn-SOD inactivation was linked to ischemia/reperfusion, inflammation, and in human kidney allograft rejection and human pancreatic ductal adenocarcinoma [38] , tyrosine nitration being identified in more than 50 human diseases.
5.2.3. SOD3
Extracellular superoxide dismutase (ECSOD or SOD3) is localized in extracellular fluids such as lymph, synovial fluid, and plasma [39] . ECSOD is highly expressed in blood vessels, particularly arterial walls, and is the predominant form of SOD in the aortas of humans [40] . Extracellular superoxide dismutase is synthesized in vascular smooth muscle cells and secreted into the extracellular environment where it binds to endothelial surface components [41] . Its activity keeps in control superoxide level in order to prevent nitric oxide inactivation in the vasculature region [42] .
Recent evidence shows that plasma ECSOD presents three distinct domains (when subjected to chromatography): domain A (with no heparin affinity), domain B (with weak heparin affinity), and domain C (with strong heparin affinity) [43] [44] . These findings suggest that C domain anchors the protein to the endothelial cell surfaces and the extracellular matrix of blood vessels. The major portion of ECSOD in the vasculature primarily exists in the extracellular matrix and, to a lesser extent, on endothelial cell surfaces linked to heparan sulfate proteoglycans, but a small fraction of ECSOD exists in equilibrium between cell surfaces/matrix and plasma [40] . The hypotheses about ECSOD binding and/or releasing from endothelium surface is thoroughly presented in [45] .
Taking into account ECSOD localization it is not surprising that ECSOD deficiency is linked to many pathological processes related to vascular injury: hypertension, atherosclerosis, diabetes, and ischemia/reperfusion.
Many studies notice the extracellular SOD deficiency is involved in hypertension and atherosclerosis [46] [47] . Surprisingly genetic deletion of ECSOD had not consequences as severe as mitochondrial SOD deletion (which causes neonatal lethality in mice) and there was found no significant effect on the size of the aortic lesion generated in response to a pro-atherosclerotic diet [48] .
Hypertension onset in diabetes may be induced by the loss of ECSOD from the vasculature ECSOD. In diabetes high levels of glucose trigger the non-enzymatic glycosylation of ECSOD. This cleaves heparin-binding domain leading to the loss of linked ECSOD from endothelium and a decrease in ECSOD capacity of scavenging . As a consequence available NO• diminishes due to superoxide-mediated inactivation of nitric oxide and vasoconstriction installs [49] .
. As a consequence available NO• diminishes due to superoxide-mediated inactivation of nitric oxide and vasoconstriction installs [49] .
Homocysteine is the late added cardiovascular risk factor and is supposed to have an influence on ECSOD activity. Indeed a positive correlation between levels of total plasma homocysteine and plasma ECSOD was found [50] . Literature present three possible mechanisms for homocysteine influence on ECSOD: first homocysteine auto-oxidation generates superoxide and/or peroxynitrite that may up-regulate ECSOD expression in the vasculature. A second mechanism considers that homocysteine alters the binding domain C (heparin high affinity) and thus ECSOD binds to endothelial cells [51] . The third mechanism considers that homocysteine alters disulfide bond formation/glycosylation of ECSOD and as a consequence it alters the proper assembling and secretion of ECSOD from endoplasmic reticulum [52] .
6. Reactive Species in Cellular Signaling
Radicals were looked initially as “harmful” molecules and this is true for large quantities but in small or even moderate amounts these molecule prove to have physiological role.
Reactive species are highly reactive and as a consequence are short living species. Theirs impact is supposed to be limited in the proximity area of their formation. Instead recent evidences indicate their implications in cellular signaling suggesting that individual chemical properties of reactive species make difference in their biological role [53] .
Signal transduction pathways use cascades of protein and lipid kinase mediated phosphorylation to elicit specific responses to distinct extracellular stimuli. This chain put into contact the extracellular and intracellular environment. I this chain transduction the most closed to the membrane is a class of proteins named mitogen-activated protein kinases (generally noted MAPK 1-4 upon the location in the cascade, MAPK4 or MAPKKKK being the upper in the hierarchy). These proteins are activated by the growth factor receptors, small G proteins (Ras, Rac, Cdc42) situated in the upstream of the cascade [54] .
