Journal of Cancer Therapy
Vol.4 No.1(2013), Article ID:27907,9 pages DOI:10.4236/jct.2013.41030
Cell Cycle Arrest Mediates Global DNA Methylation Patterns in Normal Human Keratinocytes, Epidermoid Carcinoma Cells and Murine Embryonic Fibroblasts
![]()
1Bioderm Technologies Inc., Trenton, USA; 2Division of Cancer Prevention and Control, Moffitt Cancer Center, Tampa, USA.
Email: *jjwille@aol.com
Received December 7th, 2012; revised January 9th, 2013; accepted January 17th, 2013
Keywords: Anoikis; Epitheliod Carcinoma Cells Cell Cycle; DNA Methylation; G1 Phase; Restriction Points
ABSTRACT
The 5-methylationcytosine (5-MC) DNA content of murine embryonic fibroblasts arrested in G1 by four growth conditions (Gc, Gn, Gd, and Gs) were hypermethylated relative to rapidly growing (RG) fibroblasts. Normal human keratinocytes (NHK) arrested in G1 by suspension were hypermethylated relative to RG cultures. Four RG cultures of epidermoid carcinoma cells (ECC) were hypomethylated relative to RG NHK cultures, and two cultures (SCC25 and A431) were further hypomethylated by SUS-induced arrest. Linear regression analyses established a positive linear correlation between growth rate and 5-MC content for three murine fibroblasts lines, and a negative correlation for both NHK and ECC lines.
1. Introduction
DNA methylation is an epigenetic modification of mammalian cell DNA that plays a major role in health and disease [1,2]. It occurs predominantly at carbon 5’-position of CpG dinucleotides by the action of one of two enzymatic activities, a maintenance and a de novo DNA methyltransferase [3]. Alterations of DNA methylation in cancer development has been extensively reviewed [4-12] with examples of more than 40 different hypermethylated genes found in human tumor cell genomes. Promoter methylation is an important mechanism of inactivating tumor suppressor gene, accomplished by hypermethylation of CpG islands of promoter regions. Conversely, when methyl groups are removed from a cluster of methylated CpGs associated with gene promoters, transcription of that gene are reactivated. Many proteins interact with DNMT1 maintenance methyltransferase including PCNA, p53, EZH2, HP1 and the RAS family of GTPases that promote cell proliferation by their oncogenic nature [12]. A second alteration of DNA methyllation associated with carcinogenesis is global hypomethylation, the loss of methyl groups attached to the DNA of highly repeated sequences in the cell genome [13-15]. This loss results is associated with chromosomal instability and loss of embryonic imprinting [16-20]. Hypomethylation of specific DNA repeat elements, satellites and retroelements associated with disease-specific cancers were reported [21] that may be used to predict disease onset or progression. A list of gene specific demethylation related to seven different cancer types including breast, colon, lung, ovarian, pancreatic, prostate and testicular has been reported [22]. A complete map of the human methylome is now available for two human cell types, a fibroblast and a pluripotent stem cell, providing insight into the epigenetic mechanisms underlying the interface between a changing environment and the stable genome [23,24].
Temporal and regional changes in DNA methylation occur during early embryonic development [25]. Gametogenesis is associated with a profound genomic demethylation followed by global de novo methylation of CpG island DNA sequences. A massive demethylation occurs during induced differentiation of mouse teratocarcinoma cells [26]. This dramatic demethylation occurs to remove gamete specific regulatory region, which were related to gamete function and allow facilitating embryogenesis process [27].
Recently, Choi et al. [28] compared differential methylation patterns of genes between early and late-passage cells and estimated the relationship between sensecence and DNA methylation patterns. They identified 2739 hypermethylated genes at early or late passages, and 1205 hypermethylated genes at early and late passages, respectively. These genes are involved in DNA replication, cell cycle and the PPAR signaling pathways. These results suggested that hypermethylation in global DNA increases due to long-term culture, which may in part affect senescence and aging.
Here, we examine the physiological mechanism underlying variation in genomic DNA methylation patterns associated with different growth arrest states, and variable growth rates obtainable with normal and tumor cells cultured in vitro under a range of controlled culture conditions. For this purpose we employed mouse embryonic fibroblasts cells lines, their virallyand chemicallytransformed cell line counterparts, as well as, normal human epidermal keratinocytes (NHK) and several of their epithelioid carcinoma (ECC) line counterparts.
