Paper Menu >>
Journal Menu >>
 J. Software Engineering & Applications, 2008, 1: 76-82 Published Online December 2008 in SciRes (www.SciRP.org/journal/jsea) Copyright © 2008 SciRes JSEA An Algorithm for Generation of Attack Signatures Based on Sequences Alignment Nan Li, Chunhe Xia, Yi Yang, Hai-Quan Wang State Key Laboratory of Virtual Reality Technology, Key Laboratory of Beijing Network Technology, School of Computer Science and Engineering, Beihang University, Beijing, China Email:{linan,yangyi}@cse.buaa.edu.cn, {xch,whq}@buaa.edu.cn Received November 11 th ; 2008; revised November 19 th , 2008; accepted November 24 th , 2008 ABSTRACT This paper presents a new algorithm for generation of attack signatures based on sequence alignment. The algorithm is composed of two parts: a local alignment algorithm-GASBSLA (Generation of Attack Signatures Based on Sequence Local Alignment) and a multi-sequence alignment algorithm-TGMSA (Tri-stage Gradual Multi-Sequence Alignment). With the inspiration of sequence alignment used in Bioinformatics, GASBSLA replaces global alignment and constant weight penalty model by local alignment and affine penalty model to improve the generality of attack signatures. TGMSA presents a new pruning policy to make the algorithm more insensitive to noises in the generation of attack signatures. In this paper, GASBSLA and TGMSA are described in detail and validated by experiments. Keywords: Attack Signatures Generation, Sequence Local Alignment, Affine Penalty, Intrusion Detection, Pruning Policy 1. Introduction Network worms, viruses and malicious codes are still the top threat against the current Internet and enterprise security, and they cause a loss of hundreds of millions dollars every year [1].Intrusion detection based on attack signatures is the most effective solution of this issue currently, but the continuous emergence of new types of attacks and polymorphic engines such as PHolyP [2] are great challenges to the existing intrusion detection technologies. To solve this problem, automatic generation of attack signatures has been concerned by more and more researchers and has become a new hotspot in intrusion detection since 2003 [3]. Algorithms for generation of attack signatures can be divided into two categories: one is based on string mode and the other is based on semantics. However, the latter relies on prior semantic analysis of a certain type of attacks, so it is incompetent for generating signatures of unknown attacks automatically. Currently the research on algorithms for generation of attack signatures is mainly based on string mode, including the following categories: algorithms based on the LCS (longest common substring), algorithms based on the Token (the strings appearing frequently in suspicious datum and containing more than one character) [4], algorithms based on sequence alignment, algorithms based on finite automaton and algorithms based on protocol field and length [5]. The algorithms for generation of attack signatures based on Token is considered as the most effective and approbatory method currently. But in [3], the authors point out that signatures generated by this kind of algorithm are not precise and give out an algorithm based on sequence alignment. In this paper, we present a new algorithm for generation of attack signatures based on sequence alignment through analyzing