International Journal of Clinical Medicine
Vol. 3 No. 6 (2012) , Article ID: 24741 , 6 pages DOI:10.4236/ijcm.2012.36097
Diagnostic Strategies and Treatment for Ewing’s Sarcoma
![]()
1Institute of Biophysics and Biomedical Engineering, Bulgarian Academy of Sciences, Sofia, Bulgaria; 2Department of Physics and Biophysics, Medical Faculty, Thracian University, Stara Zagora, Bulgaria.
Email: *todorova@bio21.bas.bg, atanastod@abv.bg
Received August 19th, 2012; revised September 22nd, 2012; accepted October 24th, 2012
Keywords: Ewing’s Sarcoma Family of Tumors; Cancer Stem Cells; Immunotherapy; Hematopoietic Progenitor Cell Transplant; Diagnostic Strategies; ESFT Therapy
ABSTRACT
Ewing’s sarcoma is an enigmatic malignancy of progenitor cell origin, driven by transcription factor oncogenic fusions. About 85% of ESFT cases harbor the t(11;22) translocation and express the fusion protein EWS-FLI. Both bone marrow-derived human Mesenchymal stem cells and Neural crest stem cells are permissive for EWS-FLI1 expression that initiates transition to ESFT-like cellular phenotype. Diagnosis of Ewing’s tumor is based on pathologic and molecular findings. The hypoxia enhances the malignancy of ESFT invasive capacity. An ALDHhigh subpopulation of Ewing’s sarcoma cells, capable of self-renewal, tumor initiation and resistant to chemotherapy in vitro, are not resistant to YK-4-279. Intensive high-dose chemotherapy followed by stem-cell reconstitution was used for ESFT patients in second remission. Plerixafor in combination with G-CSF is an effective enhance stem cell mobilization regimen for stem cell collection with lowest success rate in patients with neuroblastoma. The ESFT-derived antigens EZH2(666) and CHM1(319) are suitable targets for protective allo-restricted human CD8(+) T-cell responses against non-immunogenic ESFT. Primitive neuroectodermal features and MSC origin are both compatible with G(D2) aberrant expression and explore G(D2) immune targeting in ESFT.
1. Introduction
Ewing’s sarcoma family of tumors (ESFT) are aggressive bone tumors in adolescents and young adults, but chemotherapy-sensitive and patients with metastatic disease often achieve remission.
The EWS Fusion Proteins are potent promoter-specific transcriptional activators, due to EWS-Activation-Domain (EAD) and a DNA binding domain from the fusion partner. They interact with other proteins required for mRNA biogenesis and induce tumorigenesis by perturbing gene expression. Around 85% of Ewing’s tumours carry the most frequent EWSR1-FLI-1 fusion (Table 1).
Anti-cancer agents (DHR-related peptides and other small molecules) targeted against the N-terminal part of EAD, may possess therapeutic potentialities against Ewing’s sarcoma as inhibitors of EAD-mediated trans-activation and also as immunogenic agents [1].
2. Diagnostic Strategies for Ewing’s Sarcoma
Ewing’s sarcoma family tumors (ESFTs) are aggressive tumors of putative stem cell origin for which prognostic biomarkers and novel treatments are needed [2]. Diagno-
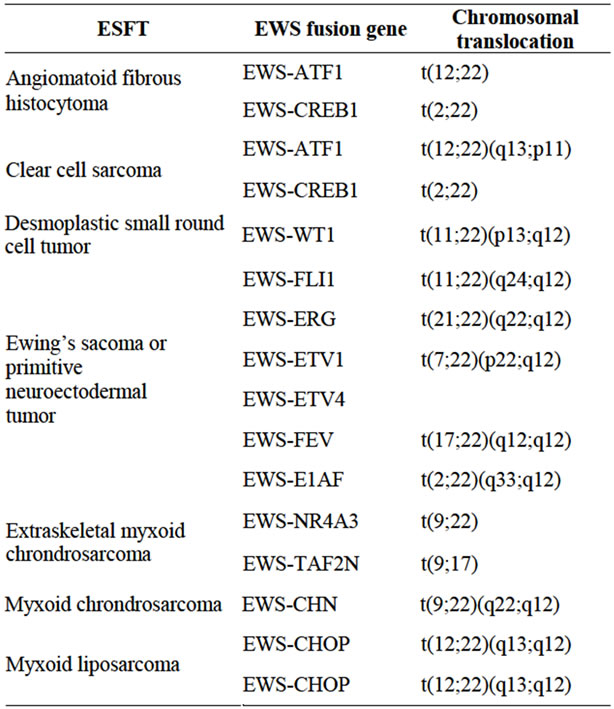
Table 1. EWS associated chromosomal translocations in ESFT.
