 J. Biomedical Science and Engineering, 2010, 3, 291-299 doi:10.4236/jbise.2010.33039 Published Online March 2010 (http://www.SciRP.org/journal/jbise/ JBiSE ). Published Online March 2010 in SciRes. http://www.scirp.org/journal/jbise Covariation of mutation pairs expressed in HIV-1 protease and reverse transcriptase genes subjected to varying treatments David King, Roger Cherry, Wei Hu* Department of Computer Science, Houghton College, Houghton, USA. Email: wei.hu@houghton.edu. Received 7 October 2009; revised 27 November 2009; accepted 4 December 2009. ABSTRACT A previous study, focused on the correlation of muta- tion pairs of synonymous (S) and asynonymous (A) mutations, distinguished only between the treated and untreated data of protease and reverse tran- scriptase (RT) of HIV-1 subtype B. It is well known that single mutation patterns in HIV-1 are treat- ment-specific. It logically follows that covariation between mutations will also be treatment specific. Thus, our motivation is to give a more in depth study of the covariation between mutation pairs, analyzing not only treated and untreated, but what specific treatments were used, and how they affected the co- variation between the mutations differently. We in- tended to further deepen this study by analyzing the covariation of mutations in protease and RT in dif- ferent subtypes of HIV-1. We found that virus sam- ples subjected to antiretroviral Protease- and RT- inhibitors do show different patterns of mutation covariation in B-subtype protease and RT of HIV-1, while maintaining the same overall trend. <A, A> covariation will tend to be higher and more distinct from <A, S> and <S, S> covariation after treatment. The same trend continues in protease and RT re- gardless of subtype. We also found the highly cova- ried codon positions, position pairs, and position- covariation clusters in protease, affected by different treatments. Most of them are well known major drug-resistance sites for these treatments. Keywords: HIV; Covariation; Synonymous Mutation; Asynonymous Mutation; Protease; Reverse Transcriptase; Drug Resistance 1. INTRODUCTION Analysis of mutations in human immunodeficiency virus type one (HIV-1) has become a vital component of treatment development. This is largely due to the ability of mutations to alter the effectiveness of retroviral drugs in treatment. In particular, the study of correlation, or covariation, between mutations has been a focus. A particularly strong correlation between amino acids can be seen as evidence of a functional link between those amino acids. Studying the covariation of these mutations will help both our understanding of the HIV-1 virus, and our ability to treat it. There is more than one type of mutation which HIV undergoes. However, the changes in the HIV-1 genome, which is a string of nucleotides, do not necessarily lead to changes in the amino acids a particular portion of the genome generates. Asynonymous mutations, or muta- tions that affect the viral amino acid sequences, have been the focus of much research. In a previous study [1], there has been shown to be a significant increase in the co- variance of asynonymous (A) mutations after treatment. The other mutation type, synonymous (S), those muta- tions which do not affect the viral amino acid sequence, has not shown as extreme change due to treatment. Pre- vious studies [1] have also shown that on the average, the correlation between two mutations decreases as the physical distance between the mutations increased. These studies are hindered by the scarcity of data for many subtypes of HIV and several varieties of antiretro- viral drugs, since clinical tests are administered according to the needs of the patients, not the desire for data. Ge- netic records primarily focus on subtype B HIV, the most prevalent variety of the virus in the western world, so most research in turn focuses on mutations in HIV-1, subtype B. Previous studies [1,2,3,4,5] in this field have been limited in scope, focusing mainly on sequences of sub- type B, and mainly distinguishing between treated and untreated sequences without considering the specific treatment involved. Our current study expands upon that research. We have run analysis of datasets of HIV-1 sequences, distin- guishing based on the specific drug administered. In addition, we have run analysis on other subtypes of HIV-1, in order to get a more complete picture of the ways treatment, protease inhibitors (PIs) and nucleotide  292 D. King et al. / J. Biomedical Science and Engineering 3 (2010) 291-299 Copyright © 2010 SciRes JBiSE reverse transcriptase inhibitors (NRTIs), affects the co- variation of HIV-1 mutations. 2. METHODS 2.1. HIV-1 Sequence Datasets We used datasets from the Stanford HIV Drug Resistance Database (http://hivdb.stanford.edu/). All reference se- quences were taken from the Los Alamos HIV Sequence Database (http://www.hiv.lanl.gov /content/index). All data were in FASTA-format nucleotide sequences. A reference sequence, in this study, is a consensus sequence, found to be normative of a given genomic region and subtype. We used one reference sequence for each genomic region and each subtype. A mutation is considered to be a deviation from this reference sequence. We used two categories of datasets. Our primary dataset, the treatment-specific, consisted only of B- subtype protease and RT, downloaded exclusively from the Stanford database. Only data sets of significant size (100 or more) were used. We used two datasets of pro- tease sequences, both of subtype B, one treated with the drug IDV (642 sequences) and another treated with NFV (899 sequences). The RT datasets were also of subtype B exclusively, and included a set of sequences treated with the drug AZT (361 sequences), and one with a common combination of drugs, AZT, 3TC, and EFV (114 sequences). Our second set of datasets was of treated/untreated protease and RT of different subtypes. B-subtype, C-subtype, and recombinant subtype AE were obtained for both datasets. Of these, there were 8335 untreated B-subtype protease sequences, 8138 treated. There were 8364 treated B-subtype RT sequences, 5880 untreated. C-subtype had 1112 sequences untreated protease, 1565 treated protease, 650 treated RT, and 2202 untreated RT. Due to lack of data, Recombinant subtype AG was ob- tained for protease only. Also due to lack of data, we analyzed only the RT of subtype A (106 sequences treated, 1519 sequences untreated). 2.2. Covariation Measurements in Specific Mutation Pairs We used covariation measure D’ to determine the amount of non-random association between the mutations con- sidered in a pair. D’ is a well known measure for deter- mining non-random association, and was used in several previous studies, including [1]. The formula and com- plete procedure of computing D’ can be found in [6]. We chose D’ as a measure above other covariation measures because of its symmetry: the D’ value, which is a value between -1 and 1, provides an equal scale for evaluating both negative and positive correlation. This allows us to study both positive and negative correlation of mutation pairs. The D’ value of a given mutation pair containing mu- tations X and Y relies on a 2 × 2 contingency table con- sisting of NXY, NX, NY, and NO, where NXY is the number of sequences in the dataset which contain both mutations, NX, is the number of sequences in the dataset which contain only mutation X, NY, is the number of sequences in the dataset which contain only mutation Y, and NO, is the number of sequences in the dataset which contain neither mutation. N is the total number of sequences in the dataset. As in [1], we also used a value θ = (Nxy*NO)/(NX*NY) which is a maximum likelihood estimator for independ- ence of mutations X and Y. When θ = 1, there is complete independence of X and Y. We used this θ value as a cutoff when plotting our curves. By using this value to cutoff some of the outlier points which throw the curves off, we create more clear and reliable plots. In our plots, we only allowed data points with θ > 1.5 or θ < 0.5. In each dataset, a singular cutoff was utilized, such that mutations which occur only once in the dataset were not used in the calculation of D’. 2.3. Counting Paradigm for Specific Mutation Pairs The collection and calculation of the mutation pairs are handled at the same time by the following algorithm. Data preprocessing and alignment is just as important to the algorithm as the central process itself. In preproc- essing, we ensured that each sequence was correctly aligned to the reference sequence of the same genomic region and subtype. Each reference sequence was taken from the Los Alamos HIV Database. If an individual sequence couldn’t be aligned with the reference se- quence, it was not used, as a single unaligned sequence within a dataset can drastically affect the output of the D’ analysis. Gaps were not allowed in the reference sequences, but were allowed in the data sequences provided they aligned properly with the reference sequence. If a data sequence was properly aligned, but longer than the reference se- quence, we only analyzed the portion of the sequence which could be compared with the reference sequence. 2.4. D’ Values According to Codon Position The collection and calculation of the mutation pairs are handled by a simple counting mechanism. We compared all nucleotide sequences of our dataset against a con- sensus sequence, and made not of each nucleotide sub- stitution, and whether that substitution constituted a synonymous or asynonymous mutation. For each se- quence in the dataset, we record all valid pairs of muta- tions. Mutations pairs that involve both asynonymous mutations were labeled as <A, A>, those that involve one asynonymous and one synonymous mutation were labeled <A, S>, and those that involve two synonymous mutations are <S, S>. Then, we take frequency counts on all mutation pairs across all sequences in order to calcu- late the D’ of each mutation pair.  D. King et al. / J. Biomedical Science and Engineering 3 (2010) 291-299 Copyright © 2010 SciRes 293 JBiSE For display, we use a sliding window curve. This en- hances the readability and reliability of the curve. Sim- ply graphing this data such that each physical position is an average of all D’ values at that physical position give an unsteady curve towards the greater physical distances. As the physical distance increases, the number of data points available for that physical distance decreases, leading to greater oscillation as the plot goes on. A sliding window has the same amount of data going into each point on the graph, and is thus more reliable. Our sliding window curves each use 3% of the data in the set per window, with a 50% overlap. 2.5. D’ According to Genomic Position We analyzed D’ according to amino acid position within the genomic region as in [1]. This gives us information on how specific codon positions interact with one an- other within the gene, particularly in response to differ- ent treatments. We also performed a pair-wise analysis of these specific mutation positions in order to reveal more on the differ- ences between <A, A>, <A, S>, and <S, S> mutation pairs. Using this data, we generated covariation histograms. In these histograms, the value at each codon position is the sum of the D’ value for all mutation pairs associated with that position. Each mutation pair will contribute total D’ value to the positions of its two mutations. In this manner, positions which are either the site of great amounts of mutation or high covariance will stand out, with positions which are both high in mutation amount and covariance being seen as peeks. In order to further explore the relationships between the amino acid positions, we cast our histograms into two dimensional contour plots, which reveal clusters of co- variation. To generate these plots, we form a square two dimensional table with a length equal to the number of amino acid positions in a given dataset. Each mutation pair is then mapped to a position on this table, based on the position of that pair’s mutations. For example, the mutation pair L10I and Q20V would be mapped to posi- tion x = 10, y = 20. The value of each position in the table is the sum of all D’ values of the pairs assigned to that position. This provides a visual representation of the relationships be- tween positions, with higher values representing posi- tions which are highly correlated with one another, and the lower values representing unrelated positions. 3. RESULTS 3.1. Effects of Specific Treatment on the Covariation of B-Subtype Protease and RT First, in order to discover the effects of specific treat- ments on the covariation of HIV-1 mutation pairs, we ran a D’ analysis on data sets of B-subtype protease and RT with known treatment types. For reference, we also included the generically treated and untreated datasets of B-subtype protease and RT, in order to see how the dif- ferent treatments effected the genomes and, and how the compare to the effects of overall treatments. Our findings revealed that covariation between muta- tions is, as we expected, treatment dependent. In com- paring IDV- and NFV-treated protease, these results become clear. The plots in Figure 1, show the results of the analysis according to physical distance, display clearly different patterns in their covariation. The average D’ values of <A, A> pair covariation are just at 0.3 for both datasets, however we can clearly see peaks of high covariance in different positions on the <A, A> curves. If we compare these two treatment-specific plots against D’ values generated from the set of generically treated HIV-1 sequences (those sequences that have received treatment of any sort, plot not pictured), we can see that the differences even more pronounced. The average D’ of <A, A> mutation pairs in protease which has seen any sort of treatment whatsoever is much higher—a value just at 0.4, and yet a different set of peaks within the curve. We see similar results in RT. The curves generated by RT treated with the drug AZT are considerably different than the generically treated RT, as can be seen in Figure 2. The generic trends of covariation, however, were largely the same despite what specific treatments were used. <A, A> covariation tended to be higher than <A, S> or <S, S> covariation in all datasets. In addition, we also noticed that on average, <A, S> covariation tended to be higher than <S, S>. This separation was even present in the untreated dataset. Figure 1. Different treatments lead to different patterns of covariation. These two sliding window plots display the D’ analysis of two different treatment types. The top displays results derived from IDV-based treatment, and the bottom plot displays those derived from NFV. Clearly, the different treat- ments induce quite different <A, A> covariation patterns in the sliding window curves. The different treatments do not seem to significantly affect the <A, S> or <S, S> curves. 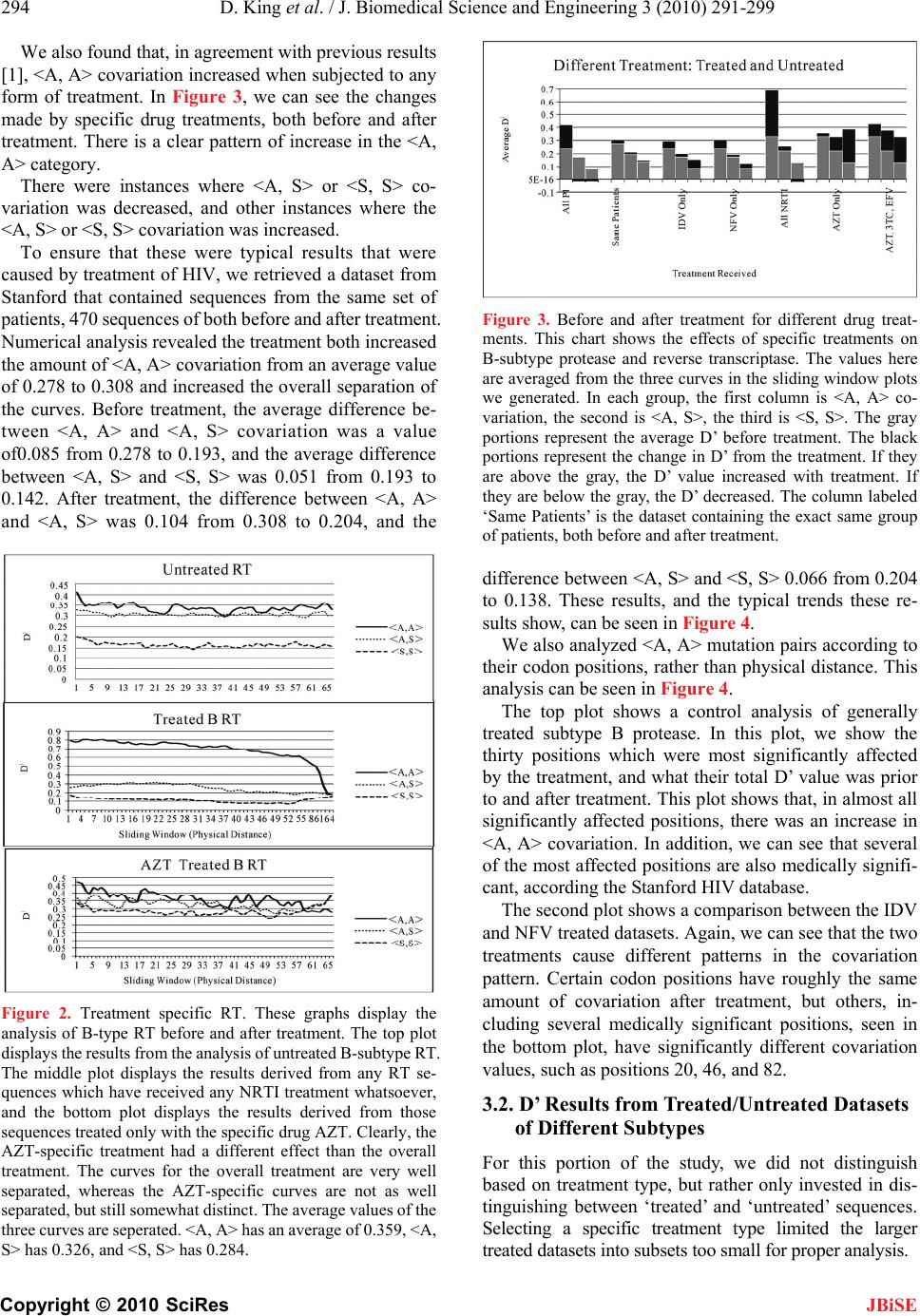 294 D. King et al. / J. Biomedical Science and Engineering 3 (2010) 291-299 Copyright © 2010 SciRes JBiSE To ensure that these were typical results that were caused by treatment of HIV, we retrieved a dataset from Stanford that contained sequences from the same set of patients, 470 sequences of both before and after treatment. Numerical analysis revealed the treatment both increased the amount of <A, A> covariation from an average value of 0.278 to 0.308 and increased the overall separation of the curves. Before treatment, the average difference be- tween <A, A> and <A, S> covariation was a value of0.085 from 0.278 to 0.193, and the average difference between <A, S> and <S, S> was 0.051 from 0.193 to 0.142. After treatment, the difference between <A, A> and <A, S> was 0.104 from 0.308 to 0.204, and the We also found that, in agreement with previous results [1], <A, A> covariation increased when subjected to any form of treatment. In Figure 3, we can see the changes made by specific drug treatments, both before and after treatment. There is a clear pattern of increase in the <A, A> category. There were instances where <A, S> or <S, S> co- variation was decreased, and other instances where the <A, S> or <S, S> covariation was increased. Figure 2. Treatment specific RT. These graphs display the analysis of B-type RT before and after treatment. The top plot displays the results from the analysis of untreated B-subtype RT. The middle plot displays the results derived from any RT se- quences which have received any NRTI treatment whatsoever, and the bottom plot displays the results derived from those sequences treated only with the specific drug AZT. Clearly, the AZT-specific treatment had a different effect than the overall treatment. The curves for the overall treatment are very well separated, whereas the AZT-specific curves are not as well separated, but still somewhat distinct. The average values of the three curves are seperated. <A, A> has an average of 0.359, <A, S> has 0.326, and <S, S> has 0.284. Figure 3. Before and after treatment for different drug treat- ments. This chart shows the effects of specific treatments on B-subtype protease and reverse transcriptase. The values here are averaged from the three curves in the sliding window plots we generated. In each group, the first column is <A, A> co- variation, the second is <A, S>, the third is <S, S>. The gray portions represent the average D’ before treatment. The black portions represent the change in D’ from the treatment. If they are above the gray, the D’ value increased with treatment. If they are below the gray, the D’ decreased. The column labeled ‘Same Patients’ is the dataset containing the exact same group of patients, both before and after treatment. difference between <A, S> and <S, S> 0.066 from 0.204 to 0.138. These results, and the typical trends these re- sults show, can be seen in Figure 4. We also analyzed <A, A> mutation pairs according to their codon positions, rather than physical distance. This analysis can be seen in Figure 4. The top plot shows a control analysis of generally treated subtype B protease. In this plot, we show the thirty positions which were most significantly affected by the treatment, and what their total D’ value was prior to and after treatment. This plot shows that, in almost all significantly affected positions, there was an increase in <A, A> covariation. In addition, we can see that several of the most affected positions are also medically signifi- cant, according the Stanford HIV database. The second plot shows a comparison between the IDV and NFV treated datasets. Again, we can see that the two treatments cause different patterns in the covariation pattern. Certain codon positions have roughly the same amount of covariation after treatment, but others, in- cluding several medically significant positions, seen in the bottom plot, have significantly different covariation values, such as positions 20, 46, and 82. 3.2. D’ Results from Treated/Untreated Datasets of Different Subtypes For this portion of the study, we did not distinguish based on treatment type, but rather only invested in dis- tinguishing between ‘treated’ and ‘untreated’ sequences. Selecting a specific treatment type limited the larger treated datasets into subsets too small for proper analysis.  D. King et al. / J. Biomedical Science and Engineering 3 (2010) 291-299 Copyright © 2010 SciRes 295 Figure 4. Site-specific Analysis for B-subtype Protease. The top plot shows the thirty postitions who’s covariation was most affected by the application of generic treatment ot B-subtype protease. The positions with * or ^ next to them are the major or minor positions respectively that are associated with drug resistance according to the Stanford HIV Database. The bottom plot contrasts the difference between the IDV- and NFV-treated datasets on medically significant sites. 3.3. Pair-Wise Mutation Analysis and Clustering For use in the analysis of protease and RT, we selected HIV-1 subtypes A, B, and C, as well as recombinant subtypes AE, and AG. Subtype AG was only analyzed for protease, and subtype A was only analyzed for RT, due to lack of data. Results of the pair-wise analysis revealed that there is a clear-and-distinct difference between the position-based covariance of <A, A> mutation pairs, <A, S> mutation pairs, and <S, S> mutation pairs. With the treatment-specific datasets, we analyzed all datasets, and generated sliding window curves for all of them, mapping the relationship of D’ values of mutation pairs and their physical distances. There tended to be far greater <A, A> covariance at certain positions than <A, S> or <S, S> covariance in general. Additionally, these peaks of high <A, A> covari- ance tended to be close to one another, creating clusters or areas of high <A, A> covariance within the genome. By contrast, <A, S> covariation was less clustered, and <S, S> not clustered at all. This can be seen in Figure 6. Our results showed that different subtypes yield dif- ferent patterns of covariation, and that once again the typical trends were maintained on average. There was a clear seperation of <A, A>, <A, S> and <S, S> covara- tion, both before and after treatment, although treatment in most cases improved the separation. There was one exception to this: subtypes A and C RT displayed a sig- nificant increase in<A, A> covariation, but similar in- creases in <A, S> and <S, S> covariation lead to them having less-separated curves after treatment. There was also increase in <A, A> covariation after treatment in all datasets. While this increase in <A, A> covariation is consistent for all datasets, we did notice that subtype-B protease and RT had a consid- erably larger increase in covariation than any other subtype. Figure 5 shows a summary of the findings in this section. Figure 5. Before and after treatment for different subtypes. This chart shows the effects of treatment on different subtypes of protease and RT. For the most part, data followed expected pat- terns. Subtype A RT does not have a clear distinction between <A, A>, <A, S> and <S, S>, but beyond that, plots behave normally. JBiSE  296 D. King et al. / J. Biomedical Science and Engineering 3 (2010) 291-299 Copyright © 2010 SciRes JBiSE Figure 6. Treated/Untreated covariation comparison for protease. These three plots show the thirty positions whose covariation was most effected by treatment. These positions were selected because they had the largest difference in their total D’ values between treated and untreated. The <A, A> mutation show that frequently D’ values were higher after treatment, trend that was not as clear in <A, S> and <S, S> plots. D’ values for <A, A> covariation are higher than those of <A, S> covariation, and much higher than those of <S, S> covariation. <S, S> covariation seems not to have been effected by treatment very much: the highest difference between before and after was less than 6.5. Casting these histograms into a 2D contour plot re- vealed further information about the relationships be- tween specific positions: we can see that covariation between positions is clearly related to the amount of covaration at a specific position. Two positions having high D’ values will very likely have a high correlation. Both the histogram and the contour mapping of generi- cally treated protease are shown in Figure 7. Based on the position-covariation histogram of gener- ally-treated subtype B protease as in Figure 4, we  D. King et al. / J. Biomedical Science and Engineering 3 (2010) 291-299 Copyright © 2010 SciRes 297 JBiSE Figure 7. Treated B Protease. The top plot is a position relationship chart, with bright colors showing positions which are highly correlated with one another, and dark colors showing positions which are not. The shade of the grid at the left is representative of total D’, the sum of the covariance values for all mutation pairs at that position. The bottom plot is a histogram of D’ values for gen- erally-treated B-subtype protease. Each column in the histogram is the sum of all values for a particular position in the 2D chart. These charts were generated from the statistically significant mutation pairs with a Fisher Test P value less than 0.05 and a ChiSQ Test P value less than 0.05. selected the 20 most correlated statistically significant <A, A> positions according to D’ value, which are: 10+^, 13+, 20+^, 33+^, 34, 36+^, 46+*, 48+*, 54+*, 62+, 63+^, 67, 71+^, 72, 73+^, 82+*, 84+*, 89, 90+*, and 95, with + positions having also been found in [1] using the θ value, and positions with * or ^ being sites of major or minor drug resistance respectively according to the Stanford HIV database. The D’ analysis has an advan- tage of being able to find negative correlation effectively. We also found the negatively correlated positions: 3, 30+, 64, 88+, 96. We find clusters of covariation to occur near positions 10, 20, 37, 50, 73, and 90. We also found top statistically significant correlated mutation pairs in our Treated Protease dataset. In order to do this, we sorted all <A, A> pairs according to the Fisher Test P value followed by the ChiSQ Test P value, 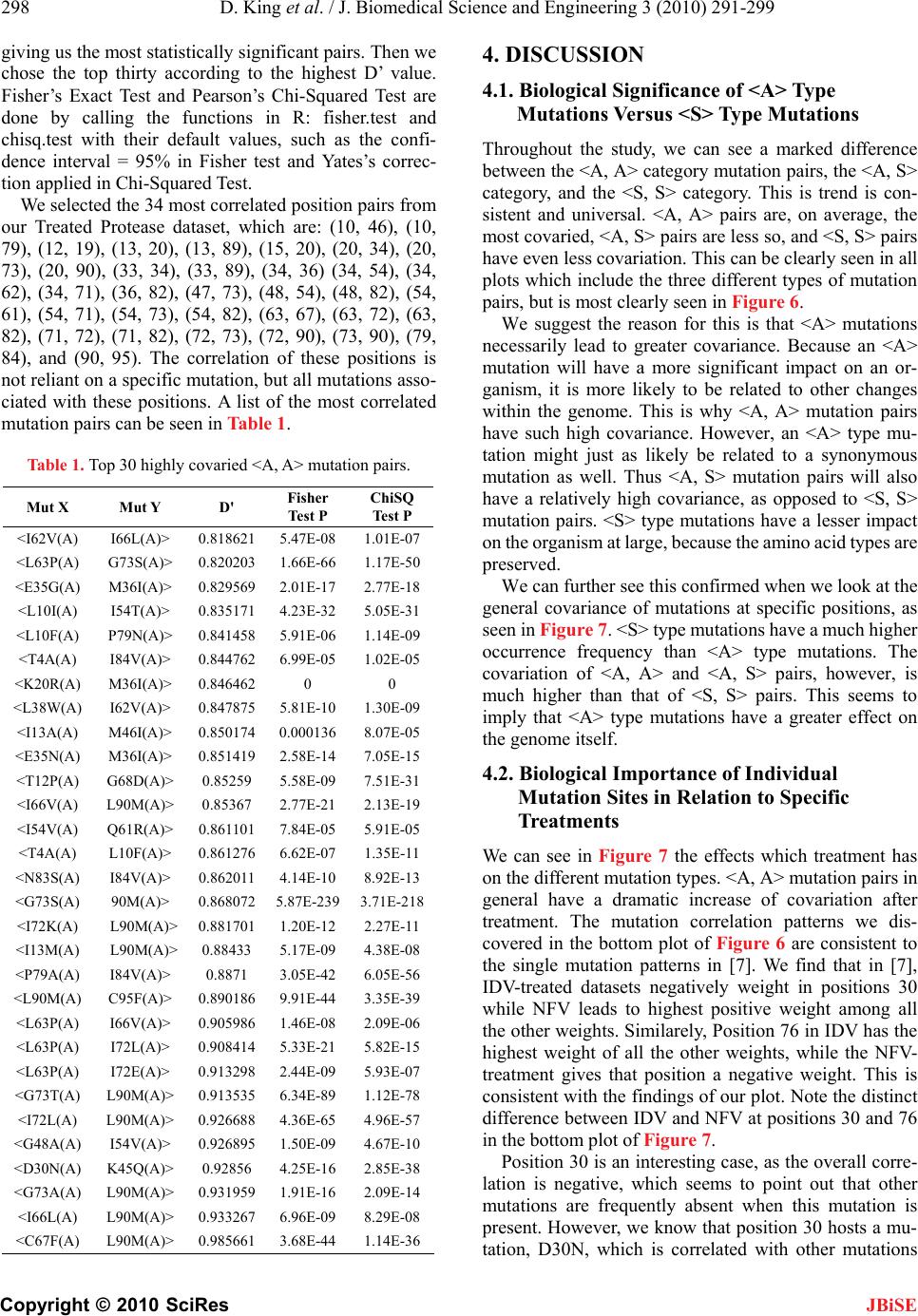 298 D. King et al. / J. Biomedical Science and Engineering 3 (2010) 291-299 Copyright © 2010 SciRes JBiSE giving us the most statistically significant pairs. Then we chose the top thirty according to the highest D’ value. Fisher’s Exact Test and Pearson’s Chi-Squared Test are done by calling the functions in R: fisher.test and chisq.test with their default values, such as the confi- dence interval = 95% in Fisher test and Yates’s correc- tion applied in Chi-Squared Test. We selected the 34 most correlated position pairs from our Treated Protease dataset, which are: (10, 46), (10, 79), (12, 19), (13, 20), (13, 89), (15, 20), (20, 34), (20, 73), (20, 90), (33, 34), (33, 89), (34, 36) (34, 54), (34, 62), (34, 71), (36, 82), (47, 73), (48, 54), (48, 82), (54, 61), (54, 71), (54, 73), (54, 82), (63, 67), (63, 72), (63, 82), (71, 72), (71, 82), (72, 73), (72, 90), (73, 90), (79, 84), and (90, 95). The correlation of these positions is not reliant on a specific mutation, but all mutations asso- ciated with these positions. A list of the most correlated mutation pairs can be seen in Table 1. Table 1. Top 30 highly covaried <A, A> mutation pairs. Mut X Mut Y D' Fisher Test P ChiSQ Test P <I62V(A) I66L(A)> 0.818621 5.47E-08 1.01E-07 <L63P(A) G73S(A)> 0.820203 1.66E-66 1.17E-50 <E35G(A) M36I(A)> 0.829569 2.01E-17 2.77E-18 <L10I(A) I54T(A)> 0.835171 4.23E-32 5.05E-31 <L10F(A) P79N(A)> 0.841458 5.91E-06 1.14E-09 <T4A(A) I84V(A)> 0.844762 6.99E-05 1.02E-05 <K20R(A) M36I(A)> 0.846462 0 0 <L38W(A) I62V(A)> 0.847875 5.81E-10 1.30E-09 <I13A(A) M46I(A)> 0.850174 0.000136 8.07E-05 <E35N(A) M36I(A)> 0.851419 2.58E-14 7.05E-15 <T12P(A) G68D(A)> 0.85259 5.58E-09 7.51E-31 <I66V(A) L90M(A)> 0.85367 2.77E-21 2.13E-19 <I54V(A) Q61R(A)> 0.861101 7.84E-05 5.91E-05 <T4A(A) L10F(A)> 0.861276 6.62E-07 1.35E-11 <N83S(A) I84V(A)> 0.862011 4.14E-10 8.92E-13 <G73S(A) 90M(A)> 0.868072 5.87E-239 3.71E-218 <I72K(A) L90M(A)> 0.881701 1.20E-12 2.27E-11 <I13M(A) L90M(A)> 0.88433 5.17E-09 4.38E-08 <P79A(A) I84V(A)> 0.8871 3.05E-42 6.05E-56 <L90M(A) C95F(A)> 0.890186 9.91E-44 3.35E-39 <L63P(A) I66V(A)> 0.905986 1.46E-08 2.09E-06 <L63P(A) I72L(A)> 0.908414 5.33E-21 5.82E-15 <L63P(A) I72E(A)> 0.913298 2.44E-09 5.93E-07 <G73T(A) L90M(A)> 0.913535 6.34E-89 1.12E-78 <I72L(A) L90M(A)> 0.926688 4.36E-65 4.96E-57 <G48A(A) I54V(A)> 0.926895 1.50E-09 4.67E-10 <D30N(A) K45Q(A)> 0.92856 4.25E-16 2.85E-38 <G73A(A) L90M(A)> 0.931959 1.91E-16 2.09E-14 <I66L(A) L90M(A)> 0.933267 6.96E-09 8.29E-08 <C67F(A) L90M(A)> 0.985661 3.68E-44 1.14E-36 4. DISCUSSION 4.1. Biological Significance of <A> Type Mutations Versus <S> Type Mutations Throughout the study, we can see a marked difference between the <A, A> category mutation pairs, the <A, S> category, and the <S, S> category. This is trend is con- sistent and universal. <A, A> pairs are, on average, the most covaried, <A, S> pairs are less so, and <S, S> pairs have even less covariation. This can be clearly seen in all plots which include the three different types of mutation pairs, but is most clearly seen in Figure 6. We suggest the reason for this is that <A> mutations necessarily lead to greater covariance. Because an <A> mutation will have a more significant impact on an or- ganism, it is more likely to be related to other changes within the genome. This is why <A, A> mutation pairs have such high covariance. However, an <A> type mu- tation might just as likely be related to a synonymous mutation as well. Thus <A, S> mutation pairs will also have a relatively high covariance, as opposed to <S, S> mutation pairs. <S> type mutations have a lesser impact on the organism at large, because the amino acid types are preserved. We can further see this confirmed when we look at the general covariance of mutations at specific positions, as seen in Figure 7. <S> type mutations have a much higher occurrence frequency than <A> type mutations. The covariation of <A, A> and <A, S> pairs, however, is much higher than that of <S, S> pairs. This seems to imply that <A> type mutations have a greater effect on the genome itself. 4.2. Biological Importance of Individual Mutation Sites in Relation to Specific Treatments We can see in Figure 7 the effects which treatment has on the different mutation types. <A, A> mutation pairs in general have a dramatic increase of covariation after treatment. The mutation correlation patterns we dis- covered in the bottom plot of Figure 6 are consistent to the single mutation patterns in [7]. We find that in [7], IDV-treated datasets negatively weight in positions 30 while NFV leads to highest positive weight among all the other weights. Similarely, Position 76 in IDV has the highest weight of all the other weights, while the NFV- treatment gives that position a negative weight. This is consistent with the findings of our plot. Note the distinct difference between IDV and NFV at positions 30 and 76 in the bottom plot of Figure 7. Position 30 is an interesting case, as the overall corre- lation is negative, which seems to point out that other mutations are frequently absent when this mutation is present. However, we know that position 30 hosts a mu- tation, D30N, which is correlated with other mutations  D. King et al. / J. Biomedical Science and Engineering 3 (2010) 291-299 Copyright © 2010 SciRes 299 when the specific PI treatment is neflinavir. This seems to hint that other treatment types have a steep inverse correlation at this mutation site. At the very least, we see that the treatment IDV gives a negative correlative weight at position 30 [7]. JBiSE 4.3. Differences in Covariation in Different Treatments and Subtypes While the general trends we found were largely consis- tent throughout our comparison between the different treatments and subtypes, we found the differences in the covariation patterns between the subtypes and treatments interesting. In Figure 4, we see that the increase in covariation between the untreated sequences and the the sequences which received any treatment whatsoever is far larger than the increase in covariation present in those se- quences only treated with individual drugs. For example, the two drugs, NFV and IDV, have the most data within the Stanford database. In spite of this, neither the co- variation increase from NFV or IDV alone is enough to cause the dramatic increase we see from generic treat- ment of B-subtype protease. The same is true of AZT- treated RT compared with generically treated RT. The generically treated datasets accounts for sequences treated with single drugs, such as NFV or IDV or AZT, as well as those treated with combinations of drugs. Our results, then, suggest that combinations of treatments lead to greater covariance than single treatments. This is further supported by the results of the RT treated with the combination of drugs, AZT, 3TC, and EFV, which have a greater increase in covariation than any single- treatment, but still not as much as the generically-treated sequences. 5. ACKNOWLEDGEMENTS We would like to thank Houghton College for providing the funding for this research through the Summer Research Institute at Houghton, as well as Qi Wang for responding to our e-mails regarding his paper [1] both promptly and helpfully. REFERENCES [1] Wang, Q. and Lee, C. (2007) Distinguishing functional amino acid covariation from background linkage disequi- librium in HIV protease and reverse transcriptase. PLoS ONE, 2(8), 814. [2] Liu, Y., Eyal, E. and Bahar, I. (2008) Analysis of corre- lated mutations in HIV-1 protease using spectral cluster- ing. Bioinformatics, 24, 1243-1250. [3] Gilbert, P.B., Novitsky, V. and Essex, M. (2005) Covari- ability of selected amino acid positions for HIV type 1 subtypes C and B. AIDS Research and Human Retrovi- ruses, 21(12), 1016-1030 [4] Hoffman, N.G., Schiffer, C.A. and Swanstrom, R. (2003) Covariation of amino acid positions in HIV-1 protease. Virology, 314, 536-548. [5] Rhee, S.Y., Liu, T.F., Holmes, S.P. and Shafer, R.W. (2007) HIV-1 subtype B protease and reverse transcrip- tase amino acid covariation. PLoS Computational Biol- ogy, 3(5), 87. [6] Hedrick, P. (1987) Gametic disequilibrium measure: proceed with caution. Genetics, 117, 331-341. [7] Rhee, S.Y., Taylor, J., Wadhera, G., Ben-Hur, A., Brutlag, D. and Shafer, R.W. (2006) Genotypic predictors of hu- man immunodeficiency virus type 1 drug resistance. Proceedings of the National Academy of Sciences USA. 103, 17355-17360.
|