Journal of Modern Physics
Vol.4 No.8(2013), Article ID:36331,9 pages DOI:10.4236/jmp.2013.48154
Quantitative Phase Analysis Based on Rietveld Structure Refinement for Carbonate Rocks
1Faculty of Education, Zirve University, Gaziantep, Turkey
2Department of Physics, Faculty of Science, Gazi University, Ankara, Turkey
Email: mehmet.tamer@zirve.edu.tr
Copyright © 2013 Mehmet Tamer. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received April 18, 2013; revised May 23, 2013; accepted June 15, 2013
Keywords: Quantitative Phase Analysis; Rietveld Structure Refinement; Reference Intensity Ratio; Calcite; Dolomite; TOPAS
ABSTRACT
QPA (Quantitative Phase Analysis) of carbonate rocks bearing calcite and dolomite using X-ray diffractometry (XRD) can be performed using the combined Rietveld Structure Refinement (RSR) and semiquantitative Reference Intensity Ratio (RIR) methods, providing an estimation of crystalline phase in a mixture. Different ratios of five samples were prepared by mixing these crystal minerals with high sensitively. The scan speeds, adequate to determine mineral phases in rock samples, were used as 6 degrees per minute with 0.08 steps. The XRD analysis with commercial TOPAS 3.0 program, defined by a new generation of profile and structure analysis software, based on RSR provides Bragg reflection profiles and the dimension of the unit cell of a phase. The weight fractions of each phase were found by Le Bail and Pawley methods in RSR using a pseudo-Voigt peak shape model. The samples were also characterized by using X-ray fluorescence (XRF) and atomic absorption spectrometer (AAS). These analyses were conducted to compare these results with those obtained from the RSR. In addition to these, RSR of phases is very important to improve the goodness of fit (GOF). Therefore, the discussions of refinement of the carbonate mixing were made and a refinement procedure was given for these mixing in detail. Taking advantage of the RSR with the addition of an internal standard, the phase fraction of all the crystalline phases as well as the amorphous component, has been accurately determined. RSR technique offers a valid support for the characterization of marble in the light of industrial products.
1. Introduction
X-ray diffraction (XRD) has long been known as a powerful tool for the determination of phase composition [1]. RSR method of based quantitative analysis indicates the capable of yielding accurate results. Most of the advantageous properties of RSR which were included are the textural description in positive definite spherical harmonics, the refining of polymers, amorphous phases, molecules and the calculation of parameter errors. The main advantage of RSR for quantitative phase analysis is the excellent stability of refinement, even if minor phases are presented. Several methods have been used to analyze the quantify of phases determined by X-ray diffraction data. For instance, RSR method used widely in Xray laboratory has been utilized in the quantification of geological and large used industrial samples [2]. The Rietveld method (Rietveld, 1969) is uniquely versatile for quantitative phase analysis (QPA), providing that the structural models of the phases are known [3,4]. The Rietveld method is able to accurately decouple phases with high degrees of reflection overlap, unlike traditional methods such as the reference intensity ratio (RIR) method [5,6]. Cases with minimal or no peak overlap of the measured reflections. This restricts usage of the RIR method to low numbers of high symmetry phases which yield low reflection densities. The Rietveld method has other advantages over the RIR method, such as the clear illustration of unknown impurities by differences between the observed and calculated patterns (Reid, 2006). In recent years, comprehensive round-robin studies have been undertaken examining the various methodologies employed for QPA, of which Rietveld-based methods are the most widely used [7,8]. An individual unknown or partially unknown crystalline phase can be quantified through PONKS method with RSR [9].
In this work, we employed to estimate the crystalline phase compositions of carbonate mixtures containing calcite and dolomite with RSR and RIR methods. The mixtures in the different ratio were made ready by mixing these crystal minerals. Results of every two methods were compared with weight values of obtained high sensitive balance from XRF and AAS methods.
2. Sample Preparation and Characterization
Five carbonate mixing compositions have been examined by using XRD. The carbonate samples were ground, sieved below 80 mesh sieve (opening 177 µm, 0.0070 inches, No.80, US standard sieve series) and put into the PMMA, which is the thermoplastic and transparent plastic covering by polymethyl 2-methylpropenoate [10-12]. The length, thickness, position and homogeneity of samples are very important. The X-ray analyses have been carried out by using Bruker-D8 advance XRD calibrated with corundum tablet sample including graphite monochromator very sensitively. In particular, mixing ratios of calcite and dolomite which were more or less, were prepared by increasing or decreasing in quantities. The 40 KV (voltage) and 40 mA (current) values for diffraction have been selected for a Cu target tube. Samples were scanned over the range of 2˚ - 70˚ 2θ with CuKα radiation, NaI scintillation detector and a diffracted beam graphite monochromator crystal. For the whole rocks analysis, the setting of 2 mm (1˚) divergence, antiscatter slits, 0.5 mm receiving slit and incident-beam soller slit were necessary with a step size of 0.08˚ and a time of 6˚ per minute.
For quantitative analysis, TOPAS (Total Pattern Analysis Solution) PROGRAM was used based on Rietveld analysis defines a new generation of profile and structure analysis software by integrating employed profile fitting techniques as well as related applications: Single Line Fitting, Indexing (LSI and LP-Search methods), whole powder pattern decomposition (Pawley and Le Bail methods), Abinitio structure determination in direct space from powder and single crystal data, RSR and quantitative Rietveld analysis. This study is concerned with Rietveld structure refinement and quantitative Rietveld analysis with Le Bail and Pawley method [13].
