Advances in Bioscience and Biotechnology
Vol.3 No.3(2012), Article ID:20032,7 pages DOI:10.4236/abb.2012.33040
The cloning of non-structural-1 (NS1) gene of H9N2 subtype of avian influenza virus in pGEX-4T-1 and pMAL-c2X plasmids and expression in Escherichia coli DH5α strain
![]()
1Department of Poultry Diseases, Faculty of Veterinary Medicine, University of Tehran-Iran, Tehran, Iran
2Medical Faculty and Cellular and Molecular Research Center, Mazandaran University of Medical Sciences, Sari, Iran
3School of Animal and Veterinary Sciences, The University of Adelaide, Adelaide, Australia
Email: *mvmarand@ut.ac.ir
Received 19 February 2012; revised 26 March 2012; accepted 17 April 2012
Keywords: Avian Influenza Virus; H9N2 Subtype; Recombinant Protein; Non-Structural Protein 1; DIVA
ABSTRACT
Avian influenza is a viral contagious disease that affects poultry industry and human health. Vaccination has been considered as a preventive tool in the eradication of AI, but it causes some limitations including trade embargoes and interfering with serologic surveillance in differentiation between infected and vaccinated animals (DIVA strategy). Several distinct DIVA strategies have been presented to conquer these limitations. In this study, the open reading frame of NS1 gene of a H9N2 subtype of AI virus was amplified by polymerase chain reaction. After extraction and purification of NS1 gene from agarose gel, it was inserted into two different pGEX-4T-1 and pMAL-c2X plasmids and transferred in DH5α strain of Escherichia coli by using electroporation procedure. The E. coli colonies possessing recombinant NS1 gene were screened using PCR, restriction mapping and sequencing analysis. The expressed rNS1 protein was purified using affinity chromatography based on MBP (pMALc2X) and GST (pGEX-4T-1). The MBP-NS1 and GSTNS1 proteins on SDS-PAGE had bands with molecular weight of 68 and 52 kDa respectively. Western blotting with MBP-NS1 protein showed positive reaction using antisera obtained from chickens challenged with a H9N2 subtype strain. But, the most sera prepared from H9N2 vaccinated chickens were negative in WB. These findings indicated that the MBP-rNS1 protein of 26 kDa expressed by pMAL-c2X plasmid can be used in a DIVA for differentiation of AI infected and vaccinated chickens.
1. INTRODUCTION
Influenza viruses belonging to the family Orthomyxoviridae are enveloped viruses with segmented and negative stranded RNA genome. These viruses are classified into three types of A, B and C [1-3]. All types of influenza viruses are characterized in humans, but only the type A viruses that are termed as avian influenza viruses (AIV), are found in birds. According to the surface glycoproteins including haemagglutinin (H) and neuraminidase (N), the type A influenza viruses are divided into H and N subtypes. At present sixteen haemagglutinin subtypes (H1-H16) and nine neuraminidase subtypes (N1-N9) have been recognized [2-4].
The AIV are further divided into two distinct pathotypes on the basis of their ability to cause disease in chickens. Highly pathogenic avian influenza (HPAI) viruses are restricted to subtypes H5 and H7 in which mortality may be as high as 100%. Low pathogenic avian influenza (LPAI) viruses pathotype are included all subtypes of AIV isolated from birds. This pathotype causes a mild to severe respiratory disease and reductions in egg production in infected poultry [2].
From 2003 to 2011, several outbreaks of H5N1 with HPAI pathotype have been involved in poultry and wild birds in 63 countries and territories in Asia (18 countries), Europe (26 countries), Africa (12 countries) and near East (7 countries). According to the latest news in the world health organization (WHO), among 565 people infected since 2003 to present, 331 people have died from bird influenza illness. The majority of cases occurred in Indonesia, Vietnam, Egypt and People’s Republic of China [5].
The HPAI can cause considerable losses in egg and meat producing chickens that affect national and international trade. Since 2003 H5N1 has globally killed or forced the culling of more than 400 million domestic poultry and caused an estimated $20 billion of economic damage worldwide, before it was eliminated from most of 63 countries infected at its peak in 2006 [5].