2. Material & Methods
Chemicals: The following chemicals were ordered from Sigma Chemical Co., (St. Louis, MO): Cytosine, 5-methylcytosine, proteinase K, calf thymus DNA, sodium duponal, sodium hydroxide, hydrochloric acid, potassium hydroxide, Tris-HCl buffer, perchloric acid, and trichloroacetic acid. Sterile phosphate buffered saline solution, pH 7 was ordered from Grand Island Biologicals, Grand Island, NY). Precoated thin layer plates (EM #:5757-9H) (20 × 20 cm), isopropanol, n-butanol, methanol and ammonium hydroxide were ordered from Curtin-Matheson Scientific, Inc (Elk Grove, IL). Uridine, [5, 6-3H]-(Lot 2083-015), specific activity (38.9 Ci/mmol) was obtained from New England Nuclear (Boston, MA).
Mouse cells and cell culture: The following Balb/c mouse embryonic fibroblasts cell lines 3T3A31 and 3T3 T were obtained from Dr. Robert E. Scott (Mayo Medical School, Rochester, MN). 3T3-SV is a simian virus transformed derivative of the Swiss 3T3 mouse embryo fibroblasts, and 3T3-MCA is a methylcholanthracene derivative of the Swiss mouse embryo fibroblasts were also a gift from Dr. Robert E. Scott’s laboratory. Fibroblast cell lines were grown in DMEM-10% FCS medium. Growth arrest in the G1 phase of the cell cycle was achieved by various media substitutions as previously described [29]. Briefly, density-dependent growth arrest (Gc) occurs in well fed cultures refed growth medium beyond confluent cell density; serum-depletion arrest, Gs, occurs in low cell density cultures deprived of serum; nutrient arrest also occurs in low cell density cultures deprived of the amino acid, isoleucine. Finally, differentiation arrest, GD, occurs in low cell density cultures refed medium containing dialysed human platelet-poor plasma as described [30], and is a necessary prelude to the induction of adipocyte differentiation in Balb/c 3T3 proadipocytes. The values for the average population doubling time were obtained from log-linear curves plotting daily cell densities (cells/cm2) over five days of rapid exponential cell growth.
Human foreskin neonatal epidermal keratinocytes (NHK): Primary and secondary cultures of neonatal foreskin epidermal keratinocytes were prepared and cultured in a growth factor supplemented serum free medium [31] (MDCB 153 medium). Suspension-induced G1 arrest occurs when cell dispersions of cultured keratinocytes are resuspended in 1.3% methylcellulose as previously describe [32]. The values for the average population doubling time were obtained from log-linear curves plotting daily cell densities (cells/cm2) over five days of rapid exponential cell growth.
Human epithelioid tumor cell culture: The following epithelioid carcinoma cell lines were obtained from American Tissue Type Culture (ATTC) collection: KB, a human epithelioid carcinoma of nasopharnyx, D562, a human immortalized pharyngeal epithelial tumor line, HeLa, a human cervical carcinoma cell line, A431, a human epidermal cell carcinoma cell line. SCC9 and SCC25 are human tongue squamous carcinoma cell lines obtained from Dr. James G. Rhienwald of the Dana Farber Cancer Lab of Harvard University, Cambridge, MA. All tumor cells were maintained in a modified DMEM medium (YL) containing sodium pyruvate and L-glutamine, and supplemented with 10% dialyzed fetal calf serum and cultured at 37˚C in a humidified tissue culture incubator with a gas mixture of 5% dry carbon dioxide and 95% compressed air. The methods of suspension culture of the above tumor cell lines is the same as that described above for NHK cultures. The values for the average population doubling time were obtained from log-linear curves plotting daily cell densities (cells/cm2) over five days of rapid exponential cell growth.
Detection of genomic 5-methylcytosine: Procedures for the detection of genomic 5-methylcytosine are modifications of previous publications [33]. Briefly, cells were labeled for 16 hours with 6-3H-uridine (15 µCi/ml). The cells were then washed with 1X PBS, pH 7 and lysed by treatment with 0.5% sodium dupanol in the presence of 0.3 N NaOH and incubated overnight (>16 hours) at 37˚C. The lysate was neutralized with NaOH and HCl and an equal volume of 0.25 M Tris-HCl (pH 7.6) and proteinase K (0.1 mg/ml) was added, and the mixture incubated overnight at 37˚C. The 3H-labelled DNA was precipitated with calf thymus DNA (1 mg/ml) as carrier with 10% trichloroacetic acid (TCA, final concentration). The precipitate was washed extensively with cold 5% TCA. The precipitate was dried and then digested overnight with 10% perchloric acid at 65˚C. The digest was neutralized with 1 N KOH and clarified by low speed centrifugation.