Both calcite and dolomite crystallizing in the trigonalrhombohedral system are the carbonate minerals, unless the calcite is the most stable polymorph of calcium carbonate (CaCO3). Both have the same physical properties, but dolomite does not rapidly dissolve in dilute hydrochloric acid. Calcite exhibits an unusual characteristic called retrograde solubility in which it becomes less soluble in water as the temperature increases. Calcite is white or none in color. On the other hand; dolomite forms white, gray, green, brown to pink in colors, commonly curved crystals. Calcite occurs in sedimentary, volcanic and metamorphic rocks. Dolomite which was formed at temperatures of greater than 100˚C, is the source of magnesium oxide [14,15]. Pure crystal calcite and dolomite minerals were collected from Çanakkale Karabiga region and Yildiz mounts in Trakya of Turkey, respectively.
3. Results and Discussions
Quantitative analysis with RSR must be started by a few parameters because of providing a very important advantage for developing in fitting. Therefore, RSR procedures and their results will be given in step by step for precision of fitting through paper. Five samples prepared with mixing calcite and dolomite crystal minerals are given in the ratios of 0.9; l99.1, 30; 70, 62; 38, 97.5; 2.5 and 99.1; 0.9 respectively, and they are labeled with #1, #2, #3, #4 and #5, respectively. Data were collected on a Bruker D8 advance diffractometer. The diffraction profiles are shown in order in Figure 1. Increasing or decreasing weighted amounts (wt%) of dolomite or calcite can be observed from this figure. Also peaks in the diffraction profiles have the preferred orientations in the direction of (104) with higher intensity and sharp peak. These preferred peaks with perfect crystal plane createserious problem for RIR method because they are not proportional with accurate weighted amount of dolomite or calcite.
Owing to the background appeared in X-ray measurement data, we use a Chebychev polynomial of 4th order and with or without the 1/X B kg function used for Le Bail method. The latter accounts for increasing background due to air scattering when coming close to the primary beam, and also allows using a Chebychev polynomial with fewer coefficients [16].
Instrument part composed of three parts, goniometer radii, equatorial convolutions and axial convolution, respectively. In the first part, the primary and secondary radiuses of the goniometer are determined. In the second and third parts, the primary and secondary solar slits eliminating unphysical broadening in peaks and slit systems are basically used. All of these instrument properties were fixed to instrument parameters of Bragg-Brentano Geometry (primary and secondary goniometer Radius are 250 mm, Detector slit is 0.1 mm, Fixed Divergence and Anti-scatter slits are 0.5 mm, Primary and Secondary solar slits are 2˚). In addition, these properties determining a fit function for the reflection peaks are known as Bragg-Brentano geometry is shown in Figure 2. Also additional convolution functions (Gaussian, lorentzian and hat) can be used together with it if they are necessary. Here additional convolutions functions were not used because the reflection peaks for dolomite and calcite minerals exhibit very high isotropic behavior showing the evidence of the pure and high crystal mineral phases. These values can be also changed to describe

Figure 1. X-ray diffraction patterns of the mixtures in different ratio.

Figure 2. Bragg-Brentano geometry with Rigaku definition.
the phase owing to the quality of sample.
In corrections part of the program, there are three main head as peak shift, intensity corrections and sample convolutions. Peak shift composes of the zero error and sample displacement. The zero error comes from the shift of “d” value which is described as the spacing between the planes in the atomic lattice. Sample displacement is the height of the sample above or below the axis of the goniometer and continues to be a major problem in the preparation of flat sample mounts for XRD analysis. Sample displacement can also be a problem when environmental conditions such as heating, cooling, or changing the humidity cause the sample to expand or contract during analysis (http://www.dxcicdd.com/05/PDF/John_Neil_1.pdf). Because of the well alignment of the height of the sample, the sample displacement is zero but the shift of reflection peaks for the mixture was refined [17].
In intensity corrections, the polarization effects coming from the secondary Graphite monochromator have to be accounted for. Therefore, we consider LP factor as well and set the monochromator angle to 26.4˚ 2θ. Also in sample convolutions, the absorption coefficient (1/cm) can be refined to correct the significant profile shape distortion caused by the low mass absorption of the sample. The absorption correction accounts for the profile distortion due to the sample transparency effect inherent to the Bragg-Brentano geometry. The absorption parameter provides the effective mean absorption coefficient of the sample. For the Bragg-Brentano geometry this parameter is a fundamental parameter. However, absorption results are only meaningful, if the instrument and the microstructure properties of all phases in the sample have been described properly. If these requirements cannot be fulfilled or if a different instrument geometry has been used, the absorption correction can be applied as an additional, empirical function to describe peak asymmetry. In this case the refined absorption parameter does not have a physical meaning.
The maximum peak’s “d” values of the dolomite and calcite phases in “structure/hkl phase” of program are controlled from JPDS cards. Peak shift can be the problem for lattice parameters therefore it must be corrected with Eva or similar programs. In all phases, values of the scale factor must be refined to minimize to 0.001. Preferred orientations in scan must be determined in PO March-Dollase page of the cif extension file menu of the program [18]. Preferred orientation correction for up to two directions. The TOPAS Structure Database contains the typical preferred orientation direction for most of its entries. Here, for calcite and dolomite crystal minerals, this direction (1 0 4) is included and therefore automatically entered into the hkl field of the PO March-Dollase page. This correction decreases importantly the RWP value. In case of doubt the March-Dollase parameter and its refinement code should be set to 1 and “Fix”, respectively, unless preferred orientation has always to be considered for the present phase.