sis of Ewing’s tumor is based on pathologic and molecular findings [3]. Thus ESFTs are diagnosed by needle biopsy (and expression using immunohistochemistry), and not resected until after administration of neoadjuvant chemotherapy, such that adequate numbers of fresh cells could not be obtained in primary patient samples [4].
The RT-qPCR or RT-cPCR detection of tyrosine hydroxylase (TH) is used to detect Ewing’s sarcoma, nephroblastoma and rhabdomyosarcoma [5]. Pediatric undifferentiated soft tissue sarcomas (USTSs) are composed predominantly of primitive round cell sarcomas, the histogenesis of which is uncertain and their diagnosis and therapy remain a challenge. Stem cell markers were applied to pediatric sarcomas, including Ewing’s sarcoma/primitive neuroectodermal tumors (ESFTs) and USTSs, indicating that they are closely related and may share a common histogenesis. Stem cell antibodies included 3 mesenchymal stem cell markers (CD44, CD105 and CD166) and 5 neural stem cell markers (CD15, CD29, CD56, CD133 and nestin) [6] Morphologically was observed a soft white tumor with hemorrhagic areas, where the tumorous cells expressed cytokeratins AE1/ AE3, EMA, and CD99 (expression of vimentin is not relevant). pPNET was detected and EWS/FLI1 diagnosis in an unusual location was performed. The uncommon adrenal involvement could be explained by the Ewing cell’s origin as a mesenchymal stem cell, which may derive from neural crest cell [3]. Patients had CD99+ small blue round cell tumours diagnosed as Ewing’s sarcomas. GD2 expression was determined by staining with FITC-labeled 14.G2a antibody and subsequent flow cytometry of cell cultures or immunofluorescence analysis of tissue sections (Table 2) [7].


Table 2. Clinical characteristics and GD2 expression in tumour cell cultures and tissue sections [7]: [BM = bone marrow metastases; CR = complete remission; DOD = dead of disease; F = female; M = male; mo = months; o = osseous metastases; P = pulmonary metastases; PD = progressive disease].
3. Cancer Stem Cell Model as a Strategy in the Treatment of Bone Cancer
Recent experimental evidences support either primitive neural crest cells or mesenchymal stem cells (MSCs) as candidate cells of origin for Ewing’ sarcoma, while the MSCs can arise from neural crest progenitors [7].
Cancer stem cells (CSCs) are a chemotherapy-resistant population of cells capable of self-renewal and of regenerating the bulk tumor, resulting in relapse and patient death. Thus the Ewing’s sarcoma follows a clinical pattern consistent with the Cancer Stem Cell model in which remission is easily achieved, even for patients with metastatic disease, but relapse remains frequent and is usually fatal [4]. The response of bone tumor stem cell to chemotherapy only shrinks tumor tissues and the tumor develop after some time, due to CSC presence, which self-renew and differentiate to form a new tumor. Chemotherapy together with CSCs-targeted therapy can kill most of the tumor cells and also the CSCs. Thus the non-CSC tumor will be differentiated with no proliferateing ability, followed by cell death. Chemotherapy, including Albendazole, together with CSC targeted therapy can block these stem cell-like cells and their ability to form tumor [8]. The presence of chemotherapy-resistant CSC is one explanation for the discrepancy between response to treatment and patient survival [4].
Downregulation of miRNA-145 is implicated in the development of CSC in ESFT. Let-7a and miRNA-145 repression may play a critical role in EWS-FLI-1-mediated CSC generation and provides a means to control the ESFT growth. Let-7a target genes, relevant to transformation and subsequent tumor development include RAS, MYC, IGF2BP1 and HMGA2 that may constitute a therapeutic target [9].