In the downstream of the cascade are MAPK1s which target different proteins as well as transcription factors, whose activation regulates almost all critical cellular function (proliferation, apoptosis, and inflammatory genes expression). MAPK1s is a group of proteins that contains three main classes of regulatory proteins: ERK (extracellular-signaling regulating kinase, ERK1 and ERK2), JNK (SAPK/JNKs or stress—activated protein kinase/ JUN amino-terminal kinases) and p38k (α, β, γ, δ). These proteins are all activated by a dual phosphorylation at a specific site by the protein kinase cascade (MAPK 4/MAPK 3/MAPK 2) [55] .
Being highly reactive ROS can intercept cell signaling pathways within any successive steps in cascade events modulating the functions of many enzymes and transcription factors. Oxidative stress triggers cellular response by activating/inhibiting many signaling pathways (Figure 10).
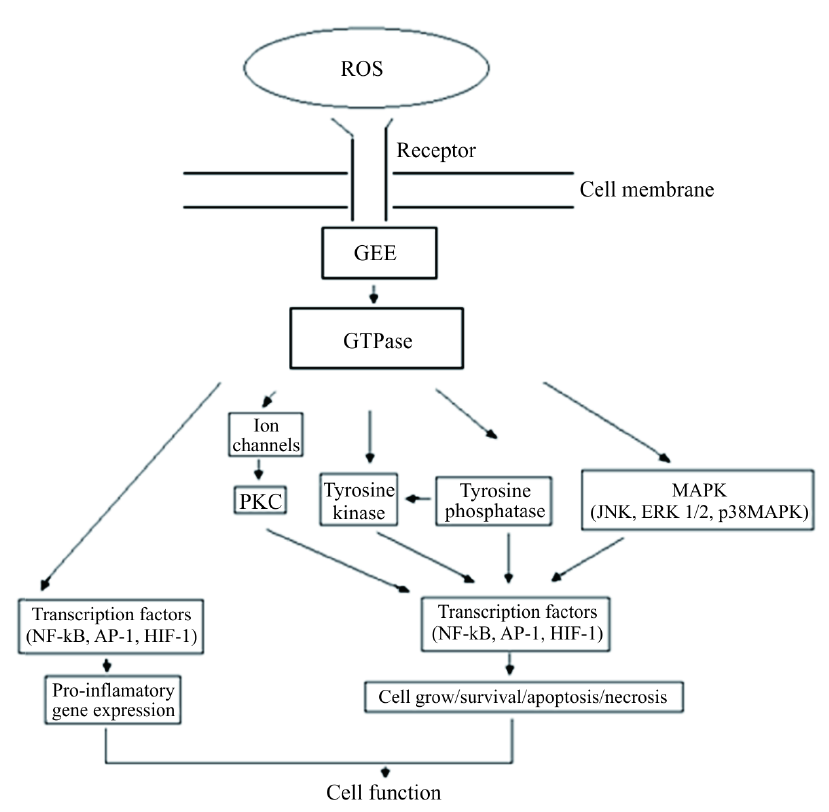
Figure 10. Major pathways activated by ROS generation, GES = guanine nucleotide exchanging factor, GTPase = protein activated by GDP changing to GTP, MAPK = mitogen-activated protein, JNK = c-Jun N-terminal kinases, ERK = extracellular signal-regulated kinases, NFκB = nuclear factor κB, AP-1 = activator protein-1, HIF-1 = hypoxia-inducible factor-1, PKC = protein kinase C.
More details about the cellular response in ROS and other radical and non-radicals species attack in oxidative events can be found in [53] [56] -[60] .
As a general conclusion, the superoxide dismutase acts for preventing reactive species accumulation. Lack of this enzyme means the shortening of lifespan. In this case medication is supposed to use antioxidant supplementation or SOD activity mimics compounds. On the opposite way super-expression/-activation of this enzyme may lead to supplementary reactive species that damage tissues. In this case medication must use inhibitory competitive drugs in order to restore oxidation-reducing balance.