the algorithms presented by [3] and referring to the idea of sequence alignment used in Bioinformatics. The algorithm is composed of two parts: GASBSLA algorithm and TGMSA algorithm. With the inspiration of sequence alignment used in Bioinformatics, GASBSLA replaces global alignment and constant weight penalty model by local alignment and affine penalty model to improve the generality of attack signatures. TGMSA presents a new pruning policy to make the algorithm more insensitive to noises in the generation of attack signatures. The rest of the paper is organized as follows. Section 2 refers to related research, which describes the algorithms for generating attack signatures in [3] and analyzes its weakness. Section 3 presents the design of GASBSLA algorithm and TGMSA algorithm, and details their relative analysis. Section 4 presents the experiments on the effectiveness and the anti-noise ability of the algorithms. Section 5 concludes the paper and mentions of some future work. This work was supported by three projects: the National 863 Project- Research on high level descripti on of network survivability model and its validation simulation platform under Grant No.2007 AA01Z407, The Co-Funding Project of Beijing Municipal E ducation Commission under Grant No.JD100060630 and National Foundation Research Project.  An Algorithm for Generation of Attack Signatures Based on Sequences Alignment 77 Copyright © 2008 SciRes JSEA 2. Related Research Sequence alignment is divided into pair-wise alignment and multi-sequence alignment, and most of multi- sequence alignment is based on pair-wise alignment. Firstly, this section introduces and analyzes a pair-wise sequence alignment algorithm CMENW (Contiguous- Matches Encouraging Needleman-Wunsch) and a multi-sequence alignment algorithm HMSA (Hierarchical Multi-Sequence Alignment) [3]. They are the most representative algorithms applied to the generation of attack signatures based on sequence alignment, and they are also the foundation of this paper. Then we introduce the most representative pair-wise local alignment algorithm-Smith-Waterman algorithm [6]. 2.1 CMENW Algorithm CMENW algorithm is a pair-wise alignment algorithm based on global alignment. It is improved on Needleman- Wunsch algorithm [7], which is the typical pair-wise alignment algorithm. The main difference between the two algorithms is: Needleman-Wunsch algorithm easily leads to fragments. In order to reduce the influence of fragments in the process of alignment, CMENW algorithm introduces contiguous-matches encouraging function ( ) enc x ( x is the number of contiguous bytes in the alignment), which is used to encourage contiguous bytes to be aligned together. The score function of CMENW algorithm is as follows: 12 3 ( ,)() S x ykmkdkencs δ δδ δ =×+× +× + =×+× +× +=×+× +× + =×+×+×+∑ ∑∑ ∑ (1) m is the score of bytes matched, d is the score of bytes unmatched, δ δδ δ is the score of empty penalty, 1 k is the number of bytes matched in the result of alignment, 2 k is the number of bytes unmatched, 3 k is the number of gaps, s is contiguous bytes. Attack signatures generated by CMENW algorithm are not effective enough while facing to polymorphic attack because of the insufficient generality. It can be improved by using multi-sequence alignment, but the number of samples is difficult to meet the requirement in real world situation. 