Outbreaks of influenza with H9 subtype virus have been considerable during the recent years. In 1995, an outbreak of AI H9N2 subtype occurred in turkeys in the USA that resulted enormous economic losses. In Europe and Asia, it was initially detected in ducks and isolated from chickens in 1994 [6]. In addition isolation of AIV H9N2 from humans in Hong Kong shown that direct transmission of H9N2 virus to humans is possible and presents potential public health risk [7]. At present AIV H9N2 is circulating widely in domestic poultry all over the world and contributed widespread outbreaks in commercial poultry [8].
Although vaccination can be considered in some cases as an option to control and eliminate of AIV, but to date fully eradication of the disease agent has not achieved [8-10]. For many reasons, including the concerns of serologic surveillance of AI by traditional serologic tests and international trade on poultry and poultry products; vaccination has not been commonly used for the control and eradication of AI. Since the most currently used AI vaccines can interfere with the serologic surveillance; hence these killed vaccines produce the measurable antibodies against the structural proteins that are completely similar to the antibodies produced by live wild AIV infection. To prevent and control of AI, the classic serological surveillance diagnostic tests based on antibodies detection against AIV are agar gel immunodiffusion (AGID), enzyme-linked immunosorbent assay (ELISA) and haemagglutinin inhibition (HI) tests. The AGID test is type A specific, based on both common nucleoprotein (NP) and matrix (M) protein antigens, but its sensitivity is lower than the ELISA and haemagglutination inhibition (HI) tests [11-13]. It also needs a large amount of antigen and antibody to be established precipitation line.
The HI test is widely used to detect HA specific antibodies, but it is time consuming and tedious. Besides, interpretation of the results is sometime different among the laboratories [14] and it was demonstrated a weak diagnostic correlation between ELISA and HI test as well [15]. The NP specific ELISA is also a reliable, sensitive and accurate test, but inactivated vaccines lead to the production of antibodies which are indistinguishable from live AIV infection with field strain. So, for both trade and surveillance purposes, differentiation between infected, vaccinated and non-infected vaccinated flocks is a new approach commonly known as the DIVA strategy. There are four different DIVA strategies to solve this problem [12-16]. A simple approach is to release of non-vaccinated sentinels birds among vaccinated birds typically 30 or more marked birds, but this method has some management problems [12]. Subunit vaccines are another preventive tool that target M and NP proteins with the most flexibility to work with the available serologic surveillance tests. Several types of subunit vaccines have been made, but just one viral vectored vaccine, recombinant fowl pox virus (rFPV), is available for H5 subtype [17,18].
Another method is heterologous NA-DIVA strategy using the same haemagglutinin and different neuraminidase killed vaccine prepared with field strain virus. In this type of NA-DIVA strategy, neuraminidase acts as an indicator of field infection. This system was used for the first time in Italy during an H7N1 outbreak in 2000, that vaccination was performed with a H7N3 vaccine [19,20]. The NS1 protein is also one option of DIVA strategy to AI [16]. This protein is synthesized in detectable scale in infected cells, but it is not possible to measure it into the virion. Therefore, the chemically inactivated influenza vaccines produced from whole viral particles do not include any NS1 protein. So, wouldn’t have any antibody response to NS1 protein. Whereas, infected chickens will produce antibodies against NS1 protein [16,21,22]. The NS1-DIVA (ELISA) strategy using of purified NS1 protein as an antigen demonstrated high level of NS1 antibody titer in infected chickens comparing to vaccinated birds [16-23]. This approach was initially suggested for monitoring equine influenza virus [21,22]. As indicated by Avellaneda, the NS1-ELISA based DIVA strategy detected HPAI virus challenge better than LPAI virus challenge [15]. It seems that this test is sensitive and suitable for assessment of AI in adult chickens [23]. The NS1-DIVA strategy has many benefits as compared to other DIVA strategies. One of the most important advantages is the possibility to work with any killed vaccine [12]. If NS1 based DIVA test to be selected as screening test for differentiation of infected vaccinated flocks from non infected vaccinated flocks, the flocks could be vaccinated with complete homologous killed vaccine regarding to H and N subtypes that improve flock protection.
Based on the limited available research, in NS1-DIVA strategy, the goal of this study was the cloning of NS1 gene of H9N2 subtype of AIV in pGEX-4T-1 and pMALc2X plasmids. Expression and purification of rNS1 protein evaluated in SDS-PAGE and western blot assays.
2. MATERIALS AND METHODS
2.1. Virus and RNA Extraction
A H9N2 subtype strain of AIV designated as A/Chicken/ Iran/ZMT-101/98, was propagated in 10 days old embryonated chicken eggs and then tittered using haemagglutination (HA) test. The RNA extraction of AIV, was undertaken by QIAquick RNeasy mini kit (No. 74104) according to manufacturer’s procedure.
2.2. Reverse Transcriptase (RT)-PCR and Cloning of the NS1 Gene Coding Sequence
To construction of recombinant plasmid vectors, two plasmids designated as pMAL-c2X and pGEX-4T-1 were used. The NS1 gene coding sequence was amplified by RT-PCR using two sets of primers:
F1-AAGAATTCATGGATTCCAACACTGTGTC, R1-ACA TCTAGATCAAACTTCTGACTCAATTGTTC,
having of EcoRI and XbaI restriction sites for pMAL-c2X plasmid and R2-TATGTCGACTCAATGGTGATGGTGATGGTGATCTTCTGACTC, having an EcoRI and SalI restriction sites for pGEX-4T-1 plasmid. The forward primer for pGEX-4T-1 plasmid was the same as forward primer for pMAL-c2X plasmid and the second reverse primer had 6xHis-tag sequence for double purification as well.
Two different PCR products were run on a 1.5% agarose gel and then purified by using a QIAquick gel extraction kit (No. 28704). These two purified DNA were digested distinctly with EcoRI, XbaI and EcoRI, SalI (Biolab) together with BSA respectively for pMAL-c2X and pGEX-4T-1 at 37˚C for 3 h.
Ligation of digested products including the first insert with pMAL-c2X and the second insert with pGEX-4T-1 were carried out with 1 µl T4 DNA ligase (Invitrogen) at 22˚C for 1 h.
2.3. Transformation and Expression
Transformation of two recombinant plasmids (pMALNS1 and pGEX-NS1) were performed into the competent cell of E. coli DH5α strain, according the electroporation procedure. The transformed E. coli cells, incubated at 37˚C for 1 h and then plated on Luria-Bertani (LB) agar containing of 50 µg/ml of ampicillin overnight. In order to analysis of transformants and recombinant plasmids, bacterial colonies were screened for NS1 inserts using several methods including PCR by vector specific primers, plasmid preparation and subsequently restriction analysis and sequencing. For protein expression, the transformants of E. coli including of NS1 insert were cultured in LB broth at 37˚C overnight in a shaking incubator at 250 rpm.
A volume of 100 µl of overnight culture was transferred to 10 ml of LB broth supplemented with 0.2% glucose and 50 µg/ml of ampicillin at the same condition as mentioned above. Isopropyl-β-D-thiogalactopyranoside (IPTG) was added to the culture to a final concentration of 1 mM when the optical density (OD) with an absorbance at 600 nm reached 0.5 - 0.8. Incubation of the culture was preformed for an additional 3 h. Then, harvesting of the culture was carried out by centrifugation at 8000 rpm for 20 min at 4˚C. After centrifugation and discarding of the supernatant, the cells were resuspended with the column buffer (20 mM Tris-HCl PH 7.4, 1 mM EDTA, 200 mM NaCl, 1 mM sodium azide and 10 mM β-ME) for pMAL-NS1 construct and with phosphate buffered saline (PBS) (140 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.3) for pGEXNS1 construct and finally stored at –20˚C.
2.4. The Recombinant NS1 (rNS1) Protein Purification
A comparative study of purification methods according to different fusion proteins including MBP, GST and His fusion proteins were applied. The cell suspensions were denatured in ice water bath by sonication in short pulses for five times. The suspensions were then centrifuged at 8000 g for 20 min at 4˚C and supernatants were collected and stored at 4˚C.
2.5. Purification of MBP-NS1 Fusion Protein
The supernatant was loaded on amylose resin (New England Biolab) at a flow rate of 1 ml/min and followed by flow-through collection. The column was washed with column buffer overnight by using of safety loop and continued by an elution step of MBP-NS1 fusion protein with column buffer and 10 mM maltose. About 20 fractions of eluted protein was collected, dialyzed overnight and stored at –20˚C after monitoring of the protein concentration in UV absorbance at 280 nm by Nanodrop spectrophotometer.