7. Therapeutic Targets in SOD Related Pathology
7.1. Gene Therapy
One of the most important clinical contexts in which SOD antioxidant protection represents the main mechanism is renal ischemia-reperfusion injury. Ischemia and reperfusion leads not only to an increase in superoxide production, but also to a rapid depletion of SOD. Therefore, ensuring the necessary amount of SOD to combat the superoxide radical byproduct of ischemia-reperfusion processes should be done from an external source. Exogenous SOD administration has a short half-life in plasma. The entering SOD gene in renal tissue is a way to ensure a continuous production of SOD to achieve protection from renal ischemia-reperfusion injury. In this regard, during experiments on animal models the effective gene delivery was established by intravenous injection of the gene vectors before the ischemic insult without toxic side effects. The use of adenovirus as a vector for kidney-directed gene therapy has made significant progress in the area of kidney biology, in particular in hereditary kidney disease and inflammatory and fibrotic disease [61] . Adenoviral vectors has some advantages which make them suitable for gene transfer into complex organs such as the kidney, it realize high titers and high expression of the transgene, it can infect both dividing and non-dividing cells and delivering genes into quiescent or terminally differentiated cells. Adenoviral vectors also have disadvantages: the expression of the transfected gene is limited to weeks or months because the adenovirus does not integrate into the host genome, the adenovirus can elicit immunological responses, therefore, the vector cannot be administered repeatedly. The indication of SOD gene therapy with adenoviral vector is recommended during emergency situations, with harmful effects maximum within a week (e.g., post-transplant acute renal failure) and in other inflammatory renal disease states.
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disorder characterized by death of motor neurons leading to muscle wasting, paralysis, and death, usually within 2 - 3 years of symptom onset. The oxidative stress is a central mechanism by which motor neuron death occurs in familial ALS and this is due to the mutations in the antioxidant enzyme superoxide dismutase 1 gene (SOD1). The most effective therapeutic effects in amyotrophic lateral sclerosis (ALS) ; mouse models have been obtained with the delivery of viral vectors to mediate expression of either growth factors such IGF-1, glial cell-derived neurotrophic factor, and VEGF (11-13) or RNAi molecules to silence SOD1 mutant gene expression [62] .
7.2. Therapy of Enhanced Efficiency of Superoxide Dismutase-Induced Cardioprotection
It has been reported that combined administration of SOD and catalase effectively reduces myocardial reperfusion injury [63] . In this case, coadministration of catalase may further enhance the cardioprotective effect of retrograde intracoronary infusion of SOD. It is known that Na+/H+ exchanger inhibitors, calcium antagonists, renin-angiotensin system antagonists, adenosine and nitric oxide donors provides cardioprotection during primary angioplasty for acute myocardial infarction [64] . The efficiency of these reagents has enhanced when they were administrated using intracoronary infusion. Anterograde intracoronary and intravenous administration of anti-P-selectin and anti-intercellular adhesion molecule (ICAM)-1 antibodies are also known to attenuate myocardial ischemia-reperfusion injury [65] . Retrograde intracoronary infusion, which has direct access to postcapillary venules, may be the ideal injection route for these antibodies.
7.3. Immunotherapy in Treatment of SOD1 Gene Mutation Disease
In amyotrophic lateral sclerosis (ALS) the toxicity of the proteins coded by mutant SOD1 gene leads to death of neurons. Using an antibody specific for mutant SOD1, such as the C4F6 monoclonal antibody, it can be neutralized or eliminated the toxicity of SOD1 species without affecting the WT SOD1 which in term confers benefits [66] .
8. Conclusion
It is generally accepted that free radicals are involved in pathological processes. The main damaging mechanism relies on the higher concentrations of reactive species that finally injures all cellular constituents. The lack or dysfunctions in the antioxidant enzymes lead to the shortening of the lifespan. As a consequence restoring the normal function of the antioxidant enzymes and SOD also must be the first therapeutic approach.
References
- Vijayvergiya, C., Beal, M.F., Buck, J. and Manfredi, G. (2003) Mutant Superoxide Dismutase 1 Forms Aggregates in the Brain Mitochondrial Matrix of Amyotrophic Lateral Sclerosis Mice. Journal of Neuroscience, 25, 2463-2470. http://dx.doi.org/10.1523/JNEUROSCI.4385-04.2005
- Wei, L. and Dirksen, R.T. (2012) Mitochondrial Superoxide Flashes: From Discovery to New Controversies. The Journal of General Physiology, 139, 425-434. http://dx.doi.org/10.1085/jgp.201210790
- Xu, S.P. and Touyz, R.M. (2006) Reactive Oxygen Species and Vascular Remodeling in Hypertension: Still Alive. Canadian Journal of Cardiology, 22, 947-951. http://dx.doi.org/10.1016/S0828-282X(06)70314-2
- Paravicini, T.M. and Touyz, R.M. (2006) Redox Signaling in Hypertension. Cardiovascular Research, 71, 247-258. http://dx.doi.org/10.1016/j.cardiores.2006.05.001
- Lunderg, J.O. and Weitzber, E. (2005) NO Generation from Nitrite and Its Role in Vascular Control. Arteriosclerosis, Thrombosis, and Vascular Biology, 25, 915-922. http://dx.doi.org/10.1161/01.ATV.0000161048.72004.c2
- Stuehr, D.J. (1999) Mammalian Nitric Oxide Synthases. Biochim. Biophys. Acta, 1411, 217-230. http://dx.doi.org/10.1016/S0005-2728(99)00016-X
- van Faassen, E.E., et al. (2009) Nitrite as Regulator of Hypoxic Signaling in Mammalian Physiology. Medicinal Research Reviews, 29, 683-741. http://dx.doi.org/10.1002/med.20151
- Heinrich, P., Löffler, G. and Petrides, P.E. (2006) Biochemie und Pathobiochemie. 123.