2.2 HMSA Algorithm HMSA algorithm is a type of hierarchical multi-sequence alignment algorithm based on pair-wise alignment CMENW algorithm, which is suitable for attack signatures generation. This algorithm has three main features [3]: (1) hierarchical pair-wise alignment; (2) supporting wildcard characters; (3) with a pruning function. HMSA algorithm possesses the function of pruning, which accelerates its convergence and enhances the noise resisting ability. However, the effectiveness of pruning function is based on two assumptions: (1) the alignment result of any two noise will be pruned because of trivial solution; (2) the alignment result of any two samples will not be pruned and get a precise attack signature. However, in reality, it is possible that the alignment result of any two noises is not pruned, because input sequences of signatures generation algorithm are often processed by clustering algorithms. Thus the alignment results of noise that not pruned and the alignment results of sample will be easily prone to trivial solution and be pruned, and finally there is no result returned. 2.3 Smith-Waterman Algorithm Smith-Waterman algorithm is a pair-wise local alignment algorithm put forward by Smith and Waterman in 1981, which is used to find and compare the similarity in local regions in an overall view. Even today it is still a common basic algorithm in bioinformatics. Given sequence x and y as inputs, Smith-Waterman algorithm outputs a local alignment result which is global optimal. The similarity value of it is maximal according to formula (2). And the meanings of the bytes in this formula are the same as those in the formula (1) in Section 2.1. 12 3 ( ,) S x ykmkdk δ δδ δ =×+× +× =×+× +×=×+× +× =×+× +× (2) Smith-Waterman algorithm is used to find the biggest similarity value and the best alignment based on the principle of dynamic programming, and its process includes two major steps: 1. Calculate the similarity values of two given sequences, and get a similarity matrix; 2. Get the best results of sequence alignment through dynamic programming and backtracking algorithm, according to the similarity matrix got in step 1. Smith-Waterman algorithm improves Needleman- Wunsch algorithm. The main difference between them is: Smith-Waterman algorithm uses 0 to replace all the negatives in the similarity matrix; if the similarity values no longer increases when the length of alignment result increases, this algorithm will finish backtracking and output the result. According to the differences between the two algorithms, the idea of Smith-Waterman algorithm is helpful for CMENW algorithms to overcome the problem of insufficient generalization. 3. GASBSLA Algorithm and TGMSA Algorithm Through the analysis of CMENW algorithm and HMSA algorithm, we present a new algorithm for generation of attack signatures based on sequence alignment. The algorithm is composed of two parts: a local alignment algorithm—GASBSLA (Generation of Attack Signatures Based on Sequence Local Alignment) and a multi- sequence alignment algorithm—TGMSA (Tri-stage Gradual Multi-Sequence Alignment). 