2.6. Purification of GST-NS1 Fusion Protein
The supernatant was added to 1 ml of 50% slurry of glutathione Sepharose 4B and incubated with gentle shaking for 1 h at room temperature (RT). It was followed to wash three times by adding 50 ml ice-cold 1× PBS and centrifugation at 500 × g for five minutes. The beads were transferred to the column, then 1 ml of glutathione elution buffer (Tris-HCl 50 mM pH 8, reduced glutathione 10 mM) was mixed gently and incubated for 15 min at 22˚C. The elution and collection processes were repeated three times more.
The eluates were checked for GST fusion protein by SDS-PAGE. For batch purification, the purified 6× Histagged protein above was mixed with binding buffer (Na2H2PO4 50 mM pH 8, NaCl 300 mM, imidazole 5 mM) and Ni-NTA agarose resin for 1 h at RT with gentle agitation. Washing of the resin was performed with wash buffer (NaH2PO4 50 mM pH 8, NaCl 300 mM, imidazole 20 mM) after transferring of it to a column. The protein elution was then undertaken with elution buffer (NaHPO4 50 mM pH 8, NaCl 300 mM, imidazole 250 mM - 1 M) and repeated for additional three times. The protein was purified under native conditions.
2.7. SDS-PAGE
Sodium dodecyl sulfate polyacrilamide gel electrophoresis (SDS-PAGE) was used to separate the recombinant proteins based on their known molecular weights. The gel was prepared from 4% to 10% acrylamide gel gradiant. The samples were mixed and denatured with 4× sample buffer (250 µl Tris-HCl PH 6.8, 800 µl SDS, 400 µl glycerol, 80 µl Bromophenol blue 0.5%, 200 µl 2ME, 70 µl dH2O). Another denaturing step was performed via boiling for 5 min at 95˚C. After preparing of the gel, a volume of 6 µl of the denatured recombinant protein samples and the appropriate protein marker were loaded into the wells. Then the gel was run at 100 v for 2 h and subsequently stained with Coomassie Brilliant Blue (450 ml methanol, 450 ml dH2O, 100 ml acetic acid, 1 g Coomassie blue R 250) and destained with destaining solution (100 ml Methanol, 100 ml acetic acid, 800 ml dH2O) overnight.
2.8. Western Blot (WB)
The antigenicity of the rNS1 protein was determined by wb. The purified rNS1 proteins were electrophoresed and then transferred to PVDF membrane. After washing 3 times by 1× PBS, the membrane was incubated 1 h at RT and blocked with 10% BSA. The membranes were washed with washing buffer containing 0.5% PBS-T and 0.1% BSA and then incubated 1 h at RT in 1/500 dilution of five pooled positive H9N2 sera in PBS-T (0.5%) and BSA (1%). Subsequently washing step was performed three times with washing buffer. In the last step, horse radish peroxidase anti-IgY serum was added by a 1/1000 dilution and incubated for 1 h at 37˚C. The antigen and antibody complexes were then revealed using of 3 - 3’ Diaminobenzidine based (DAB) substrate diluted in TBS buffer (Tris-HCl 24 g, NaCl 8 g, dH20 1 L) plus 12 µl H2O2 for 5 - 10 min after 2 times washing with washing buffer and one time by PBS. Finally the membranes were washed with dH2O and dried by whatman paper for long term.
3. RESULTS
3.1. RT-PCR and Construction of the rNS1 Plasmids
The amplification of NS1 coding sequence of H9N2 subtype of AIV was achieved by RT-PCR with individual primers. The amplified sequences of NS1 gene with two sets of primers were 711 bp and 729 bp respectively (Figure 1).
The recombinant vectors of pMAL-NS1 and pGEXNS1, were successfully constructed by inserting of digested NS1 DNA into the linearized pMAL-c2X and pGEX-4T-1 plasmids. They were transformed separately into DH5α strain of E. coli. The transformants containing of rNS1 plasmids were selected and screened for NS1 insert by colony PCR, restriction analysis and sequencing of PCR product.
3.2. Expression and Purification of NS1 Protein
To expression of NS1 protein, a single positive colony was cultured in LB broth and induced by IPTG with final concentration of 1 mM/ml. The bacterial cell pellets were prepared after 3 h. SDS-PAGE analysis of MBP-NS1 and GST-NS1 fusion proteins showed the molecular weights of around 68 and 52 kDa respectively which were comparable with predicted bands (Figures 2 and 3).