- Abu-Soud, H.M. and Stuehr, D.J. (1993) Nitric Oxide Synthases Reveal a Role for Calmodulin in Controlling Electron Transfer. Proceedings of the National Academy of Sciences of the United States of America, 90, 10769-10772. http://dx.doi.org/10.1073/pnas.90.22.10769
- van Hinsbergh, V.W.M. (2001) NO or H2O2 for Endothelium-Dependent Vasorelaxation. Tetrahidrobiopterin Makes the Differences. Arteriosclerosis, Thrombosis, and Vascular Biology, 21, 719-772. http://dx.doi.org/10.1161/01.ATV.21.5.719
- Tojo, T., Ushio-Fukai, M., Yamaoka-Tojo, M., Ikeda, S., Patrushev, N. and Alexander, R.W. (2005) Role of gp91phox (Nox2)-Containing NAD(P)H Oxidase in Angiogenesis in Response to Hindlimb Ischemia. Circulation, 111, 2347- 2355. http://dx.doi.org/10.1161/01.CIR.0000164261.62586.14
- Raad, H., Paclet, M.-H., Boussetta, T., Kroviarski, Y., Morel, F., Quinn, M.T., Gougerot-Pocidalo, M.-A., Dang, P.M.- C. and El-Benna, J. (2009) Regulation of the Phagocyte NADPH Oxidase Activity: Phosphorylation of gp91phox/ NOX2 by Protein Kinase C Enhances Its Diaphorase Activity and Binding to Rac2. FASEB Journal, 23, 1011-1022. http://dx.doi.org/10.1096/fj.08-114553
- Metzler, D.E. (2001) Biochemistry: The Chemical Reaction of Living Cells. Elsevier Academic Press.
- Misra, H.P. and Fridovici, I. (1972) The Generation of Superoxide Radical during the Autoxidation of Hemoglobin. The Journal of Biological Chemistry, 247, 6960-6962.
- Balagopalakrishna, C., Manoharan, P.T., Abugo, O.O. and Rifkind, J.M. (1996) Production of Superoxide from Hemoglobin-Bound Oxygen under Hypoxic Condition. Biochemistry, 35, 6393-6398. http://dx.doi.org/10.1021/bi952875+
- Lang, D., Kredan, M.B., Moat, S.J., et al. (2000) Homocysteine-Induced Inhibition of Endothelium-Dependent Relaxation in Rabbit Aorta: Role for Superoxide Anions. Arteriosclerosis, Thrombosis, and Vascular Biology, 20, 422-427. http://dx.doi.org/10.1161/01.ATV.20.2.422
- Pacher, P., Beckman, J.S. and Liaudet, L. (2007) Nitric Oxide and Peroxynitrite in Health and Disease. Physiological Reviews, 87, 315-424. http://dx.doi.org/10.1152/physrev.00029.2006
- Sawyer, D.T. and Valentine, J. (1981) How Super Is Superoxide? Accounts of Chemical Research, 14, 393-400. http://dx.doi.org/10.1021/ar00072a005
- Bhagavan, N.V. (2002) Medical Biochemistry. Harcourt/Academic Press.
- Agullo, L. (2007) Ciclic GMP Advances.