3.1 GASBSLA Algorithm In Bioinformatics, local alignment has more practical significance than global alignment because two sequences are often with very high similarity just in some local regions [8]. For example, two long DNA sequences often  78 An Algorithm for Generation of Attack Signatures Based on Sequences Alignment Copyright © 2008 SciRes JSEA have relation with each other only in seldom areas (password districts); proteins belonging to different families often have some regions in the same on the structure and function. The situation in generating of attack signatures is very similar with that of Bioinformatics, so GASBSLA algorithm replaces global alignment by local alignment to improve the generality and precision of attack signature under the conditions of a small sample. In addition, to further reduce the number of fragments, GASBSLA algorithm replaces constant weight penalty model by affine penalty model [9]. The differences between affine penalty model and constant weight penalty model are: the penalty for each gap is independent in constant weight penalty model. That is, in any case, the penalty for one gap is d , and the penalty for n gaps is nd ; but in affine penalty model, the penalty for n gaps which attached together is 1 ( ) q nr +− × +− ×+− × +− × . Where q is the penalty for the first one of n gaps attached together, r is the penalty for the other gaps, and r « «« « q. We can learn from descriptions above that in affine penalty model, the penalty for the first gap is more than the other ones which means the reduction of single gaps and fragments in the attack signatures. The general idea of GASBSLA algorithm based on Dynamic Programming is: First, calculating the similarity values of two sequences and keeping them in a matrix (named similarity matrix or DP matrix); second, according to the dynamic programming backtracking algorithm, finding the optimal alignment sequence on the basis of the DP matrix. Both the time complexity and the space complexity of GASBSLA algorithm are ( ) O mn , where m and n are the lengths of the two sequences. ( ,) x y σ σσ σ is the similarity value of the alignment of x and y , where x and y are any two characters. Algorithm 1. GASBSLA algorithm Input: sequence a and b Output: the similarity value and optimal sequence alignment of a and b Initialization: a. 0 00 ( ,) T = == = b. 1 2 , ,, L ForeachiM = == = 0 0 ( ,) F i = == = , 0 0 ( ,)T i = == = c. 1 2 , ,, L For eachjN = == = 0 0 ( ,)F j = == = , 0 0 ( ,)T j = == = Main iteration: 1 2 , ,, L ForeachiM = == = 1 2 , ,, L For eachjN = == = 1 1 0 ( ,), ( ,), T ijifab i j T ij ifa b i j − −= − −=− −= − −= = == = ≠ ≠≠ ≠ 0 1 1 1 2 1 3 1 4 ( ,)max [ ] (,)(,)(( ,)) [ ] (, )(,) [ ] ( ,)(,) [ ] i j i j F ij case Fijabenc Tijcase F ijacase F ijbcase σ σσ σ σ σσ σ σ σσ σ = == = −− ++ −− ++−− ++ −− ++ − +− − +−− +− − +− − +− − +−− +− − +− 1 2 3 4 [ ] [ ] ( ,) [ ] [ ] STOP case DIAG case Ptr ijLEFT case UP case = == = 3.2 TGMSA Algorithm TGMSA algorithm presents a new pruning policy to avoid the situation of no output caused by not being pruned in the alignment process of two noises. The general idea is modifying pruning policy in the nth (n>1) layer alignment according to alignment similarity value. If the alignment similarity value is less than the threshold (that the alignment similarity value is out of confidence interval), the alignment result will not be pruned, but the two sequences will be laid aside then align respectively with the signature sequence result, which is the alignment result of other sequences. If the alignment result does not accord with pruning conditions, it will replace the original signature sequence, otherwise it will be deserted. Algorithm 2. TGMSA algorithm Input: sequence set S Output: multi-sequence alignment result Initialization: R S ← ←← ← {} W ← ←← ← {} T ← ←← ← Iteration of the first stage: while 1 R ≥ ≥≥ ≥ , do if 1 R = == = (denote the sequence by i s ) then i s W → →→ → else take out two sequences i s and j s orderly from R align i s with j s using pair-wise alignment algorithm, the alignment result is denoted by , i j S S Ali (including the similarity value and optimal sequence alignment) pruning if the number of fragments in , i j S S Ali ≥ 3 and there exists at least two fragments whose length ≥ 3 , i j S S Ali T → →→ → Iteration of the second stage: do {} V ← ←← ← while 1 T ≥ ≥≥ ≥ , do  An Algorithm for Generation of Attack Signatures Based on Sequences Alignment 79 Copyright © 2008 SciRes JSEA if 1 T = == = (denote the sequence by i s ) then i s V → →→ → else take out two sequences i s and j s randomly from R align i s with j s using pair-wise alignment algorithm, the alignment result is denoted by , i j S S Ali pruning if the similarity value of , i j S S Ali falls in confidence interval(the calculation of similarity value confidence interval will be specified in Section 3.3.) , i j S S Ali V → →→ → else , i j S S Ali W → →→ → T V ← ←← ← until 1 V ≤ ≤≤ ≤ Iteration of the third stage: if 1 V = == = while 0 W ≠ ≠≠ ≠ take out single sequence from W orderly, then align it with the alignment result Ali in the second stage respectively to generate a new alignment result ' Ali if the number of fragments in ' Ali ≥ 3[10] and there exists at least two fragments whose length ≥ 3 [11,12] then Ali = ' Ali else Ali = Φ = Φ= Φ = Φ 3.3 The Selection of Alignment Similarity Confidence Interval Central limit theorem holds that regardless of the statistics population on the subject obeying whatever distribution, the distribution of sample mean is close to a normal distribution, the mean of normal distribution equals that of population distribution, and the variance equals that of population distribution divided by the Sample size. Therefore, we can estimate the average signature alignment similarity based on a certain attack by the average of the similarity value samples. We use all the alignment similarity values calculated in the first stage as a sample to calculate the similarity value confidence interval which is the judgement condition of pruning in the second stage. Assume 1 2 ( ,,,) LL n F FF is a sample of the alignment similarity value population F , so the sample mean and sample standard variance are as follows: 1 1 1 1 i n n F i F n + ++ + + ++ + ∑ ∑∑ ∑ = == = = == = + ++ + (3) ( (( ( ) )) ) ( (( ( ) )) ) 1 1 12 1 1 1 2 121 1 n i i n n SF F ni nFn F ni + ++ + + ++ + + ++ + = − = −= − = − ∑ ∑∑ ∑ = == = + ++ + =− + =− +=− + =− + ∑ ∑∑ ∑ = == = (4) According to the small probability event theory of normal distribution: the most datum of normal population (99.7%) falls in the range of 3 µ σ ± , and those cases out of the range are called small probability events. Statistics holds that small probability events occur almost impossibly, and they can be ignored. The confidence interval of alignment similarity value is as follows: 1 1 1 1 3 3 1 1 , n n n n S S F F n n + + + ++ + + + + + + ++ + + + − + − +− + − + + + + ++ + + + (5) That is: ( (( ( ) )) ) ( (( ( ) )) ) ( (( ( ) )) ) ( (( ( ) )) ) 