The following methodology was undertaken to estimate the phase composition of two-phase mixtures. Rietveld structure refinements for the crystal structure of the minerals were performed on a reduced 2θ range between 25˚ and 46˚. These limits of the analysis were chosen to encompass the main reflections each phase, which are illustrated only for the sample #2 in Figure 3. In the figure, the waved peaks and the solid line peaks show X-ray pattern, yobs, and the calculated profile, ycal, respectively. The differences under the main peaks of calcite and dolomite are caused by adjustment difficulties based on the crystallinity of the phases. Also the peaks of calcite and dolomite in Figure 3 are separated by “|” symbol. Pawley and Le Bail fitting methods using a pseudo-Voigt peak shape model in the RSR were used in analyses of all samples. All samples were refined assuming the possible presence of two phases, and refined structural models from the characterization of the single-phase standards were used for refinement of the two-phase standard mixtures. Pattern backgrounds were initially modeled automatically by the TOPAS, then the method refines the in-plane and out-planes lattice parameters, the zero offsets, scale factors, and crystal sizes for best goodness of fit and after the RSR, the obtained parameters for two fitting methods were given in the Tables 1 and 2. They all show an increasing or decreasing fluctuation for different phases due to the non uniform particle size. However when the values of dolomite show decreasing behavior, in calcite exhibits increasing due to the increasing or decreasing weight amount of these phases.
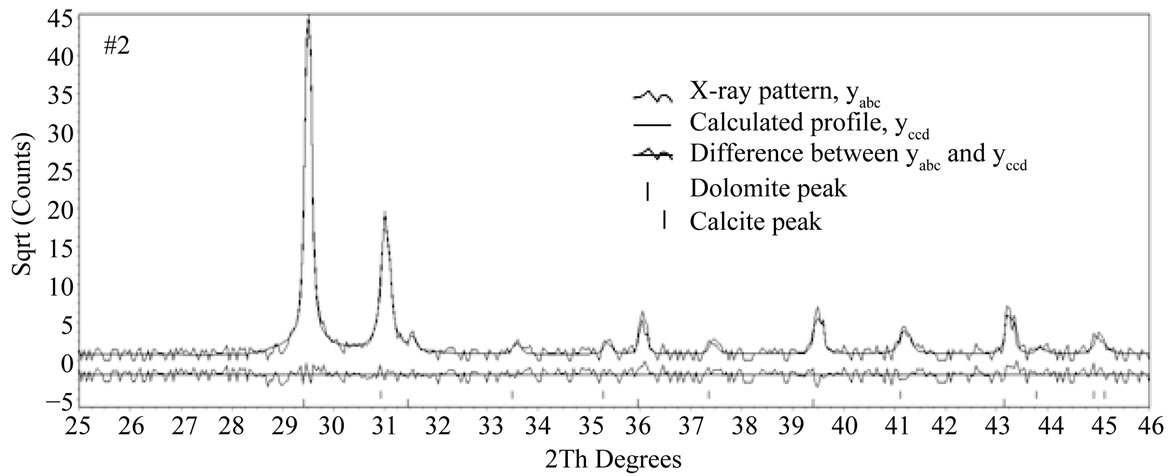
Figure 3. The result of quantitative Rietveld analyses made with commercial TOPAS program for the sample #2.
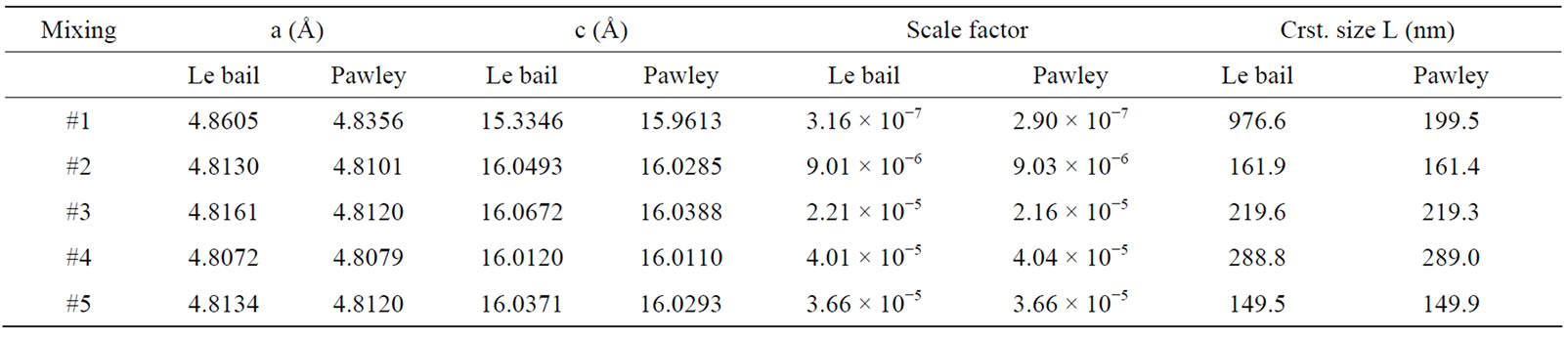
Table 1. Dolomite structure data using Le bail and Pawley methods after RSR.