- Guzy, R.D. and Schumacker, P.T. (2006) Oxygen Sensing by Mitochondria at Complex III: The Paradox of Increased Reactive Oxygen Species during Hypoxia. Experimental Physiology, 91, 807-819. http://dx.doi.org/10.1113/expphysiol.2006.033506
- Powers, S.K., Talbert, E.E. and Adhihetty, P.J. (2011) Reactive Oxygen and Nitrogen Species as Intracellular Signals in Skeletal Muscle. Journal of Physiology, 589, 2129-2138. http://dx.doi.org/10.1113/jphysiol.2010.201327
- Stroes, E., Hijmering, M., van Zandvoort, M., Wever, R., Rabelink, T. and van Faassen, E. (1998) Origin of Superoxide Production by Endothelial Nitric Oxide Synthase. FEBS Letters, 438, 161-164. http://dx.doi.org/10.1016/S0014-5793(98)01292-7
- Stroes, E., Rabelink, T. and van Faassen, E. (2002) Vascular Protection: Molecular Mechanisms, Novel Therapeutic Principles and Clinical Applications.
- Denicola, A., Souza, J.M. and Radi, R. (1998) Diffusion of Peroxinitrite across Erythrocytes Membrane. Proceedings of the National Academy of Sciences of the United States of America, 95, 3566-3571. http://dx.doi.org/10.1073/pnas.95.7.3566
- Miller, A.F. (2004) Superoxide Dismutases: Active Sites that Save, but a Protein that Kills. Current Opinion in Chemical Biology, 8, 162-168. http://dx.doi.org/10.1016/j.cbpa.2004.02.011
- Miller, A.F. (2003) Superoxide Processing. In: Que Jr., L. and Tolman, W., Eds., Coordination Chemistry in the Biosphere and Geosphere, Pergamon, Oxford, Amsterdam, New York and Tokyo, 479-506.
- Bull, C. and Fee, J.A. (1985) Steady-State Kinetic Studies of Superoxide Dismutases: Properties of the Iron Containing Protein from Escherichia coli. Journal of American Chemical Society, 107, 3295-3304. http://dx.doi.org/10.1021/ja00297a040
- Miller, A.F., Padmakumar, K., Sorkin, D.L., Karapetian, A. and Vance, C.K. (2003) Proton-Coupled Electron Transfer in Fe-Superoxide Dismutase and Mn-Superoxide Dismutase. Journal of Inorganic Biochemistry, 93, 71-83. http://dx.doi.org/10.1016/S0162-0134(02)00621-9
- Han, W.G., Lovell, T. and Noodleman, L. (2003) Coupled Redox Potentials in Manganese and Iron Superoxide Dismutases from Reaction Kinetics and Density Functional/Electrostatics Calculations. Inorganic Biochemistry, 41, 205-218. http://dx.doi.org/10.1021/ic010355z
- Immenschuh, S. and Baumgart-Vogt, E. (2005) Peroxiredoxins, Oxidative Stress, and Cell Proliferation. Antioxidants & Redox Signaling, 7, 768-777. http://dx.doi.org/10.1089/ars.2005.7.768
- James R Stone Lab-Projects. www.vascularpath.org
- Paravicini, T.M., Chrissobolis, S., Drummond, G.R. and Sobey, C.G. (2004) Increased NAD(P)H-Oxidase Activity and Nox4 Expression during Chronic Hypertension Is Associated with Enhanced Cerebral Vasodilatation to NAD(P)H in Vivo. Stroke, 35, 584-589.
- Filip, C., Albu, E., Zamosteanu, N., Jaba Irina, M., Silion, M., Jerca, L., Gheorghita, N. and Costel, M.O. (2010) Hyperhomocysteinemia’s Effect on Antioxidant Capacity on Rats. Central European Journal of Medicine, 5, 620-626. http://dx.doi.org/10.2478/s11536-010-0032-7
- Lebovitz, R.M., Zhang, H., Vogel, H., Cartwright Jr., J., Dionne, L., Lu, N., Huang, S. and Matzuk, M.M. (1996) Neurodegeneration, Myocardial Injury, and Perinatal Death in Mitochondrial Superoxide Dismutase-Deficient Mice. Proceedings of the National Academy of Sciences of the United States of America, 93, 9782-9787. http://dx.doi.org/10.1073/pnas.93.18.9782
- Duttaroy, A., Paul, A., Kundu, M. and Belton, A. (2003) A Sod2 Null Mutation Confers Severely Reduced Adult Life Span in Drosophila. Genetics, 165, 2295-2299.