1 1 1 1 1 1 2 2 2 2 1 1 1 1 1 1 3 3 1 1 1 1 , i ni n i i n n n n Fn FFn F F F i i i i n n n nn n + + + ++ + + + + + + ++ + + + + + + ++ + + + − +− + − +− +− +− + − +− + ∑ ∑ ∑ ∑∑ ∑ ∑ ∑ ∑ ∑ ∑ ∑∑ ∑ ∑ ∑ = = = == = = = = = = == = = = − + − +− + − + + + + ++ + + + + + + ++ + + + (6) 4. Experimental Results In this section we verify the effectiveness and the noise resisting ability by practical results. In our experiments, CMENW algorithm and HMSA algorithm are implemented to verify pertinently the effectiveness of improvement gave out in GASBSLA algorithm and TGMSA algorithm. 4.1 Experiments Environment Hardware environment: Dawning Server (Intel® Xeon® CPU, 4G internal storage); Software environment: Linux Red Hat 9.0 Operating System(the version of kernel is 2.4.20-8). 4.2 Algorithm Validity Verification For the purpose of comparison, we selected the same experimental method as [3]. We generate signatures for polymorphic versions of four real-world exploits: Apache-Knacker [13], CodeRed Ⅱ [14], IISPrinter [15] and TSIG [16]. The Apache-Knacker exploit, the CodeRed Ⅱ exploit and the IISPrinter exploit use the text-based HTTP protocol. The TSIG exploit uses the binary-based DNS protocol. We use polymorphic engine to generate 150 samples for each exploit attack include 50 samples used to generate signatures and 100 samples used to detect false negatives. In order to simulate an ideal polymorphic engine, we fill wildcard and code bytes for each exploit with values chosen uniformly at random. In addition, we select 10,000 data samples without attacks from the MIT Lincoln Laboratory intrusion detection system test set — DARPA99 (the third week data sets) [17] to detect False positives. In our experiments, we set the matching score 1 m = == = , the mismatching score 0 2 . d = − = −= − = − , the penalty for the first gap 1 δ δδ δ = − = −= − = − , the penalty for the other gaps 0 05 ' . δ δδ δ = − = −= − = − . The Contiguous-Matches encouragement function is set as: 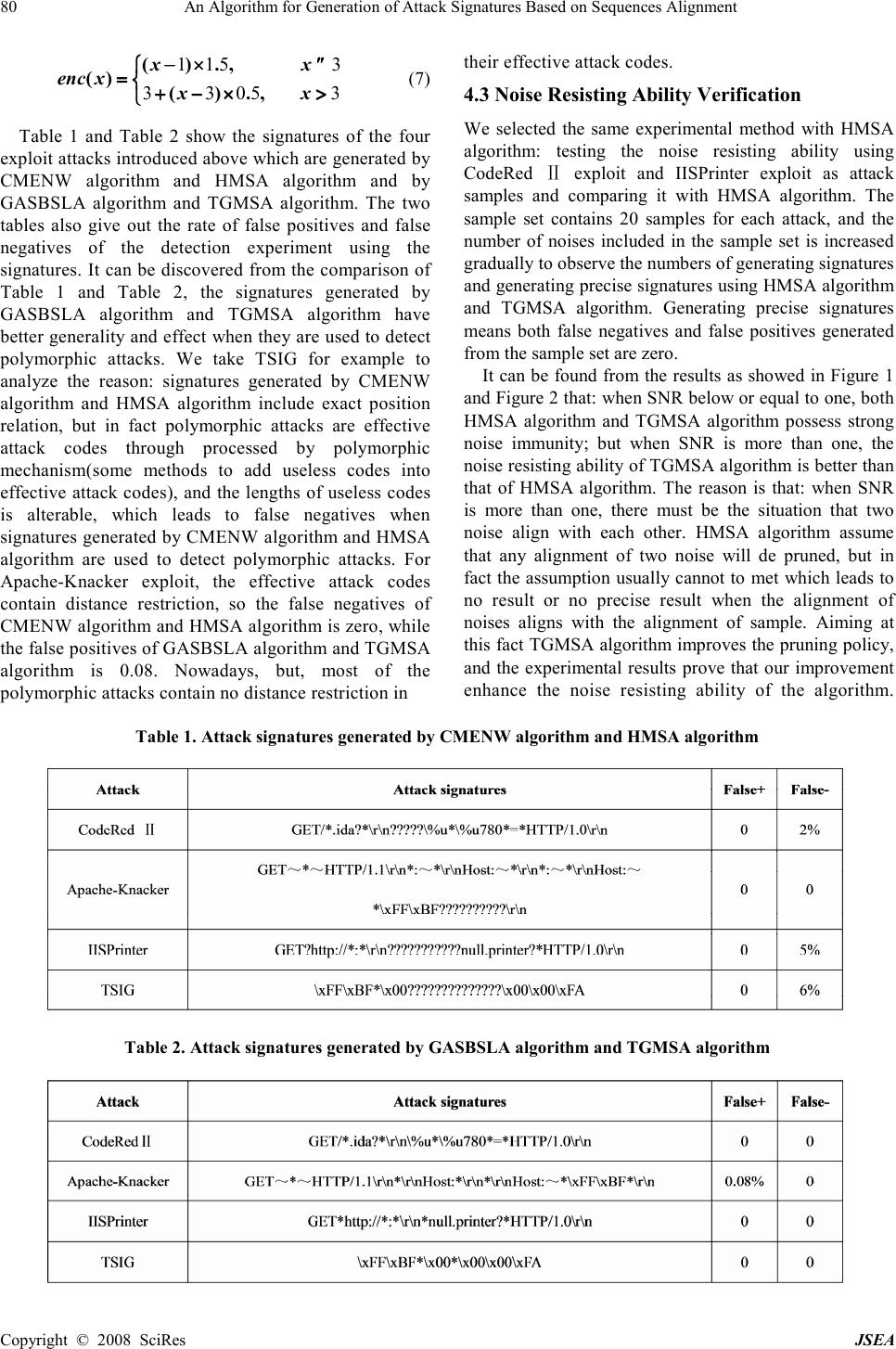 80 An Algorithm for Generation of Attack Signatures Based on Sequences Alignment Copyright © 2008 SciRes JSEA 1153 33053 (). , ( )(). , x x enc xxx − ×≤ − ×≤− ×≤ − ×≤ = == = +−×> +−×>+−×> +−×> (7) Table 1 and Table 2 show the signatures of the four exploit attacks introduced above which are generated by CMENW algorithm and HMSA algorithm and by GASBSLA algorithm and TGMSA algorithm. The two tables also give out the rate of false positives and false negatives of the detection experiment using the signatures. It can be discovered from the comparison of Table 1 and Table 2, the signatures generated by GASBSLA algorithm and TGMSA algorithm have better generality and effect when they are used to detect polymorphic attacks. We take TSIG for example to analyze the reason: signatures generated by CMENW algorithm and HMSA algorithm include exact position relation, but in fact polymorphic attacks are effective attack codes through processed by polymorphic mechanism(some methods to add useless codes into effective attack codes), and the lengths of useless codes is alterable, which leads to false negatives when signatures generated by CMENW algorithm and HMSA algorithm are used to detect polymorphic attacks. For Apache-Knacker exploit, the effective attack codes contain distance restriction, so the false negatives of CMENW algorithm and HMSA algorithm is zero, while the false positives of GASBSLA algorithm and TGMSA algorithm is 0.08. Nowadays, but, most of the polymorphic attacks contain no distance restriction in their effective attack codes. 4.3 Noise Resisting Ability Verification We selected the same experimental method with HMSA algorithm: testing the noise resisting ability using CodeRed exploit and IISPrinter exploit as attack Ⅱ samples and comparing it with HMSA algorithm. The sample set contains 20 samples for each attack, and the number of noises included in the sample set is increased gradually to observe the numbers of generating signatures and generating precise signatures using HMSA algorithm and TGMSA algorithm. Generating precise signatures means both false negatives and false positives generated from the sample set are zero. It can be found from the results as showed in Figure 1 and Figure 2 that: when SNR below or equal to one, both HMSA algorithm and TGMSA algorithm possess strong noise immunity; but when SNR is more than one, the noise resisting ability of TGMSA algorithm is better than that of HMSA algorithm. The reason is that: when SNR is more than one, there must be the situation that two noise align with each other. HMSA algorithm assume that any alignment of two noise will de pruned, but in fact the assumption usually cannot to met which leads to no result or no precise result when the alignment of noises aligns with the alignment of sample. Aiming at this fact TGMSA algorithm improves the pruning policy, and the experimental results prove that our improvement enhance the noise resisting ability of the algorithm. Table 1. Attack signatures generated by CMENW algorithm and HMSA algorithm Table 2. Attack signatures generated by GASBSLA algorithm and TGMSA algorithm 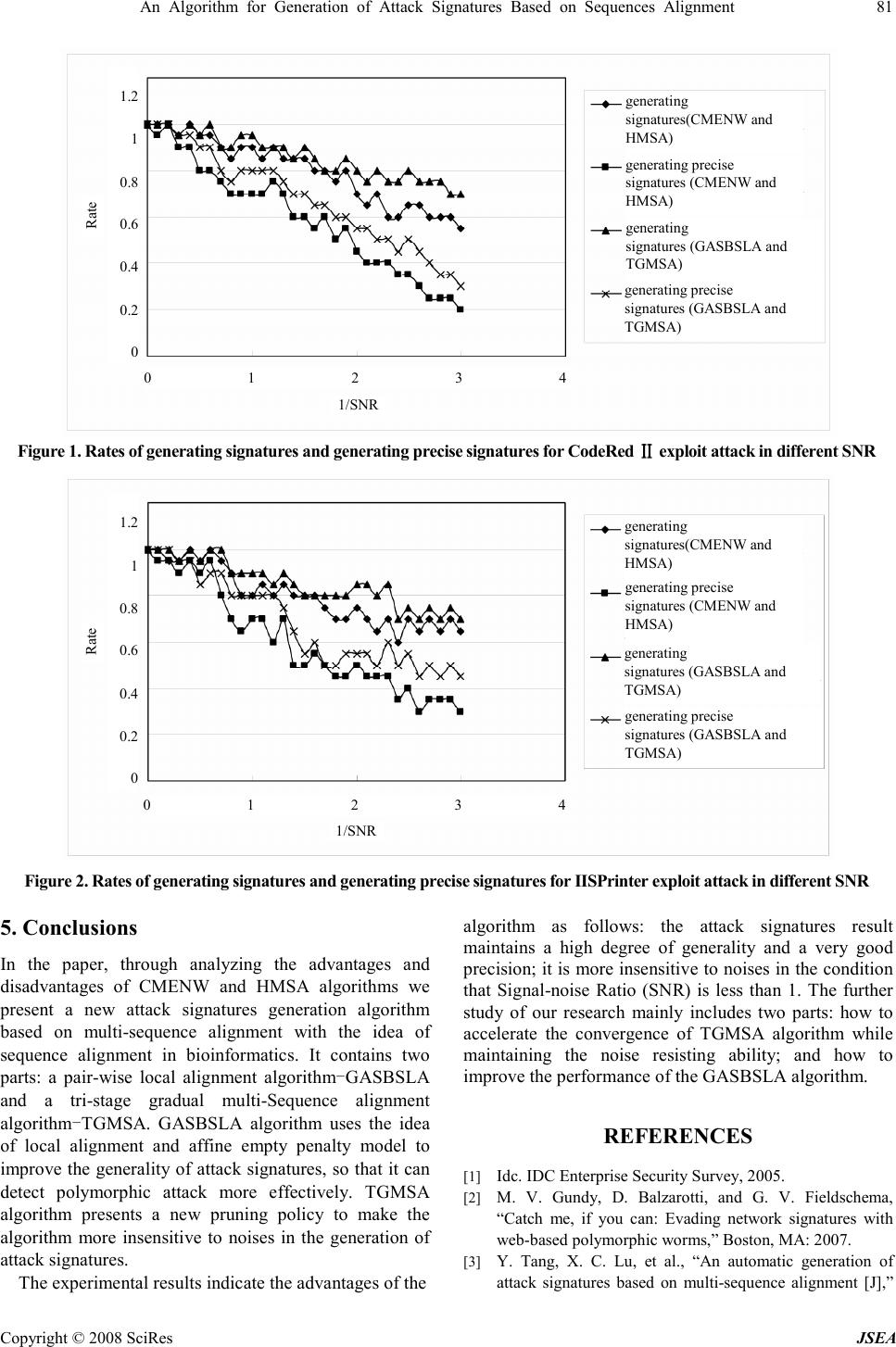 An Algorithm for Generation of Attack Signatures Based on Sequences Alignment 81 Copyright © 2008 SciRes JSEA Figure 1. Rates of generating signatures and generating precise signatures for CodeRed exploit attack in different SNRⅡ ⅡⅡ Ⅱ Figure 2. Rates of generating signatures and generating precise signatures for IISPrinter exploit attack in different SNR 5. Conclusions In the paper, through analyzing the advantages and disadvantages of CMENW and HMSA algorithms we present a new attack signatures generation algorithm based on multi-sequence alignment with the idea of sequence alignment in bioinformatics. It contains two parts: a pair-wise local alignment algorithm - GASBSLA and a tri-stage gradual multi-Sequence alignment algorithm - TGMSA. GASBSLA algorithm uses the idea of local alignment and affine empty penalty model to improve the generality of attack signatures, so that it can detect polymorphic attack more effectively. TGMSA algorithm presents a new pruning policy to make the algorithm more insensitive to noises in the generation of attack signatures. The experimental results indicate the advantages of the algorithm as follows: the attack signatures result maintains a high degree of generality and a very good precision; it is more insensitive to noises in the condition that Signal-noise Ratio (SNR) is less than 1. The further study of our research mainly includes two parts: how to accelerate the convergence of TGMSA algorithm while maintaining the noise resisting ability; and how to improve the performance of the