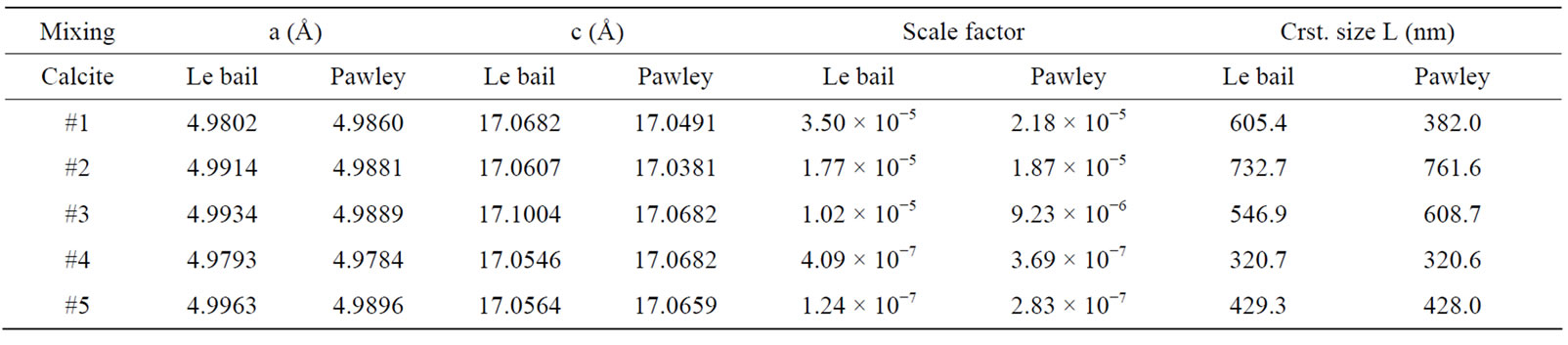
Table 2. Calcite structure data using Le Bail and Pawley methods after RSR.
Scale factors for every two phases are controlled during fit progress. The purpose of scale factors is to scale the calculated intensities of each phase to the observed intensities of the pattern. It is therefore obvious, that scale factors are directly depended on external factors such as the intensity of the X-ray beam and measurement time. Consequently it is impossible to provide default values for scale factors, which are globally valid. Scale factors are linear and very stable parameters and can be off for even some orders of magnitude. However, additional refinement cycles will be required to bring the scale factors in while complex refinements may become jeopardized. In any case poor scale factors hamper a visual check of the calculated data quality. It is therefore advisable not to stick with poor scale factors but to determine and to apply better start values before proceeding with the refinement [1]. Physical characteristics of the sample may give rise to microabsorption or extinction which influences the final value of SCal. and SDol.. In addition, the specimen preparation procedure may favour preferential orientation, especially when powder crystallites have anisotropic shape. Other sources of error in the estimated scale factor depend only on statistical considerations. Accepting that the diffraction profile has been measured with a wise combination in the selected step size with measuring time per step [19]. The relative weight fractions WCal./WDol. do not influence much the estimated errors in SCal. and SDol.. However, the low or high peak/ background intensity ratio led to high errors in SCal. and SDol., respectively [20]. Therefore, it is trick during the fitting that if the calculated intensities for all phases are very low or high, the refinement is canceled and increased all scale factors from the old value to 0.001.
The difference plot in Figure 3 does not show any significant misfits. The differences under the main peaks of calcite and dolomite are caused by adjustment difficulties based on the crystallinity of the phases. For all refined patterns, the weighted profile R factor, Rwp values lie from 22.54 to 29.91 and from 25.21 to 29.92 for Le bail and pawley fitting methods, respectively. The quality of the quantification is independent of the Rwp value ([21]. but the Rwp value is another indicator for misfits between the measured and calculated data. These values are sufficient for the purpose of this investigation because goodness of the observed X-ray, Rexp value shows similar behaviors and they lie from 21.39 to 29.34 and from 21.36 to 29.37 for Le bail and Pawley fitting methods, respectively. Therefore for the quality of fit one needs looking the GOF values are the vicinity of 1 value for both Le bail and Pawley fitting methods. All of these values are shown in Table 3. After the refinement or fitting with best Rexp, Rwp and GOF, the optimizing values of cell mass, cell volume, crystal linear absorption coefficient and crystal density obtained by using the Le bail and Pawley methods for dolomite and calcite phases are given in Tables 4 and 5. When the cell masses of the samples for every two phases are constant, the values of other physical parameter of samples for these phases show a fluctuation or less change. Therefore, weight fraction for each phase is very sensitive with scale factor more than these parameters. Also the optimized crystal densities for every phase determine the crystal densities of mixtures calculated as 2.814 (2.775), 2.784 (2.786), 2.790 (2.781), 2.794 (2.789) and 2.7845 (2.7845) g/cm3 for Le bail and Pawley methods, respectively.