Thin layer chromatography: Separation of DNA bases was accomplished by a thin layer chromatographic procedure in two-dimensions. As Rf markers 20 µl of unlabelled uridine is spotted on site 1of the baseline and a 1:1 equimolar (1 × 10−2 M) mix of unlabelled cytosine and 5-methylcytosine is spotted at position 2 on the baseline. The plates are placed vertically in battery jars baseline down with sufficient volume of solvents (isopropanol: n-butanol:methanol:NH4OH) to achieve capillary mobility of solvents and separation allowed to continue over a 24 hours period. Clean separation occurs in the second dimension rotated 90˚ away from the first dimension with Rf values of 5.5 cm for cytosine, and 6.5 cm for 5-metylcytosine. For labeled uridine and labeled and separated DNA bases the spots were eluted with 0.1 N HCl and their radioactive contents determined by scintillation spectrophotometric analysis.
Statistical method: The 5-methylcytosine between different cultures was compared using t-test using the SPSS (ver.18).
3. Results
Genomic DNA methylation of rapidly growing (RG) mouse embryo fibroblast cell lines.
Table 1 shows the extents of genomic DNA methylation between two embryonic mouse fibroblasts cell lines (Balb/c and Swiss) are compared with a SV-40 viral transformed and a methylcholanrtacene-transformed strain of the Swiss A31 3T3 fibroblasts. The differentiationcapable proadipocyte (Balb/c 3T3 T) cell line is less methylated relative to the differentiation-incapable, “initiated” A31 cell line (3.4% vs. 4.6%). Both transformed and tumorigenic cell lines (3T3-SV and 3T3-MCA) are hypomethylated relative to the A31 parent cell line (4.0% and 3.9% respectively).
Effect of growth rate on extent of mouse fibroblast genomic DNA methylation.
Figure 1 is a histogram plot of the growth rate plotted as the reciprocal (f) of the average population generation time (GT) versus the extent of genomic DNA methylation (5-MC%) for several mouse embryonic fibroblast cell lines. Figure 2 is a scatter plot of the above data. It was employed to statistically analyze the data by linear regression analysis. There is a strong positive correlation for the three normal embryonic fibroblast lines between growth rate (GT) and 5-MC content (r = 0.9). A weak linear correlation was found when the two outliers representing the transformed cells were included (r = 0.53).
Effect of G1 phase growth-arrest on extent of genomic DNA methylation.
Table 2 presents data showing that four different types of G1 growth arrest states of 3T3 T proadipocyte cells achieved by various media and growth conditions resulted in hypermethylation (4.1% - 4.3% 5-MC) of genomic DNA methylation by 20% (± 0 σ) a value that was uniformly greater than that observed for rapidly cycling undifferentiated 3T3 T fibroblasts (3.4% 5-MC).
Extent of genomic DNA methylation between NHK and various ECC cell lines.
Table 3 presents data showing the extent of genomic DNA methylation of NHK (NF238P1) and six different ECC cell lines (D562, Hela, KB, SCC 25, SCC 9, and A431). The 5-MC% for NHK strain and the A431 tumor cell lines were identical (3.6% 5-MC). Three of the ECC cell lines (KB, SCC25 and SCC9) tested was significantly different from normal keratinocytes (p < 0.05). Hela appeared to be hypermethylated (4.2% ± 1 σ), but the remaining four were hypomethylated by an average of 2.5% 5-MC (± 0.6 σ) relative to NHK.
Correlation of 5-methylcytosine content of genomic DNA and cellular growth rate
Figure 3 presents a histogram plotting the pair-wise data for growth rate and 5-MC% for NHK and each of the ECC cell lines. Figure 4 presents a scatter plot of the data. Linear regression analyses showed that there was a strong negative correlation between average growth rate (GT) and 5-MC content (r = 0.9).
Table 1. Genomic DNA methylation in normal and transformed mouse fibroblast cell Lines.
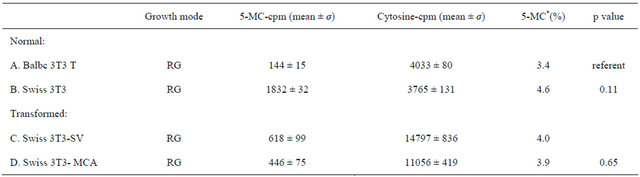
*A is significantly different than B; B is significantly different from both C and D. