Journal of Modern Physics Vol.05 No.15(2014), Article ID:50242,5
pages
10.4236/jmp.2014.515155
The Bowing Parameters of
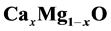 Ternary Alloys
Ternary Alloys
Sinem Erden Gulebaglan1, Emel Kilit Dogan2, Murat Aycibin2, Mehmet Nurullah Secuk2, Bahattin Erdinc2, Harun Akkus2
1Department of Electric Program, Vacational School of Van, Yuzuncu Yıl University, Van, Turkey
2Physics Department, Faculty of Sciences, Yüzüncü Yıl University, Van, Turkey
Email: sinemerden@gmail.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).


Received 8 May 2014; revised 4 June 2014; accepted 2 July 2014

ABSTRACT
On the basis of first principles calculations using density functional theory, we
explore the struc- tural and electronic properties of two binaries: CaO and MgO
in rock salt structures. Structural properties of the semiconductor
alloys are derived from total-energy minimization within the General Gradient Approximation.
The band gap bowing parameters dependence is very powerful Calcium composition.
The results offer that an average bowing parameter of
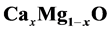 alloys is b = ~0.583$ eV. We analyzed the volume deformation, charge transfer and
structural relaxation effects of the
alloys is b = ~0.583$ eV. We analyzed the volume deformation, charge transfer and
structural relaxation effects of the
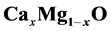 alloys.
alloys.
Keywords:
Density Functional Theory, Ternary Alloys, Band-Gap Bowing Parameter

1. Introduction
Recently, the binary compounds such as TlAs, AlAs, ScN, GaN and their mixtures such
as TlAlAs, ScGaN have been studied theoretically [1] -[3] , because the wide range
of the band gap is important for microelectronics devices. Generally, monoxide compounds
are known as rock salt structures (B1) at room temperature and under pressure. Y.
Duan et al. [4] have been handled the electronic properties of XO (X = Be, Mg, Ca,
Sr, Ba, Zn and Cd) for wrutzite, zincblende and rock salt structure. Ponce et al.
[5] have analyzed theoreticaly and experimentaly electronic and optic properties
of CaS nad CaO. Albuquerque and Vasconcelos [6] are reported structual, electronic
and optical properties of CaO. Karki et al. [7] [8] have inversigated structual,
dynamic and electronic properties of liquid MgO via density functional theory. Makaremi
and Nourbakhsh have declerated structual, electronic and magnetic properties of
Mgo nanolayers. Nishi et al. [9] have investigated experimentally metastable
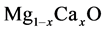 solid solutions film ZnO layers. Stolbov and Cohen [10] have studied the electronic
structure for equilibrium MgO-CaO. Miloua et al. [11] have calculated the electronic
properties of
solid solutions film ZnO layers. Stolbov and Cohen [10] have studied the electronic
structure for equilibrium MgO-CaO. Miloua et al. [11] have calculated the electronic
properties of![]() ,
theoretically. Besides, A. Srivastava et al. [12] calculated phase translations
in
,
theoretically. Besides, A. Srivastava et al. [12] calculated phase translations
in
![]() alloys.
alloys.
In this paper, we represent the bowing parameter of
![]() alloys by first principles density functional theory. To the best of our knowledge,
no theoretical as well as experimental work has been performed thus far for the
bowing parameter of
alloys by first principles density functional theory. To the best of our knowledge,
no theoretical as well as experimental work has been performed thus far for the
bowing parameter of![]() .
The aim of this paper is to understand the attitude of the bowing parameters and
contribution to the gap bowing parameters. The paper is methodized as follows: computational
methodology is given in Section 2. Results and discussion are represented in Section
3. The study is concluded in Section 4.
.