GASBSLA algorithm. REFERENCES [1] Idc. IDC Enterprise Security Survey, 2005. [2] M. V. Gundy, D. Balzarotti, and G. V. Fieldschema, “Catch me, if you can: Evading network signatures with web-based polymorphic worms,” Boston, MA: 2007. [3] Y. Tang, X. C. Lu, et al., “An automatic generation of attack signatures based on multi-sequence alignment [J],” 1.2 1 0.8 0.6 0.4 0.2 0 Rate 0 1 2 3 4 1/SNR generating signatures(CMENW and HMSA) generating precise signatures (CMENW and HMSA) generating signatures (GASBSLA and TGMSA) generating precise signatures (GASBSLA and TGMSA) 1/SNR 0 1 2 3 4 1.2 1 0.8 0.6 0.4 0.2 0 Rate generating signatures(CMENW and HMSA) generating precise signatures (CMENW and HMSA) generating signatures (GASBSLA and TGMSA) generating precise signatures (GASBSLA and TGMSA)  82 An Algorithm for Generation of Attack Signatures Based on Sequences Alignment Copyright © 2008 SciRes JSEA Chinese Journal of Computers, 2006, 29 (9): 153321541. [4] J. Newsome, B. Karp, and D. Song, “Polygraph: Automatically generating signatures for polymorphic worms,” in: Proceedings of the IEEE S &P 2005, Oakland, California, pp. 226-241, 2005. [5] Z. Li, M. Sanghi, Y. Chen, et al., “Network-based and attack-resilient length signature generation for zero-day polymorphic worms[C],” 2007. [6] T. Smith and M. Waterman, “Identification of common molecular subsequences,” Journal of Molecular Biology, 147 (1): pp. 195-197, 1981. [7] S. B. Needleman and C. D. Wunsch, “A general method applicable to the search for similarities in the amino acid sequence of two proteins,” Journal of Molecular Biology, 48(3): pp. 443-453, 1970. [8] P. K. Murphy, “Biological sequence comparison: An overview of techniques,” Technical Report, University of Arizona, Department of Computer Science, 1994. [9] S. Uliel, A. Fliess, A. Amir, and R. Unger., “A simple algorithm for detecting circular permutations in proteins,” Bioinformatics, Vol. 15, No. 11: pp. 930-936, 1999. [10] J . R. Crandall, S. F. Wu, and F. T. Chong, “Experiences using Minos as a tool for capturing and analyzing novel worms for unknown vulnerabilities,” in: Proceedings of the GI SIG SIDAR Conference on Detection of Intrusions and Malware and Vulnerability Assessment, Vienna, pp. 32-50, 2005. [11] J. R. Crandall, Su Zhen Dong, S. F. Wu, and F. T. Chong, “On deriving unknown vulnerabilities from Zero Day polymorphic and metamorphic worm exploits,” in: Proceedings of the ACM CCS 2005, Alexandria, Virginia, pp. 235-248, 2005. [12] J. Xu, P. Ning, C. Kil, Y. Zhai, and C. Bookholt, “Automatic diagnosis and response to memory corruption vulnerabilities,” in: Proceedings of the ACM CCS 2005, Alexandria, Virginia, pp. 223-234, 2005. [13] Symantec Security Response: CodeRed Worm. http://www. sarc.com/avcenter/venc/data/codered.worm.html. [14] C. CAN-2003-0245. Apache apr-psprintf memory corruption vulnerability. http://www.securityfocus.com/bi-d/7723/ discussion/. [15] Viruslist.com: Net-Worm. Linux. Adm. http://www.viruslist.com/en/viruses/encyclopedia?virusid =23854. [16] SANS Institute: Lion worm. http://www.sans.o-rg/y2k/ lion.htm. [17] R. P. Lippmann, D. J. Fried, I. Graf, et al., “Evaluating intrusion detection systems: The 1998 DARPA offline intrusion detection evaluation,” in: Proceedings of the 2000 DARPA Information Survivability Conference and Exposition, Hilton Head, SC, 2: pp. 1012-1035, 2000. |