4. Hypoxia and Ewing’s Sarcoma
Hypoxia is a pathological condition in which the body is deprived of adequate oxygen supply and is important in development and growth of solid tumors, driving tumor bone metastases. Normal oxygen level is critical and play vital role in regulating stem or tumor cells behavior. Thus targeting hypoxia would be a novel strategy to eliminate CSCs, curing bone tumor. Hypoxia can induce the non-CSC tumor to become stem cell type, which have self-renewing and proliferating ability and can form tumor. HIF is highly expressed in CSCs and blockage of HIF pathway provides therapeutic advantage in bone tumor patients. A feature of Ewing’s sarcoma is the presence of blood lakes linked by tumor cells that correlates with poor clinical outcome. Oxygen tension plays a significant role in tube formation in vitro. EWS-FLI1 protein was up-regulated in a HIF-1α-dependent manner and HIF-1α induced its accumulation. The regulation of EWS-FLI1 in hypoxic environments may occur at posttranscriptional level, supported by HIF-1α-activation of genes VEGF, Aldolase-C, GLUT-1, CA9, and IGFBP3, increased under hypoxia. EWS-FLI1 RNA expression remains unchanged and thus hypoxia enhances the malignancy of ESFT mainly through increasing invasive capacity. HIF-1α and EWS-FLI1 may function together in both synergistic and antagonistic cross-talk under conditions of hypoxia, and their targeting may prove to be of therapeutic benefit [8].
5. YK-4-279 Treatment
The binding of RNA helicase A (RHA) to EWS-FLI1 promotes its oncogenic function. YK-4-279 (C17H13Cl2 NO4) is an inhibitor of protein-protein interactions between EWS-FLI1 and RHA, inducing apoptosis of ESFT lines and represents a different approach to targeted therapy for Ewing’s sarcoma. YK-4-279 reduces the growth of Ewing’s sarcoma orthotopic xenografts in mice after treatment with the inhibitor for two weeks [10].
A subpopulation of Ewing’s sarcoma cells (less than 5%) with high expression of aldehyde dehydrogenase (ALDHhigh) are capable of self-renewal and of tumor initiating activity in immune deficient mice, both characteristics of tumor stem cells. The ALDHhigh cells grow much more rapidly than the ALDHlow cells do and show tumor initiating activity when grown in culture, form colonies in soft agar and grow in “sarcospheres”. Fewer ALDHhigh cells than ALDHlow cells are needed to grow a tumor in mice. These cells are resistant to standard chemotherapy drugs and if they became more sensitive, this would dramatically improve the outcomes for patients with Ewing’s sarcoma [11]. ALDH itself contributes to chemoresistance, functioning as a detoxification enzyme for chemotherapeutic drugs such as cyclophosphamide. A subpopulation of Ewing’s sarcoma cells was isolated, from both human cell lines and human xenografts grown in immune deficient mice, which express ALDHhigh activity and are enriched for clonogenicity, sphere-formation, and tumor initiation. The ALDHhigh cells are resistant to chemotherapy in vitro, but this can be overcome by the ATP binding cassette transport protein inhibitor verapamil. These cells are not resistant to YK-4-279 that is selectively toxic to Ewing’s sarcoma cells both in vitro and in vivo. The ALDHhigh population is relatively resistant to cytotoxic chemotherapeutic agents, including doxorubicin and etoposide, but retains sensitivity to the EWS-FLI1 targeted inhibitor, YK-4-279. Although Ewing’s sarcoma stem cells (ESSC) are resistant to standard cytotoxic agents, they retain sensitivity to YK-4-279 [4].
6. Medical Cases in Stem Cell Treatment of Ewing’s Sarcoma
New treatment strategies are needed to cure disseminated Ewing sarcoma [7].
The concepts for high-dose therapy followed by hematopoietic cell transplantation in Ewing’s family of tumors are based on dose-response and dose-intensity relationships [12]. This type of treatment is used in patients with Ewing’s tumors that are unlikely to be cured with other treatments (with metastatic disease or those whose disease comes back after the standard treatment). It involves giving very high doses of chemotherapy, and then replacing the body’s bone marrow cells, which were killed by the treatment. Possible early complications and side effects are low blood cell counts (with increased risk of infection and bleeding), nausea, vomiting, loss of appetite, mouth sores, and hair loss, increased risk of infection, low red blood cell and platelet counts that may require blood product transfusions or other treatments [13].
Intensive chemotherapy followed by stem-cell reconstitution (high-dose chemotherapy [HDT]) was used as consolidation therapy for patients with ESFTs in second remission. The patients receiving HDT have an improved progression-free and overall survival. HDT did not improve the outcome for patients with metastatic ESFTs. Tumor-cell contamination seems to play a role in recurrent disease for patients with metastatic neuroblastoma and acute myeloid leukemia and, therefore, remains a considerable concern for ESFTs. The European Bone Marrow Transplant Registry reported that HDT might be beneficial for a number of patients with recurrent ESFTs [14]. The HDT was added to conventional chemotherapy in Ewing’s sarcoma family tumor patients, poor responders to induction chemotherapy in order to improve their survival. High-dose therapy added to the VACA-IE regimen in poor responder patients is feasible and effective. Selected groups of patients with Ewing’s sarcoma can benefit from HDT [15].
Plerixafor or chemotherapy in combination with granulocyte-colony-stimulating factor (G-CSF) is an effective enhance stem cell mobilization regimen with the potential of successful stem cell collection. The lowest success rate with plerixafor remobilization was found in patients with neuroblastoma. High-dose chemotherapy followed by autologous hematopoietic progenitor cell transplant (HPCT) has become a well-established therapy for disorders of the hematopoietic system and also for other chemosensitive, radiosensitive, immunosensitive, or even refractory (germ cell tumors) malignancies in children, adolescents, and adults. A successful HPCT is dependent on transplantation of a sufficient amount of CD34+ cells to ensure prompt and durable engraftment [16].
7. Immunotherapeutic Strategies in Stem Cell Treatment of Ewing’s Sarcoma
Low-dose IL-2 was applied in children with malignancies at very high risk of relapse who underwent highly T cell and B cell depleted HLA-identical (MUD). Full haplotype mismatched related hematopoetic stem cell transplantation [HSCT] was used in patients with relapsed or progressive Ewing’s sarcoma who received prophylactic IL-2 treatment for a high probability of disease recurrence after allo-HSCT [17].
In some cancers tumour antigen-specific antibodies or T cells show promising activity. Immune targeting of Ewing’s sarcoma was limited by the lack of adequate antigens. Although the breakpoint region of the characteristic EWS-FLI1 fusion protein contains unique peptide sequences specifically recognized by T cells, clinically tested fusion peptides failed to induce high-avidity T-cell responses [7].
The successful immunotherapy is limited by an ineffective T-cell repertoire against tumour antigens and the inability of the patient’s immune system to overcome tolerance-inducing mechanisms. The specific recognition and lytical potential of allo-restricted CD8(+) T cells were tested against ESFTs associated antigens Enhancer of Zeste, Drosophila Homolog 2 (EZH2), and Chondromodulin-I (CHM1). The ESFTs-derived antigens, EZH2 (666) and CHM1(319) are suitable targets for protective allo-restricted human CD8(+) T-cell responses against non-immunogenic Ewing’s tumours. These new therapeutic strategies may benefit the ESFT patients treated with allogeneic stem cell transplantation [18].