The aim of this paper is to understand the attitude of the bowing parameters and
contribution to the gap bowing parameters. The paper is methodized as follows: computational
methodology is given in Section 2. Results and discussion are represented in Section
3. The study is concluded in Section 4.
2. Computational Details
The calculations for CaO, MgO and
![]() in the rock salt structure were investigated within the generalized gradient approximation
(GGA) of density functional theory (DFT) using the PWSCF code [13] . In Quantum
Espresso, the examining is performed by utilizing the Kohn-Sham [14] formation established
on the DFT. Total energies have been calculated by using ultrasoft pseudopotentials
and plane-wave basis sets. The exchange- correlation potentials in the GGA [15]
is separately used in the calculations. The electronic configurations used for the
pseudo potentials were Ca(3p64s), Mg(2p63s) and O(2s22p4).
The Khon-Sham [14] orbitals were described using a plane wave basis set. The highest
kinetic energy of a plane wave in the chosen fundamental set is known the cutoff
energy. Specialize assignation of the cutoff energy is important for achieving accurate
results with available computational process. The values of cutoff energies used
in our calculations are summarized in Table 1.
in the rock salt structure were investigated within the generalized gradient approximation
(GGA) of density functional theory (DFT) using the PWSCF code [13] . In Quantum
Espresso, the examining is performed by utilizing the Kohn-Sham [14] formation established
on the DFT. Total energies have been calculated by using ultrasoft pseudopotentials
and plane-wave basis sets. The exchange- correlation potentials in the GGA [15]
is separately used in the calculations. The electronic configurations used for the
pseudo potentials were Ca(3p64s), Mg(2p63s) and O(2s22p4).
The Khon-Sham [14] orbitals were described using a plane wave basis set. The highest
kinetic energy of a plane wave in the chosen fundamental set is known the cutoff
energy. Specialize assignation of the cutoff energy is important for achieving accurate
results with available computational process. The values of cutoff energies used
in our calculations are summarized in Table 1.
The plane wave energy cut off is selected 90 Ry. Accurate Brillounin zone investigations
are carried out using the standard special k-points technique of Monkhorst and Pack
[16] . The Brillouin zone investigation was performed over a
![]() mesh points. Our calculations involve an 16 atom for
mesh points. Our calculations involve an 16 atom for
![]() alloys in a supercell. We start at MgO cluster and finish at CaO cluster.
alloys in a supercell. We start at MgO cluster and finish at CaO cluster.
3. Results and Discussion
3.1. Structural Properties of Binary Compounds
The ternary compounds
![]() are bordered by two binary compounds of CaO and MgO. In order to be able to analyze
the energy band gaps and bowing parameters of
are bordered by two binary compounds of CaO and MgO. In order to be able to analyze
the energy band gaps and bowing parameters of
![]() ternary alloys, it is wholesome to study the CaO and MgO binary compounds in terms
of their structural and electronic properties. By lessening the total energy with
regards to the atomic positions and lattice parameters we carried out the structural
optimization. Equilibrium lattice parameters are obtained by fitting the total energy
with the different volumes according to the Birch equation of states.
ternary alloys, it is wholesome to study the CaO and MgO binary compounds in terms
of their structural and electronic properties. By lessening the total energy with
regards to the atomic positions and lattice parameters we carried out the structural
optimization. Equilibrium lattice parameters are obtained by fitting the total energy
with the different volumes according to the Birch equation of states.
The Birch equation of states [17] can be seen in the Equation (1):
![]() (1)
(1)
where
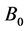 and
and
 are the bulk modulus and its pressure derivative at the equilibrium volume
are the bulk modulus and its pressure derivative at the equilibrium volume . The calcu-
. The calcu-

Table 1. Ground state energies for equilibrium MgO and CaO with various cutoff energies.
lated values for the equilibrium of CaO and MgO are 4.805 Å and 4.263 Å,
respectively. We represented and compered the equilibrium lattice parameter, bulk
modulus
 and bulk modulus derivation
and bulk modulus derivation
 in Table 2.
in Table 2.