In this study two rNS1 proteins were generated. So, one of them was considered to produce NS1 protein containing MBP fusion protein at N-terminal and another one containing both GST fusion protein at N terminal and 6 × His-tag at C-terminal. Therefore, rNS1 protein purification was undertaken by amylose resin for MBPNS1 and glutathione sepharose 4B and Ni-NTA resin for GST-NS1.
MBP-NS1 protein was successfully purified and detected on SDS-PAGE. Whereas, nanodrop expectrophotometry of the second purified protein (GST-NS1) demonstrated a very low quantities of the protein and nothing
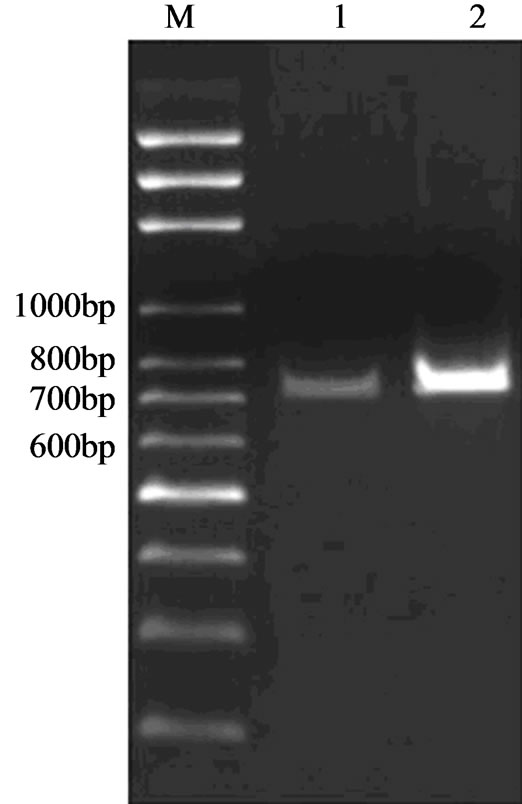

Figure 1. Agarose gel electrophoresis of NS1 PCR product. Lane M: marker; Lane 1: NS1 with the EcoRI and XbaI restriction site; Lane 2: NS1 with the EcoRI and SalI restriction site.
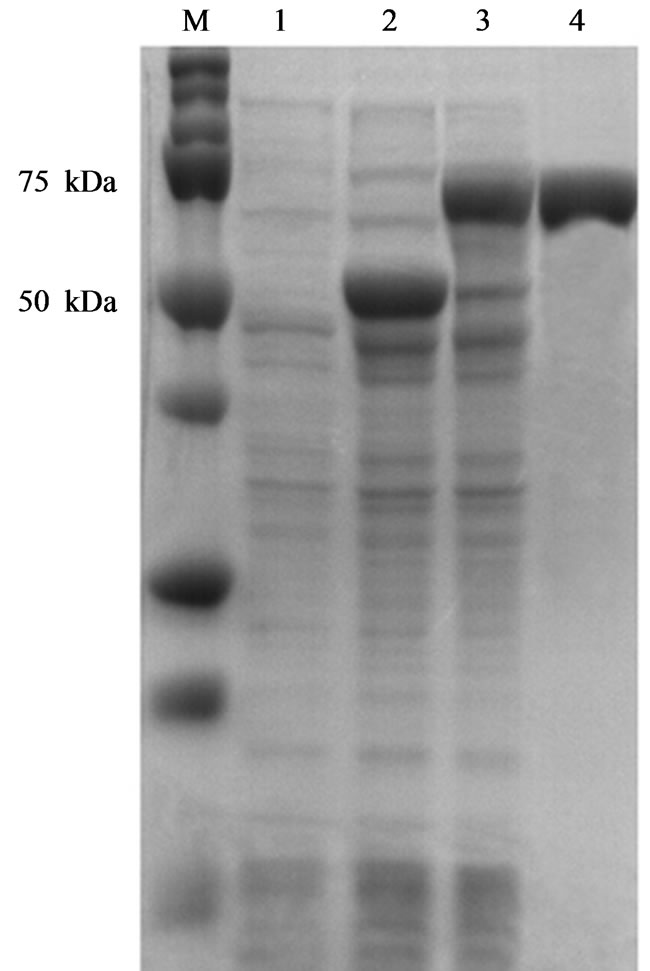
Figure 2. SDS-PAGE analysis of MBP-NS1 protein expressed in E. coli. Lane M: protein marker; Lane 1 and 2: crude extracts of transformed pMAL-c2X vector in E. coli. before and after being induced with IPTG respectively; Lane 3: Recombinant pMAL-NS1 expressed in E. coli; Lane 4: Purified MBPNS1 protein.