Structural properties given in Tables 1, 2, 4 and 5 for
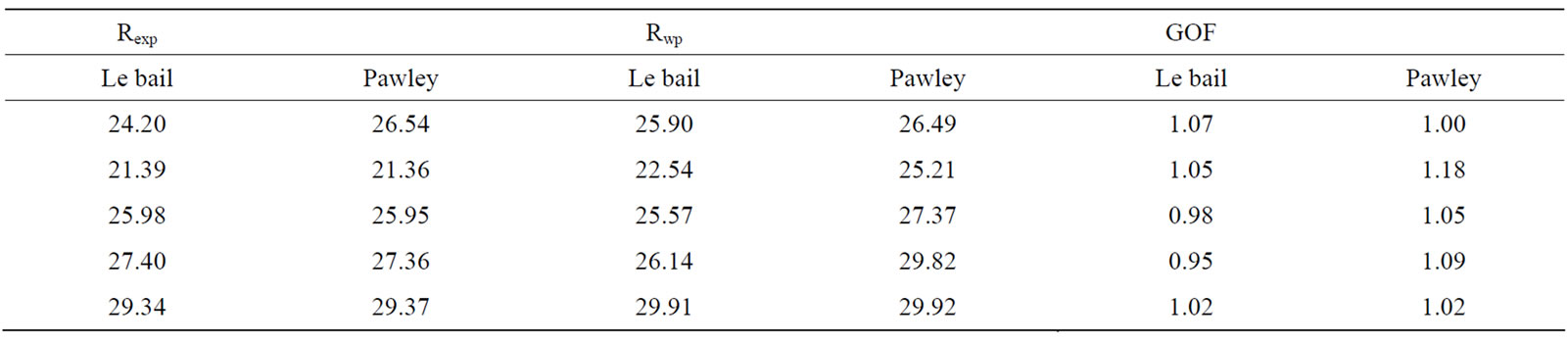
Table 3. R-expected, weighted profile R factor and goodness of fit for Lebail and Pawley method after RSR.

Table 4. Cell mass, cell volume, cry linear absorption coefficient and cry density of dolomite mineral calculated using Lebail and Pawley methods after RSR.

Table 5. Cell mass, cell volume, cry linear absorption coefficient and cry density of calcite mineral calculated using Lebail and Pawley methods after RSR.
every phase are used to calculate the weight amounts of phase in the mixture with the Equation (4). The calculated values with RSR method for dolomite and calcite are presented in Tables 6 and 7 and they are compared with weight amounts in the different ratios of five samples were prepared by mixing these crystal minerals with high sensitivity in second column of Tables 6 and 7. The measured values of dolomite in amounts are ranging from 0.9 to 99.1 as percentages. The calculated ones are from 1.15 to 99.58 in percentages in Le bail method. In Pawley method, the calculated values are from 0.1 to 98.8 in amounts. For calcite, the measured values are from 0.9 to 99.1 in percentages. In Le bail method the calculated values are ranging from 0.49 to 98.85, but in Pawley these are from 0.91 to 98.94 as percentages (Tables 6 and 7). The calculated values are in agreement with these sensitive values between ±1.0 and ±1.5 error limits. These errors are due to impurity, goodness of fit, high noise in X-ray scan, very little anisotropic effects, the calibration problems of X-ray devices etc. Also it is observed in Tables 6 and 7 that there are a perfect harmony between Le bail and Pawley methods and in low ratios of phase per percent, the fitting for every two method is very successful within error limits.
The RIR, AAS and XRF methods were used to compare with both RSR results and sensitive values for quantitative analysis of these samples. The semi quantitative RIR methods are used commonly in many areas and it is very successive if it is used correctly, even contrariwise error exceeds 20 percent, which originates from favored directions of peaks. Samples are adapted to this case. Therefore, Figure 4 shows RIR fitting with commercial EVA program of BRUKER firm for sample #2 and their results are given in Tables 6 and 7. In EVA program, RIR method takes into account all peaks and depends on peak intensities. In Figure 4, there are the preferred peaks for both dolomite and calcite minerals at the vicinity of 30˚ 2θ. Therefore if one increases RIR intensities

Table 6. Weighed amount (wt%) of dolomite obtained using RSR (Lebail and Pawley) with ±1% error and RIR methods.
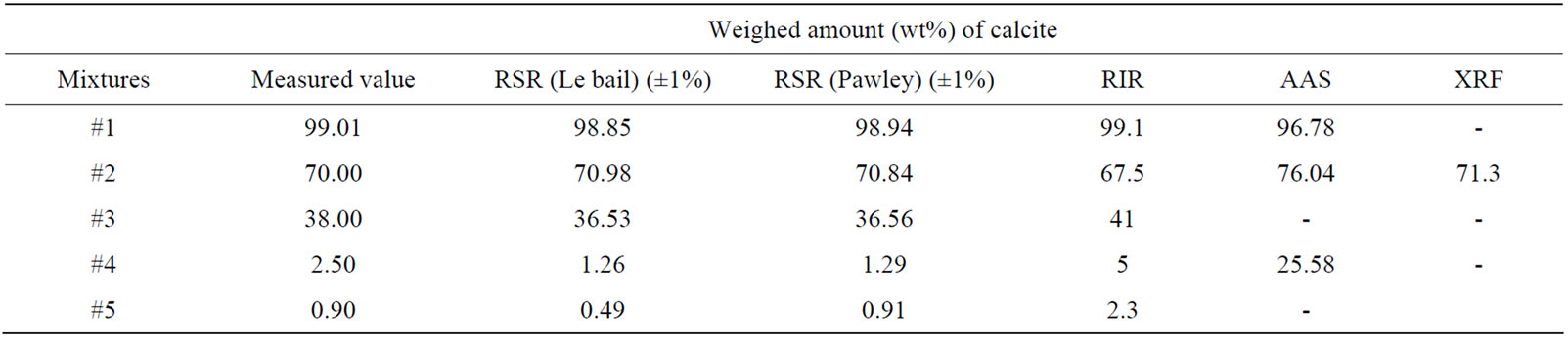
Table 7. Weighed amount (wt%) of calcite obtained using RSR (Le bail and Pawley) with ±1% error and RIR methods.