3.2. Structural Properties of

In this paper, we examined the effectiveness of the Vegard’s law for rocksalt
 alloys in its ground states. Ca composition
alloys in its ground states. Ca composition
 dependent lattice constant of
dependent lattice constant of
 compounds is expressed as:
compounds is expressed as:
 (2)
(2)
where ,
,
 and
and
 are the lattice constants of the
are the lattice constants of the ,
CaO and MgO, respectively. The volve of the deflection from Vegard’s law can be
calculated by Equation (2). The numerical calculations are fulfilled for the different
situations. Seven compositions of
,
CaO and MgO, respectively. The volve of the deflection from Vegard’s law can be
calculated by Equation (2). The numerical calculations are fulfilled for the different
situations. Seven compositions of
 alloys were checked: 0.125, 0.25, 0.375, 0.5, 0.625, 0.75 and 0.875. The calculated
results of lattice parameters with respect to
alloys were checked: 0.125, 0.25, 0.375, 0.5, 0.625, 0.75 and 0.875. The calculated
results of lattice parameters with respect to
 compositions are itemized in Table 3 from which
we find that the lattice parameter increases with increasing Calcium composition
because these results are due diognosis atomic radius. The atomic radius of Calcium
and Magnesium are 194 and 145 picometers, respectively. We represented and compered
the equilibrium lattice parameter of
compositions are itemized in Table 3 from which
we find that the lattice parameter increases with increasing Calcium composition
because these results are due diognosis atomic radius. The atomic radius of Calcium
and Magnesium are 194 and 145 picometers, respectively. We represented and compered
the equilibrium lattice parameter of
 ternary alloys in Table 3.
ternary alloys in Table 3.
The results also propose that the composition-dependent lattice parameter of the
 ternary alloys can be represented by a third-order polynomial equation,
ternary alloys can be represented by a third-order polynomial equation,
 .
.
The bowing parameter,
 ,
is significant for investigating the band gap energy ternary alloys. The band gap
energy of ternary alloys describe by the band gap energy of binary compounds, a
quadratic interpolation of
,
is significant for investigating the band gap energy ternary alloys. The band gap
energy of ternary alloys describe by the band gap energy of binary compounds, a
quadratic interpolation of
Table 2. Itemized lattice
parameter a, bulk modulus
 and bulk modulus derivation
and bulk modulus derivation
 for the binary compounds MgO and CaO in rocksalt structure at equilibrium volume.
for the binary compounds MgO and CaO in rocksalt structure at equilibrium volume.
Table 3. Itemized lattice
parameter a for the ternary alloys
 in rocksalt structure at equilibrium volume. All values are Å.
in rocksalt structure at equilibrium volume. All values are Å.
composition amount
 and the bowing parameter.
and the bowing parameter.
The band gap energy of ternary alloys
 given by
given by
 . (3)
. (3)
Here,
 is
the band gap energy of the ternary
is
the band gap energy of the ternary
 compound,
compound,
 is
the band gap energy of the CaO compound,
is
the band gap energy of the CaO compound,
 is
the band gap energy of the MgO compound and
is
the band gap energy of the MgO compound and
 is the band gap bowing parameter of
is the band gap bowing parameter of .
Bowing parameter is associated with the band gap energy.
Figure 1 shows bowing parameter as a function of
.
Bowing parameter is associated with the band gap energy.
Figure 1 shows bowing parameter as a function of
 for ternary alloys. We calculated nearly linear variation for
for ternary alloys. We calculated nearly linear variation for
 different composition,
different composition,
 ,
determinating powerful. Our calculations show that according to the band gap energy
of ternary alloys, bowing parameter decreases. The results are given by Table 4 and our results show that an average bowing parameter
of
,
determinating powerful. Our calculations show that according to the band gap energy
of ternary alloys, bowing parameter decreases. The results are given by Table 4 and our results show that an average bowing parameter
of
 is ~0.583 eV.
is ~0.583 eV.
The combination-dependent bowing parameter function [21]
 was
described as
was
described as
 (4)
(4)
The band gap of the ternary alloys are correlated with the band gaps of the binary
compounds. The band gap bowing parameters
 have three physically contributions [17] .
have three physically contributions [17] .
Finally, the total band gap bowing parameter can be written by resolving into its components as:
 . (5)
. (5)
The effect of volume deformation causes the first term of bowing parameter,
 ,
in Equation (5). The no-
,
in Equation (5). The no-

Figure 1. Composition dependence
of the bowing parameter for
 ternary alloys.
ternary alloys.
Table 4. Itemized bowing
parameter and contribution of the bowing parameter a for the ternary alloys
 in rocksalt structure at equilibrium volume.
in rocksalt structure at equilibrium volume.
tional response of MgO(CaO) to hydrostatical pressure states this term via the effect
of the contribution of balanced lattice constant
 to the lattice constant of the alloy value
to the lattice constant of the alloy value .
.
 (6)
(6)
A charge transfer in MgO and CaO at
 indicates the second term,
indicates the second term,
 ,
of the total band gap bowing parameter,
,
of the total band gap bowing parameter,
 (7)
(7)
The third term,
 refers
to the structural relaxation which takes place during the passing from the unrelaxed
to the relaxed alloy
refers
to the structural relaxation which takes place during the passing from the unrelaxed
to the relaxed alloy
 (8)
(8)
In Table 4, the values of bowing parameter,
 and
its volume deformation component
and
its volume deformation component ,
charge exchange component
,
charge exchange component
 and structural relaxation component
and structural relaxation component
 are expressed for
are expressed for
 ternary alloys.
ternary alloys.
The results also propose that the combination-dependent bowing parameter of the
 alloys can be represented by a second-order polynomial equation,
alloys can be represented by a second-order polynomial equation,
 eV.
The results clearly show that
eV.
The results clearly show that
 and
and
 are weak and composition dependent. The prime contribution to the gap bowing is
from the
are weak and composition dependent. The prime contribution to the gap bowing is
from the .
The interval of the calculated band gap bowing coefficient for
.
The interval of the calculated band gap bowing coefficient for
 alloys is from 2.900
alloys is from 2.900
 to 4.234
to 4.234 .
One can note that for all contributions of
.
One can note that for all contributions of
 the main addition to the gap bowing is owing to the
the main addition to the gap bowing is owing to the
 effect. This could be correlated to the strong ionicity dissociable of the corresponding
binary compounds (CaO and MgO). The contribution of the volume deformation term
to the bowing parameter
effect. This could be correlated to the strong ionicity dissociable of the corresponding
binary compounds (CaO and MgO). The contribution of the volume deformation term
to the bowing parameter
 is gradually decreasing with
is gradually decreasing with
 concentration. Suddenly, in the case of
concentration. Suddenly, in the case of
 the addition of the
the addition of the
 has increasing. The addition of the other structural relaxation
has increasing. The addition of the other structural relaxation
 is weak. Furthermore,
is weak. Furthermore,
![]() and
and
![]() is nearly no effect to the bowing parameter. Namely, the
is nearly no effect to the bowing parameter. Namely, the
![]() is the only are component which enables the bowing parameter for
is the only are component which enables the bowing parameter for
![]() alloys.
alloys.
4. Conclusion
We have examined the electronic properties of the rocksalt
![]() ternary alloys as a function of Calcium composition
ternary alloys as a function of Calcium composition
![]() by using the GGA method within DFT. The electronic band structures, which are calculated
by using the lattice parameters composed from Vegard’s law. In the GGA, a band gap
bowing parameters are achieved for rock salt
by using the GGA method within DFT. The electronic band structures, which are calculated
by using the lattice parameters composed from Vegard’s law. In the GGA, a band gap
bowing parameters are achieved for rock salt
![]() ternary alloys. These results propose the physical condition of a composition dependent
band gap energy for
ternary alloys. These results propose the physical condition of a composition dependent
band gap energy for
![]() ternary alloys. Our calculations show that the
ternary alloys. Our calculations show that the
![]() ternary alloy’s the bowing parameters are very strong Calcium composition. The average
bowing parameter is 0.583 eV for
ternary alloy’s the bowing parameters are very strong Calcium composition. The average
bowing parameter is 0.583 eV for
![]() ternary alloys. The results clearly show that the
ternary alloys. The results clearly show that the
![]() is the only dominant component of the bowing parameter for
is the only dominant component of the bowing parameter for
![]() alloys. Additionally
alloys. Additionally
![]() and
and
![]() are weak.
are weak.
References
- Erden Gulebaglan, S. (2012) Modern Physics Letters B, 26, 1250199-8.
- Mazouza, H.M.A., Belabbesa, A., Zaouib, A. and Ferhat, M. (2010) Superlattices and Microstructures, 48, 560-568. http://dx.doi.org/10.1016/j.spmi.2010.09.012
- Moreno-Armenta, M.G., Mancera, L. and Takeuchi, N. (2003) Physica Status Solidi (B), 238, 127-135. http://dx.doi.org/10.1002/pssb.200301808
- Duan, Y., Qin, L., Tang, G. and Shi, L. (2008) European Physical Journal B, 66, 201-209. http://dx.doi.org/10.1140/epjb/e2008-00415-3
- Ponce, S., Bertrand, B., Smet, P.F., Poelman, D., Mikami, M. and Ganze, X. (2013) Optical Materials, 35, 1477-1480. http://dx.doi.org/10.1016/j.optmat.2013.03.001
- Albuquerque, E.L. and Vasconcelos, M.S. (2008) Journal of Physics: Conference Series, 042006, 1-4. http://dx.doi.org/10.1088/1742-6596/100/4/042006
- Karki Bijiya, B., Bhattarai, D. and Stixrude, L. (2006) Physical Review B, 73, 174208-1:7. http://dx.doi.org/10.1103/PhysRevB.73.174208
- Makaremi, N. and Nourbakhsh, Z. (2013) Journal of Superconductivity and Novel Magnetism, 26, 818-824. http://dx.doi.org/10.1007/s10948-012-1991-5
- Nishii, J., Ohtomo, A., Ikeda, M., Yamado, Y., Ohtani, K., Ohno, H. and Kawasahi, M. (2006) Applied Surface Science, 252, 2507-2511. http://dx.doi.org/10.1007/s10948-012-1991-5
- Stolbov, S.V. and Cohen, R.E. (2002) Physical Review B, 65, 092203-3. http://dx.doi.org/10.1103/PhysRevB.65.092203
- Miloua, R., Miloua, F., Kebbab, Z. and Benramdane, N. (2008) ISJAEE, 6, 91-95.
- Srivastava, A., Chauhan, M., Singh, R.K. and Padegaonker, R. (2011) Physica Status Solidi B, 248, 1901-1907. http://dx.doi.org/10.1002/pssb.201046508
- Baroni, S., Corso, A.D., de Gironcoli, S. and Giannozzi, P. http://www.pwscf.org
- Kohn, W. and Sham, L.J. (1965) Physical Review, 140, 1133-1138. http://dx.doi.org/10.1103/PhysRev.140.A1133
- Perdew, J.P., Burke, K. and Ernzerhof, M. (1996) Physical Review Letters, 77, 3865-3868. http://dx.doi.org/10.1103/PhysRevLett.77.3865
- Monkhorst, H.J. and Pack, J.D. (1976) Physical Review B, 13, 5188-5192. http://dx.doi.org/10.1103/PhysRevB.13.5188
- Mehl, M.J., Klein, B.M. and Papaconstantopoulos, D.A. (1995) Intermetallic Compounds: Principles and Practice, Vol. 1: Principles. 195-210.
- Fei, Y. (1999) American Mineralogist, 84, 272-276.
- Karki, B.B., Stixrude, L., Clark, S.J., Warren, M.C., Ancland, G.J. and Crain, J. (1997) American Mineralogist, 82, 51- 60.
- Richet, P., Mao, H.K. and Bell, P.M. (1988) Journal of Geophysical Research: Solid Earth, 93, 15279-15288. http://dx.doi.org/10.1029/JB093iB12p15279
- Drablia, S., Meradji, H., Ghemid, S., Labidi, S. and Bouhafs, B. (2009) Physica Scripta, 79, Article ID: 045002.