Figure 3. SDS-PAGE analysis of GST-NS1 protein expressed in E. coli. Lane M: protein marker; Lane 1 and 2: crude extracts of transformed pGEX-4T-1 vector in E. coli. before and after being induced with IPTG respectively; Lane 3: recombinant pGEX-NS1expressed in E. coli.
found on SDS-PAGE. Although expression of the protein to the different scales of the culture and also changing the method to produce more soluble protein was considered, attempts to purify NS1 protein by pGEX expression vector was greatly unsuccessful.
3.3. Western Blot Assay
MBP-NS1 protein and its antigenicity was investigated in western blot assay using mixed positive H9N2 antisera and serum samples obtained from vaccinated chickens challenged with H9N2 subtype strain. Western blot analysis revealed a 68 kDa protein band that included around 42 kDa of the fusion protein and 26 kDa NS1 protein (Figure 4). Because in a few cases, there was some reactivity between MBP protein of recombinant MBPNS1 and antiserum, the active antibody sites were primarily blocked by treating of the purified MBP protein for one hour then incubated with MBP-NS1 protein to follow the rest process. Meanwhile, twelve sera obtained from vaccinated chickens that were immunobloted on membranes, did not show detectable reactions in most of them, whereas only three samples showed a band with NS1 protein which prepared from chickens vaccinated two or three times (Table 1).

Figure 4. Western blot analysis of the recombinant NS1 protein. Lane M: protein marker; Lane 1: SPF serum; Lane 2: serum of vaccinated chicken; Lane 3: pooled positive H9N2 sera.
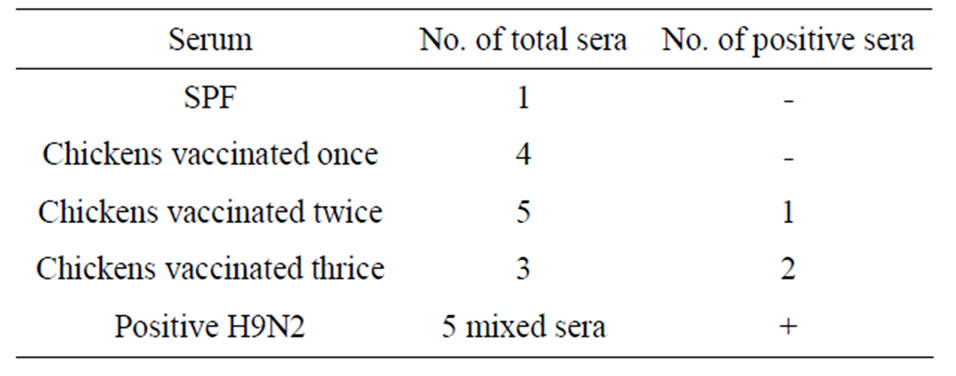
Table 1. Western blot analysis of MBP-NS1 protein using of sera from SPF, vaccinated chickens and mixed positive H9N2 samples.
4. DISCUSSION
As avian influenza outbreaks contributed progressively in the world in recent years, the global market meets the import bans on poultry product that causes great economic losses and remains as a permanent danger for poultry industry. Therefore, in order to reduce the world restriction measures on exports, protection of the public health and accurate diagnosis, the control of AI disease is necessary. Since the traditional serologic tests were not able to discriminate chickens as a positive or negative for circulating AIV, the NS1 protein was selected as a detectable marker to distinguish AIV infected from vaccinated chickens.
In this study, the open reading frame of the NS1 was cloned both into pMAL-c2X and pGEX-4T-1, and expressed in high scales in prokaryotic system which the sequencing analysis showed that was in agreement to expected sequence of NS1 with the identity of 97% - 100% to other isolates of H9N2 subtype in Iran. The NS1 protein was then purified by amylose and glutathione resin and finally analyzed for immunogenicity just for MBP-NS1 protein. The NS1 antibody detection was established using antisera from vaccinated and infected chickens by western blot. It was cleared a great reactivity of the MBP-NS1 protein with H9N2 specific antisera. Western blot was also performed using just purified MBP protein. It showed a weak reactivity with the NS1 antibody in some cases which is in disagreement with the other reports [16]. It might be due to high solubilization and concentration of MBP protein following purification. So, in order to eliminate the eventual reactivity of the MBP portion in MBP-NS1 protein with NS1 antibody, the H9N2 specific antiserum was previously treated with MBP alone.
Twelve antisera obtained from chickens immunized with a single to three doses of commercial H9N2 vaccines with two weeks interval, were used to investigate the presence of NS1 specific antibodies in western blot test. Two serum samples showed fairly strong bands and one of them reacted weakly. The aforementioned data corroborated the results gained by Avellaneda and Tumpey [15,16]. All positive sera samples were obtained from chickens immunized three times, had HI titers above 8log2. The third serum sample that reacted weakly in western blot, related to chickens vaccinated two times. This indicates that specific antibodies against NS1 protein may be produced in chickens immunized with inactivated vaccines, specially after three times injection of oil emulsion vaccine. Regarding to large amount of antibody in infected birds, compared to small amount in vaccinated birds suggesting that an optimal dilution of the sera samples may be useful for discrimination of vaccinated and infected chickens [16]. Meanwhile, it seems that increasing of chickens age more than 4 weeks, may affect the results of reaction by creating the nonspecific binding and cross reactivity specially when the rNS1 protein is not fully purified. MBP-NS1 immunoblotting using of negative sera showed no reaction as well.
In the case of GST-NS1 protein, although the rNS1 protein was expressed in E. coli and confirmed by SDSPAGE analysis, sometimes particularly when a large quantity of culture has been expressed, it was contributed to lysis of the cells and a high reduction in the NS1 protein yield. It was known GST fusion protein is quite soluble but in combination form with NS1 protein because of the charged regions and hydrophobic nature of the NS1 protein it could be retained insoluble. The similar work has been reported previously to clone NS1 gene into pGEX vector as well [21]. The cloning and expression of a small fragment of N-terminal and C-terminal hydrophilic regions of NS1 protein was also undertaken using pGEX vector to confirm this results by author. It produced more soluble GST-NS1 protein (data not shown).
As described above, purification of the GST-NS1 protein was unsuccessful and unfortunately it was not detectable on SDS-PAGE. In this regard improvement of the condition to purify and produce more soluble protein followed by adding of Triton X-100 was useless.
In this study, regarding to the results of western blot assay, the rNS1 protein, can be applied in a DIVA strategy test to control and eradicate AIV, when inactivated vaccination strategy has been considered as a tool for eradication. Besides, it seems to be of low NS1 antibody titers in birds sera vaccinated three times with the killed AI vaccines. Therefore, NS1-DIVA test such as other potential DIVA strategies may have some limitations in accurate application. However, to apply the NS1 protein as a marker in DIVA strategy, additional studies are necessary to detect fully antigenicity properties of this protein.
REFERENCES
- Bouvier, N.M. and Palese, P. (2008) The biology of influenza viruses. Vaccine, 26, 49-53. doi:10.1016/j.vaccine.2008.07.039
- Webster, R.G., Bean, W.J., Gorman, O.T., Chamber, T.M. and Kawaoka, Y. (1992) Evolution and ecology of influenza A viruses. Microbiological Reviews, 56, 152-179.
- Wright, P.E., Neumann, G. and Kawaoka, Y. (2007) Orthomixoviruses. In: Fields Virology, 5th Edition, Lippincott, Willams, Philadelphia, 1692-1731.
- Nichols, J.E. and Leduc, J.W. (2009) Influenza. In: Alan, D.T.B. and Lawrence, R.S., Eds., Vaccines for Biodefence and Emerging and Neglected Diseases, Academic Press, London, 497-525. doi:10.1016/B978-0-12-369408-9.00027-5
- FAO, AIDEnews (2011) Bird flu rears its head again: Increased preparedness and surveillance urged against variant strain. 7 September 2011, 1-8.