- Kirby, K., Hu, J., Hilliker, A.J. and Phillips, J.P. (2002) RNA Interference-Mediated Silencing of Sod2 in Drosophila Leads to Early Adult-Onset Mortality and Elevated Endogenous Oxidative Stress. Proceedings of the National Academy of Sciences of the United States of America, 99, 16162-16167. http://dx.doi.org/10.1073/pnas.252342899
- Yamakura, F. and Kawasaki, H. (2010) Post-Translational Modifications of Superoxide Dismutase. Biochimica et Biophysica Acta, 1804, 318-325. http://dx.doi.org/10.1016/j.bbapap.2009.10.010
- Fattmen, C.L., Schaefer, L.M. and Oury, T.D. (2003) Extracellular Superoxide Dismutase in Biology and Medicine. Free Radical Biology & Medicine, 35, 236-256. http://dx.doi.org/10.1016/S0891-5849(03)00275-2
- Oury, T.D., Day, B.J. and Crapo, J.D. (1996) Extracellular Superoxide Dismutase in Vessels and Airways of Humans and Baboons. Free Radical Biology & Medicine, 20, 957-965. http://dx.doi.org/10.1016/0891-5849(95)02222-8
- Stralin, P., Karlsson, K., Johansson, B.O. and Marklund, S.L. (1995) The Interstitium of the Human Arterial Wall Contains very Large Amounts of Extracellular Superoxide Dismutase. Arteriosclerosis, Thrombosis, and Vascular Biology, 15, 2032-2036. http://dx.doi.org/10.1161/01.ATV.15.11.2032
- Culotta, V.C., Yang, M. and O’Halloran, T.V. (2006) Activation of Superoxide Dismutases: Putting the Metal to the Pedal. Biochimica et Biophysica Acta, 1763, 747-758.
- Macmillan-Crow, L.A. and Cruthirds, D.L. (2001) Invited Review: Manganese Superoxide Dismutase in Disease. Free Radical Research, 34, 325-336. http://dx.doi.org/10.1080/10715760100300281
- Sandstrom, J., Karlsson, K., Edlund, T. and Marklund, S.L. (1993) Heparin-Affinity Patterns and Composition of Extracellular Superoxide Dismutase in Human Plasma and Tissues. Biochemical Journal, 294, 853-857.
- Bowler, R.P., Nicks, M., Olsen, D.A., Thogersen, I.B., Valnickova, Z., Hojrup, P., Franzusoff, A., Enghild, J.J. and Crapo, J.D. (2002) Furinproteolytically Processes the Heparin-Binding Region of Extracellular Superoxide Dismutase. Journal of Biological Chemistry, 277, 16505-16511. http://dx.doi.org/10.1074/jbc.M105409200
- Gongora, M.C., Qin, Z., Laude, K., Kim, H.W., McCann, L., Folz, J.R., Dikalov, S., Fukai, T. and Harrison, D.G. (2006) Role of Extracellular Superoxide Dismutase in Hypertension. Hypertension, 48, 473-481. http://dx.doi.org/10.1161/01.HYP.0000235682.47673.ab
- Lob, H.E., Marvar, P.J., Guzik, T.J., Sharma, S., McCann, L.A., Weyand, C., Gordon, F.J. and Harrison, D.G. (2010) Induction of Hypertension and Peripheral Inflammation by Reduction of Extracellular Superoxide Dismutase in the Central Nervous System. Hypertension, 55, 277-283. http://dx.doi.org/10.1161/HYPERTENSIONAHA.109.142646
- Sentman, M.L., Brannstrom, T., Westerlund, S., Laukkanen, M.O., Yla-Herttuala, S., Basu, S. and Marklund, S.L. (2001) Extracellular Superoxide Dismutase Deficiency and Atherosclerosis in Mice. Arteriosclerosis, Thrombosis, and Vascular Biology, 21, 1477-1482. http://dx.doi.org/10.1161/hq0901.094248
- Adachi, T., Ohta, H., Hirano, K., Hayashi, K. and Marklund, S.L. (1991) Non-Enzymic Glycation of Human Extracellular Superoxide Dismutase. Biochemical Journal, 279, 263-267.