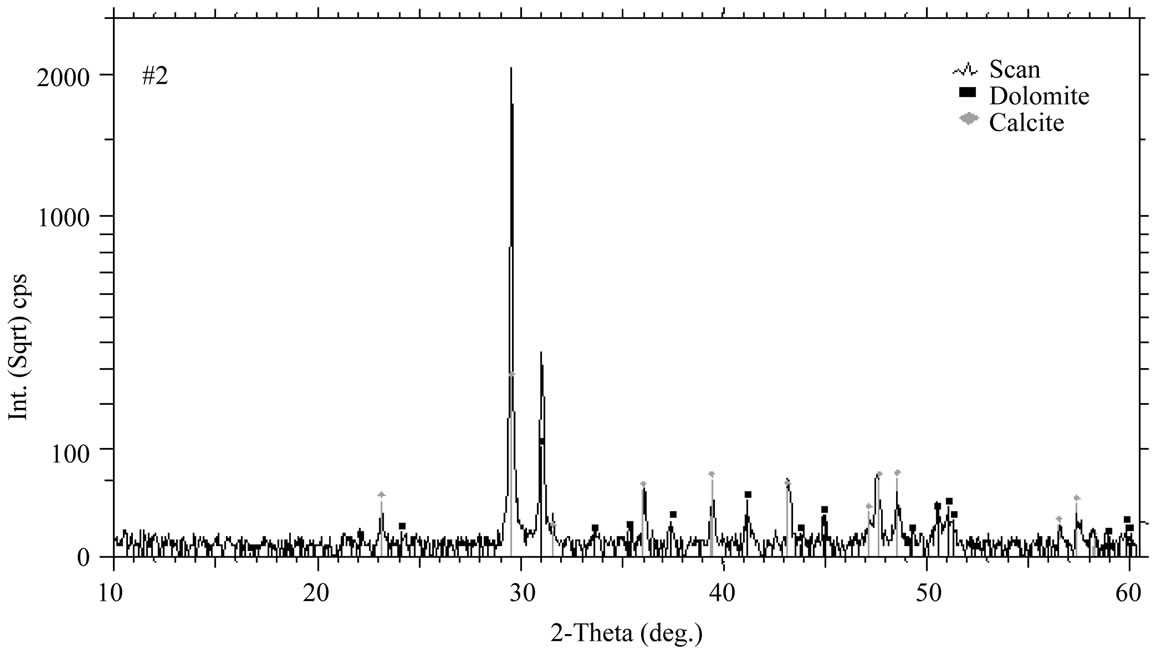
Figure 4. The result of quantitative RIR analyses made with commercial EVA program for the sample #2.
of peaks to these favored peaks, the errors in the results increase excessively. It is observed clearly in Figure 4 that the low intensities of RIR peaks for two phases are adopt with those of scan peaks unlike intensities of favored peaks. In sixth column of Tables 6 and 7, weight amounts are changed from 0.9 to 97.7 in dolomite, unless in calcite they are from 2.3 to 99.1. Results of RIR are in good agreement with other RSR and sensitive values within ±3 error limits.
Quantitative analysis with XRF is used as routine for carbonate samples and is known well as an accurate technique. Therefore in this study it was used only for one sample and element percentages are 6.23, 0.04, 0.02, 47.89, 0.01 and 45.80 for MgO, SiO2, SO3, CaO, SrO and CO2, respectively. Here CO2 element percentage was measured with lost in fire. From Mg difference in dolomite and well known ratio of 21.7 percent, one can calculate the weight amount in percentage as 28.7 value for dolomite and then for calcite is 71.3 percent found by extracting from the hundred. They are compared to Tables 6 and 7 and are in good agreement with sensitive and RSR value for sample #2.
Table 8 presents the percent values of minerals calculated with AAS for sample #1, #2 and #3. Dolomite percents in mixtures are similar to XRF which is determined using MgO percent from the ratio given above and they are given in sixth column of Tables 6 and 7. Results in Tables 6 and 7 show clearly that the RSR results accord
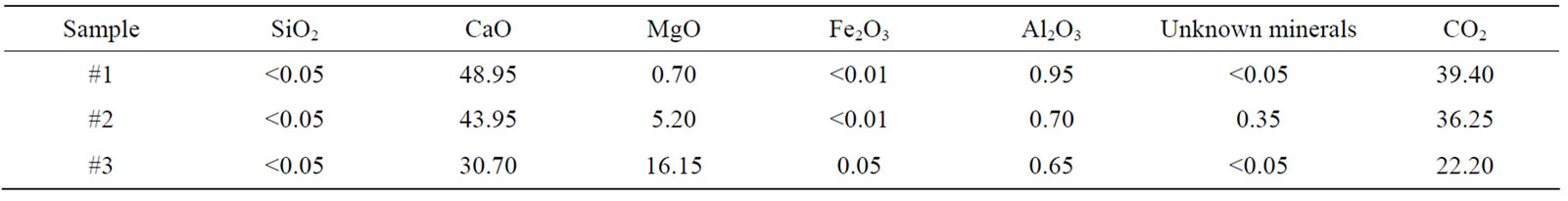
Table 8. The element percents using AAS.
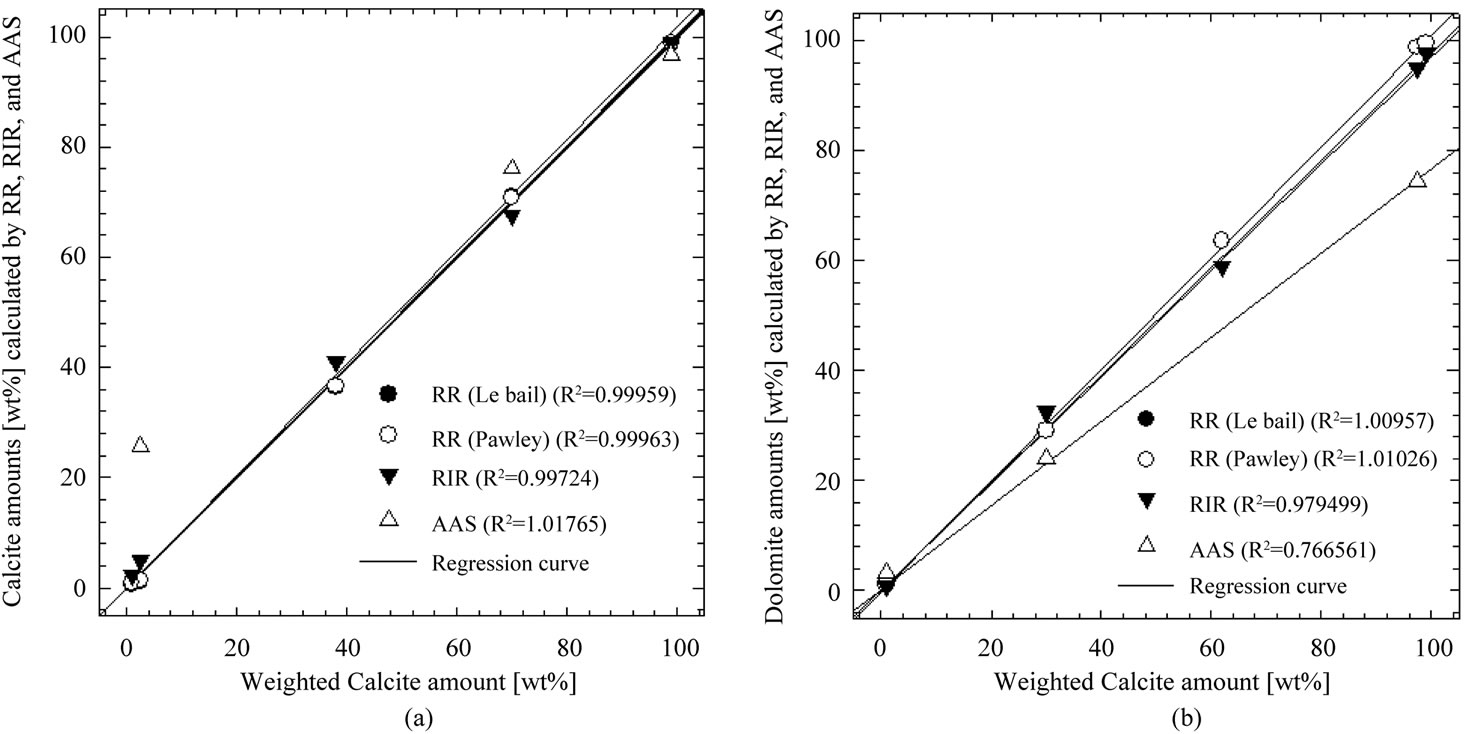
Figure 5. Regression curves of the nominal versus RSR, RIR and AAS phase compositions of the five-phase standard mixtures bearing calcite and dolomite minerals.
ing to those of this method which are used commonly, give best adopt with sensitive values [22].
Finally, QPA for all methods is compared with sensitive values in regression curve in Figure 5. Here, R2 is the coefficient of determination, the most common measure of how well a regression model describes the data. The R2 values near 1 indicate that the equation is a good description of the relation between the independent and dependent variables. R2 equals 0 when the values of the independent variable do not allow any prediction of the dependent variables, and equals 1 when you can perfectly predict the dependent variables from the independent variables. For all methods, R2 values are given for calcite and dolomite in inset of Figure 5. Among them, RSR method gives best R2 values for carbonate samples.
4. Conclusion
The X-ray powder diffraction pattern is used as a fingerprint to identify the phase. QPA of 5 carbonate samples bearing calcite and dolomite using XRD showed that the RSR method combined with the RIR, AAS and XRF methods is a suitable method for “routine” quantitative analyses. The calculation of powder diffraction pattern by means of RSR is made quantitatively by atomic arrangement of a crystal. Optimization of scale factor for every phase is very effective on QPA and it is strongly related with structural properties as the lattice parameter, and peak properties as zero position. Errors in the crystal structure file obtained from the internet (www.crystallography.net/search.html) are very important for QPA and therefore they were discussed. QPA is carried easily out if the derivations in the lattice parameter and peak shift are removed. The preferred orientations area serious problem for RIR method but RSR method overcomes easily this problem. Also RIR method gives the accurate results if one selects the peaks in the scan except the preferred ones. The RSR method with both Le bail and Pawley fitting functions for carbonate samples gives high quality results similar to those of RIR and XRF but not best results in respect of AAS.
5. Acknowledgements
I want to thank very much Dr. Mustafa Kemal Öztürk, Dr. Serap İçöz and Dr. Sezin Özaksoy for their sopport of my theses.
REFERENCES
- B. D. Cullity, “Elements of X-ray Diffraction,” 2nd Edition, Addison-Wesley, Boston, 1978.
- J. W. Reid, A. Jason and J. Hendry, “Rapid, Accurate Phase Quantification of Multiphase Calcium Phosphate Materials Using Rietveld Refinement,” Journal of Applied Crystallography, Vol. 39, 2006, pp. 536-543. doi:10.1107/S0021889806020395
- D. L. Bish and S. A. Howard, “Quantitative Phase Analysis Using the Rietveld Method,” Journal of Applied Crystallography, Vol. 21, 1998, pp. 86-91.
- R. J. Hill, G. Tsambourakis and I. C. Madsen, “Improved Petrological Modal Analyses from X-Ray Powder Diffraction Data by Use of the Rietveld Method,” Journal of Petrology, Vol. 34, No. 5, 1993, pp. 867-900. doi:10.1093/petrology/34.5.867
- F. H. Chung, “Quantitative Interpretation of X-Ray Diffraction Patterns of Mixtures. II. Adiabatic Principle of X-Ray Diffraction Analysis of Mixtures,” Journal of Applied Crystallography, Vol. 7, 1974, pp. 519-525.
- C. R. Hubbard, E. H. Evans and D. K. Smith, “The Reference Intensity Ratio, I/Ic, for Computer Simulated Powder Patterns,” Journal of Applied Crystallography, Vol. 9, 1976, pp. 169-174. doi:10.1107/S0021889876010807
- I. C. Madsen, N. V. Y. Scarlett, L. M. D. Cranswick and T. Lwin, “Outcomes of the International Union of Crystallography Commission on Powder Diffraction Round Robin on Quantitative Phase Analysis: Samples 1a to 1h,” Journal of AppliedCrystallography, Vol. 34, 2001, pp. 409- 426. doi:10.1107/S0021889801007476
- N. V. Y. Scarlett, I. C. Madsen, L. M. D. Cranswick and T. Lwin, “On-Line X-Ray Diffraction for Quantitative Phase Analysis: Application in the Portland Cement Industry,” Powder Diffraction, Vol. 16, No. 2, 2002, pp. 71- 80. doi:10.1107/S0021889801007476
- N. V. Y. Scarlett and I. C. Madsen, “Quantification of Phases with Partial or No Known Crystal Structures,” Powder Diffraction, Vol. 21, No. 4, 2006, pp. 278-284. doi:10.1154/1.2362855
- B. Beckhoff, B. Kanngieβer, N. Langhoff, R. Wedell and H. Wolff, “Handbook of Practical X-Ray Fluorescence Analysis,” Springer, Berlin, 2006.
- R. J. Hill, “Improved Petrological Modal Analyses from X-Ray Powder Diffraction Data by Use of the Rietveld Method I. Selected Igneous, Volcanic, and Metamorphic Rocks,” Powder Diffraction, Vol. 6, No. 2, 1991, pp. 74- 77. doi:10.1017/S0885715600017036
- R. A. Young, “Introduction to the Rietveld Method,” International Union of Crystallography Monographs on Crystallography, Vol. 5, Oxford University Press, Oxford, 1993, pp. 1-39.
- D. L. Bish and J. E. Post, “Quantitative Mineralogical Analysis Using the Rietveld Full-Pattern Fitting Method,” American Mineralogist, Vol. 78, 1993, pp. 932-940.
- W. A. Deer, R. A. Howie and J. Zussman, “Introduction to the Rock Forming Minerals,” Longman, London, 1966, pp. 489-493.
- C. S. Hurlbut Jr., “Dana’s Manual of Mineralogy,” 17th Edition, John Wiley and Sons, Inc., New York, 1959, pp. 330-340, 609.
- R. W. Cheary and A. A. Coelho, “A Fundamental Parameters Approach to X-Ray Line-Profile Fitting,” Journal of Applied Crystallography, Vol. 25, 1992, pp. 109- 121. doi:10.1107/S0021889891010804
- R. W. Cheary and A. A. Coelho, “Axial Divergence in a Conventional X-Ray Powder Diffractometer. II. Realization and Evaluation in a Fundamental-Parameter Profile Fitting Procedure,” Journal of Applied Crystallography, Vol. 31, 1998, pp. 862-868. doi:10.1107/S0021889898006888
- “Diffrac plus TOPAS v. 3.0 (Manual),” BRUKER AXS GmbH, Karlsruhe, 2006.
- R. J. Hill and C. J. Howard, “Quantitative Phase Analysis from Neutron Powder Diffraction Data Using the Rietveld Method,” Journal of Applied Crystallography, Vol. 20, 1987, pp. 467-474.
- F. Guirade, S. Gali and S. Chinchon, “Quantitative Rietveld Analysis of Aluminous Cement Clinker Phases,” Cement and Concrete Research, Vol. 30, No. 7, 2000, pp. 1023-1029. doi:10.1016/S0008-8846(00)00289-1
- H. M. Rietveld, “A Profile Refinement Method for Nuclear and Magnetic Structures,” Journal of Applied Crystallography, Vol. 2, 1969, pp. 65-71. doi:10.1107/S0021889869006558
- R. E. Dinnebier and S. J. L. Billinge, “Powder Diffraction: Theory and Practice,” Royal Society of Chemistry, 2008. doi:10.1039/9781847558237