- Alexander, D.J. (2007) An overview of the epidemiology of avian influenza. Vaccine, 25, 5637-5644. doi:10.1016/j.vaccine.2006.10.051
- Alexander, P.E., De, P. and Rave, S. (2009) Is H9N2 avian influenza virus a pandemic potential? Canadian Journal of Infectious Disease & Medical Microbiology, 20, 35-36.
- Swayne, D.E. (2003) Vaccines for list A poultry diseases: Emphasis on avian influenza. Developmental Biology, 114, 201-212.
- Capua, I. and Alexander, D.J. (2008) Avian influenza vaccines and vaccination in birds. Vaccine, 26S, 70-73. doi:10.1016/j.vaccine.2008.07.044
- Halvorson, D.A. (2002) The control of H5 or H7 mildly pathogenic avian influenza—A role for inactivated vaccine. Avian Pathology, 31, 5-12. doi:10.1080/03079450120106570
- Capua, I., Cattoli, G. and Marangon, S. (2004) DIVA—A vaccination strategy enabling the detection of field exposure to avian influenza. Developmental Biology, 119, 229- 233.
- Suarez, D.L. (2005) Overview of avian influenza DIVA test strategies. Biologicals, 33, 221-226. doi:10.1016/j.biologicals.2005.08.003
- Suarez, D.L., Lee, C.W. and Swayne, D.E. (2006) Avian influenza vaccination in north America: Strategies and difficulties. Developmental Biology, 124, 117-124.
- Beard, C.W. (1989) Serological procedures. In: Friedman, H., Ed., A Laboratory Manual for the Isolation and Identification of Avian Pathotypes, Kendall Hunt Publishing, Dubuque, 192-200.
- Avellaneda, G., Mundt, E., Lee, C.W., Jadhao, S. and Suarez, D.L. (2010) Differentiation of infected and vaccinated animals (DIVA) using the NS1 protein of avian influenza virus. Avian Diseases, 54, 278-286. doi:10.1637/8644-020409-Reg.1
- Tumpey, T.M., Alvarez, R., Swayne, D.E. and Suarez, D.L. (2005) Diagnostic approach for differentiating infected from vaccinated poultry on the basis of antibodies to NS1, the nonstructural protein of influenza. JC Microbiology, 43, 676-683.
- Lee, C.W. and Suarez, D.L. (2005) Avian influenza virus: Prospects for prevention and control by vaccination. Animal Health Research Reviews, 6, 1-15. doi:10.1079/AHR2005101
- Van den Berg, T., Lambrech, B.T., Marche, S., Steensels, M., Van, B.S. and Bublot, M. (2008) Influenza vaccines and vaccination strategies in birds. Comparative Immunology, Microbiology & Infectious Diseases, 31, 121-165. doi:10.1016/j.cimid.2007.07.004
- Capua, I. and Marangon, S. (2007) The use of vaccination to combat multiple introductions of notifiable avian influenza viruses of the H5 and H7 subtypes between 2000 and 2006 in Italy. Vaccine, 25, 4987-4995. doi:10.1016/j.vaccine.2007.01.113
- Capua, I., Cattoli, G., Marangon, S., Bortolotti, L. and Ortali, G. (2002) Strategies for the control of avian influenza in Italy. Veterinary Record, 150, 223.
- Birch, M.I., Rowan, A., Pick, J., Mumford, J. and Binns, M. (1997) Expression of the nonstructural protein NS1 of equine influenza A virus: Detection of anti-NS1 antibody in post infection equine sera. Journal of Virological Methods, 65, 255-263. doi:10.1016/S0166-0934(97)02189-7
- Ozaki, H., Sugiura, T., Sugita, I., Magawa, H. and Kida, H. (2001) Detection of antibodies to the nonstructural protein (NS1) of influenza A virus allows distinction between vaccinated and infected horses. Veterinary Microbiology, 82, 111-119. doi:10.1016/S0378-1135(01)00366-2
- Zhao, S., Jin, M., Li, H., Tan, Y., Wang, G., Zhang, R. and Chen, H. (2005) Detection of antibodies to the nonstructural protein (NS1) of avian influenza viruses allows distinction between vaccinated and infected chickens. Avian Diseases, 49, 488-493. doi:10.1637/7321-010405R1.1
NOTES
*Corresponding author.