- Loscalzo, J. (1996) The Oxidant Stress of Hyperhomocyst(e)inemia. Journal of Clinical Investigation, 98, 5-7. http://dx.doi.org/10.1172/JCI118776
- Yamamoto, M., Hara, H. and Adachi, T. (2000) Effects of Homocysteine on the Binding of Extracellular Superoxide Dismutase to the Endothelial Cell Surface. FEBS Letters, 486, 159-162. http://dx.doi.org/10.1016/S0014-5793(00)02260-2
- Nonaka, H., Tsujino, T., Watari, Y., Emoto, N. and Yokoyama, M. (2001) Taurine Prevents the Decrease in Expression and Secretion of Extracellular Superoxide Dismutase Induced by Homocysteine: Amelioration of Homocysteine-Induced Endoplasmic Reticulum Stress by Taurine. Circulation, 104, 1165-1170. http://dx.doi.org/10.1161/hc3601.093976
- Truong, T.H. and Carroll, K.S. (2013) Redox Regulation of Protein Kinases. Critical Reviews in Biochemistry and Molecular Biology, 48, 332-356.
- Martindale, J.L. and Holbrook, N.J. (2002) Cellular Response to Oxidative Stress: Signaling for Suicide and Survival. Journal of Cellular Physiology, 192, 1-15. http://dx.doi.org/10.1002/jcp.10119
- Dong, C., Davis, R.J. and Flavell, R.A. (2002) MAP Kinases in the Immune Response. Annual Review of Immunology, 20, 55-72. http://dx.doi.org/10.1146/annurev.immunol.20.091301.131133
- Bahorun, T., Soobratte, M.A., Luximon-Ramma, V. and Aruoma, O.I. (2006) Free Radicals and Antioxidants in Cardiovascular Health and Disease. Internet Journal of Medical Update, 1, 25-40.
- Novo, E. and Parola, M. (2008) Redox Mechanisms in Hepatic Chronic Wound Healing and Fibrogenesis. Fibrogenesis & Tissue Repair, 1, 5.
- Chen, Z., Gibson, T.B., Robinson, F., Silvestro, L., Pearson, G., Xu, B., Wright, A., Vanderbilt, C. and Cobb, M.H. (2001) MAP Kinases. Chemical Reviews, 101, 2449-2476. http://dx.doi.org/10.1021/cr000241p
- Powers, S.K., Duarte, J., Kavazis, A.N. and Talbert, E.E. (2010) Reactive Oxygen Species Are Signalling Molecules for Skeletal Muscle Adaptation. Experimental Physiology, 95, 1-9. http://dx.doi.org/10.1113/expphysiol.2009.050526
- Droge, W. (2002) Free Radicals in the Physiological Control of Cell Function. Physiological Reviews, 82, 47-95.
- Yin, M., Wheeler, M., Connor, H., Zhong, Z., Bunzendahl, H., Dikalova, A., Samulski, R., Schoonhoven, R., Mason, R., Swenberg, J. and Thurman, R. (2001) Cu/Zn-Superoxide Dismutase Gene Attenuates Ischemia-Reperfusion Injury in the Rat Kidney. Journal of the American Society of Nephrology, 12, 2691-2700.
- Urushitani, M., Ezzi, S.A. and Julien, J.P. (2007) Therapeutic Effects of Immunization with Mutant Superoxide Dismutase in Mice Models of Amyotrophic Lateral Sclerosis. Proceedings of the National Academy of Sciences of the United States of America, 104, 2495-2500. http://dx.doi.org/10.1073/pnas.0606201104
- Fukushima, S., Coppen, S., Varela-Carver, A., Brindley, G., Yamahara, K., Sarathchandra, P., Yacoub, M.H. and Suzuki, K. (2006) Enhanced Efficiency of Superoxide Dismutase-Induced Cardioprotection by Retrograde Intracoronary Administration. Cardiovascular Research, 69, 459-465. http://dx.doi.org/10.1016/j.cardiores.2005.10.008
- Moukarbel, G.V., Ayoub, C.M. and Abchee, A.B. (2004) Pharmacological Therapy for Myocardial Reperfusion Injury. Current Opinion in Pharmacology, 4, 147-153. http://dx.doi.org/10.1016/j.coph.2003.10.012
- Nikolaos, G., Frangogiannis, C., Smith, W. and Entman, M.L. (2002) The Inflammatory Response in Myocardial Infarction. Cardiovascular Research, 53, 31-47. http://dx.doi.org/10.1016/S0008-6363(01)00434-5
- Barber, S.C. and Shaw, P.J. (2010) Oxidative Stress in ALS: Key Role in Motor Neuron Injury and Therapeutic Target. Free Radical Biology & Medicine, 48, 629-641. http://dx.doi.org/10.1016/j.freeradbiomed.2009.11.018
NOTES

*Corresponding author.