The ganglioside antigen GD2 (G(D2)) is characteristic surface-expressed in Ewing’s sarcoma and allows for MHC-independent immune targeting (Table 2). Primitive neuroectodermal features and MSC origin are both compatible with G(D2) aberrant expression and explore G(D2) immune targeting in ESFT. The GD2 expression is a common feature of ESFT, including established cell lines, low-passage cell cultures, and primary biopsies, compatible with the cellular origin from either primitive neuroectodermal cells or bone marrow-derived mesenchymal progenitor cells (both cell types express GD2). Chimeric antigen receptor-engineered GD2 specific T cells efficiently interact with GD2-expressing neuroblastoma cells in vitro, resulting in specific tumour cytolysis. GD2-specific T-cell transfer was well tolerated and first evidence of in vivo persistence and antitumour activity was obtained. Specifically modified T cells express the G(D2)-specific chimeric receptor 14. G2a-28ζ efficiently interacted with ESFT cells, resulting in antigen-specific secretion of cytokines. The chimeric receptor gene-modified T cells exerted potent, G(D2)-specific cytolytic responses to Ewing’s sarcoma, including tumour cells grown as multicellular, anchorage-independent spheres. T cells, specific to G(D2) further had activity against Ewing’s sarcoma xenografts. Thus GD2-targeted immunotherapy may be a potent strategy to control or eliminate residual disease in patients with recurrent or refractory Ewing’s sarcoma. Future studies with Ewing’s sarcoma patients should address the prognostic significance of GD2 expression status and analysis at both diagnosis and relapse. In ESFTs this can be achieved by applying T-cell therapy in the context of lymphopenia after high-dose chemotherapy with autologous stem cell rescue. CAR-transduced, GD2-specific T cells exert cytolytic responses against allogeneic and autologous ESFTs targets and efficiently lyse multicellular Ewing’s sarcoma spheres. GD2-positive normal bone marrow MSCs may interfere with haematopoietic stem cell engraftment, however, no haematological toxicity was observed in any clinical trial in patients receiving GD2 mAbs or conjugates or GD2-reengineered T cells. Increasing knowledge of immune escape mechanisms will help to optimize integration of immunotherapies into the treatment of Ewing’s sarcoma [7].
Evidence for an antitumour activity of GD2-specific T cells against larger and vascularised tumours was obtained in vivo, thus supporting the translational application of GD2-targeted therapies in Ewing’s sarcoma.
8. Conclusions
The knowledge of mechanisms and the inhibition of interactions, required for the oncogenic activity of EWS fusion proteins, may help the development of uniquely effective, tumor-specific anticancer agents against Ewing’s Tumors. Primitive neuroectodermal features and MSC origin of Ewing’s sarcoma are important in immune targeting of ESFTs. New therapeutic strategies of Immune targeting of Ewing’s sarcoma and development of adequate antigens may benefit the ESFTs patients treated with allogeneic stem cell transplantation.
The finding of new EAD partners and the understanding of mechanisms of carcinogenesis are related to the treatment of ESFTs [19]. Oncogenic fusion proteins, including EWS-FLI1, are located within the tumor and thus become excellent therapeutic targets. There are currently no agents targeted toward transcription factors. Promising new therapies for the treatment of Ewing’s sarcoma are the development and the use of new molecules-inhibitors of specific protein-protein interactions, such as the small molecule YK-4-279 (still in pre-clinical studies). RNA Helicase A enhances the oncogenesis of EWS-FLI1, but the compound YK-4-279 [10] [Cayman Chemical; Tocris Bioscience], targeted against this protein-protein interaction, has a chiral center and only one functional enantiomer [20]. Although Ewing’s sarcoma stem cells are resistant to standard cytotoxic chemotherapeutic agents, including doxorubicin (anthracycline antibiotic intercalating in DNA (Adriamycin PFS and RDF, Rubex, hydroxydaunorubicin)) and etoposide (topoisomerase inhibitor (VP-16, Etopophos, Vepesid)), they retain sensitivity to (S)-YK-4-279 that impose to move this compound towards clinical trials in small molecule targeting of intrinsically disordered proteins.
REFERENCES
- R. Todorova, “In Vitro Interaction between the N-Terminus of the Ewing’s Sarcoma Protein and the Subunit of RNA Polymerase II hsRPB7,” Molecular Biology Reports, Vol. 36, No. 6, 2009, pp. 1269-1274. doi:10.1007/s11033-008-9308-2
- A. Cooper, J. van Doorninck, L. Ji, D. Russell, M. Ladanyi, H. Shimada, M. Krailo, R. B. Womer, J. H. Hsu, D. Thomas, T. J. Triche, R. Sposto and E. R. Lawlor, “Ewing Tumors That Do Not Overexpress BMI-1 Are a Distinct Molecular Subclass with Variant Biology: A Report from the Children’s Oncology Group,” Clinical Cancer Research, Vol. 17, No. 1, 2011, pp. 56-66. doi:10.1158/1078-0432.CCR-10-1417
- J. Gonin, F. Larousserie, B. Dousset, J. Rousseau, O. Delattre, C. Waintrop, V. Tsatsaris, J. Y. Pierga, M. C. Vacher-Lavenu and F. Tissier, “An Unusual Adrenal Tumor: Ewing Tumor,” Annals of Pathology, Vol. 31, No. 1, 2011, pp. 28-31. doi:10.1016/j.annpat.2010.07.043
- O. Awad, J. T. Yustein, P. Shah, N. Gul, V. Katuri, A. O’Neill, Y. Kong, M. L. Brown, J. A. Toretsky and D. M. Loeb, “High ALDH Activity Identifies ChemotherapyResistant Ewing’s Sarcoma Stem Cells That Retain Sensitivity to EWS-FLI1 Inhibition,” PLoS One, Vol. 5, No. 11, 2010, p. e13943. doi:10.1371/journal.pone.0013943
- R. Esser, W. Glienke, K. Bochennek, S. Erben, A. Quaiser, C. Pieper, A. Eggert, A. Schramm, K. Astrahantseff, M. L. Hansmann, D. Schwabe, T. Klingebiel and U. Koehl, “Detection of Neuroblastoma Cells during Clinical Follow Up: Advanced Flow Cytometry and rt-PCR for Tyrosine Hydroxylase Using Both Conventional and Real-Time PCR,” Klinical Padiatrics, Vol. 223, No. 6, 2011, pp. 326-331. doi:10.1055/s-0031-1287842
- B. Sadikovic, C. Graham, M. Ho, M. Zielenska and G. R. Somers, “Immunohistochemical Expression and Cluster Analysis of Mesenchymal and Neural Stem Cell-Associated Proteins in Pediatric Soft Tissue Sarcomas,” Pediatric and Development Pathology, Vol. 14, No. 4, 2011, pp. 259-272. doi:10.2350/10-08-0890-OA.1
- S. Kailayangiri, B. Altvater, J. Meltzer, S. Pscherer, A. Luecke, C. Dierkes , U. Titze, K. Leuchte, S. Landmeier, M. Hotfilder, U. Dirksen, J. Hardes, G. Gosheger, H. Juergens and C. Rossig, “The Ganglioside Antigen GD2 Is Surface-Expressed in Ewing Sarcoma and Allows for MHC-Independent Immune Targeting,” British Journal of Cancer, Vol. 106, No. 6, 2012, pp. 1123-1133. doi:10.1038/bjc.2012.57
- W. Zeng, R. Wan, Y. Zheng, S. R. Singh and Y. Wei, “Hypoxia, Stem Cells and Bone Tumor,” Cancer Letters, Vol. 313, No. 2, 2011, pp. 129-136. doi:10.1016/j.canlet.2011.09.023
- C. De Vito, N, Riggi, M.-L. Suvà, M. Janiszewska, J. Horlbeck, K. Baumer, P. Provero and I. Stamenkovic, “Let-7a Is a Direct EWS-FLI-1 Target Implicated in Ewing’s Sarcoma Development,” PLoS One, Vol. 6, No. 8, 2011, p. e23592. doi:10.1371/journal.pone.0023592
- H. V. Erkizan, Y. Kong, M. Merchant, S. Schlottmann, J. S. Barber-Rotenberg, L. Yuan, O. D. Abaan, T. H. Chou, S. Dakshanamurthy, M. L. Brown, A. Uren and J. A. Toretsky, “A Small Molecule Blocking Oncogenic Protein EWS-FLI1 Interaction with RNA Helicase A Inhibits Growth of Ewing’s Sarcoma,” Nature Medicine, Vol. 15, No. 7, 2009, pp. 750-756. doi:10.1038/nm.1983
- D. Loeb, “Identification and Characterization of the Ewing’s Sarcoma Stem Cell. An ESUN Study Report,” Liddy Shriver Sarcoma Initiative, 2009.
- J. Rosenthal and A. B. Pawlowska, “High-Dose Chemotherapy and Stem Cell Rescue for High-Risk Ewing’s Family of Tumors,” Expert Review of Anticancer Therapy, Vol. 11, No. 2, 2011, pp. 251-262. doi:10.1586/era.10.215
- American Cancer Society, “High-Dose Chemotherapy and Stem Cell Transplant for Ewing Family of Tumors,” 2012. http://www.cancer.org/Cancer/EwingFamilyofTumors/Detailed-Guide/ewing-family-of-tumors-treating-high-dose-chemo-stem-cell
- N. Marina and P. A. Meyers, “High-Dose Therapy and Stem-Cell Rescue for Ewing’s Family of Tumors in Second Remission,” Journal of Clinical Oncology, Vol. 23, No. 19, 2005, pp. 4262-4264. doi:10.1200/JCO.2005.12.915
- S. Ferrari, K. S. Hall, R. Luksch, A. Tienghi, T. Wiebe, F. Fagioli, T. A. Alvegard, A. B. del Prever, A. Tamburini, M. Alberghini, L. Gandola, M. Mercuri, R. Capanna, S. Mapelli, A. Prete, M. Carli, P. Picci, E. Barbieri, G. Bacci and S. Smeland, “Nonmetastatic Ewing Family Tumors: High-Dose Chemotherapy with Stem Cell Rescue in Poor Responder Patients. Results of the Italian Sarcoma Group/Scandinavian Sarcoma Group III Protocol,” Annals of Oncology, Vol. 22, No. 5, 2011, pp. 1221-1227. doi:10.1093/annonc/mdq573
- N. Worel, J. F. Apperley, G. W. Basak, K. W. Douglas, I. H. Gabriel, C. Geraldes, K. Hübel, O. Jaksic, Z. Koristek, F. Lanza, R. Lemoli, G. Mikala, D. Selleslag, R. F. Duarte and M. Mohty, “European Data on Stem Cell Mobilization with Plerixafor in Patients with Nonhematologic Diseases: An Analysis of the European Consortium of Stem Cell Mobilization,” Transfusion, 2012. doi:10.1111/j.1537-2995.2012.03603.x
- P. Schlegel, H.-M. Teltschik, M. Pfeiffer, R. Handgretinger, M. Schumm, E. Koscielniak, T. Feuchtinger, T. Klingebiel, P. Bader, P.-G. Schlegel, J. Greil and P. Lang, “Long-Term IL-2 Therapy after Transplantation of T Cell Depleted Stem Cells from Alternative Donors in Children,” Best Practice & Research in Clinical Haematology, Vol. 24, No. 3, 2011, pp. 443-452. doi:10.1016/j.beha.2011.04.007
- U. Thiel, S. Pirson, C. Müller-Spahn, H. Conrad, D. H. Busch, H. Bernhard, S. Burdach and G. H. S. Richter, “Specific Recognition and Inhibition of Ewing Tumour Growth by Antigen-Specific Allo-Restricted Cytotoxic T Cells,” British Journal of Cancer, Vol. 104, No. 6, 2011, pp. 948-956.
- R. Todorova, “Machanism of Transactivation by EAD Based on Protein-Protein Interaction Studies,” Acta Medica Bulgarica, Vol. 37, No. 2, 2010, pp. 58-64.
- J. S. Barber-Rotenberg, S. P. Selvanathan, Y. Kong, H. V. Erkizan, T. M. Snyder, S. P. Hong, C. L. Kobs, N. L. South, S. Summer, P. J. Monroe, M. Chruszcz, V. Dobrev, P. N. Tosso, L. J. Scher, W. Minor, M. L. Brown, S. J. Metallo, A. Üren and J. A. Toretsky, “Single Enantiomer of YK-4-279 Demonstrates Specificity in Targeting the Oncogene EWS-FLI1,” Oncotarget, Vol. 3, No. 2, 2012, pp. 172-182.
Abbreviations:
ESFT: Ewing’s sarcoma family of tumors;
EAD: EWS-Activation-Domain;
MSC: Mesenchymal stem cell;
ALDHhigh: High expression of aldehyde dehydrogenase;
BM: Bone marrow;
CSCs: Cancer stem cells;
ESSC: Ewing’s sarcoma stem cells;
HDT: High-dose chemotherapy;
HPCT: Hematopoietic progenitor cell transplant;
G(D2): Ganglioside antigen GD2.
NOTES
*Corresponding author.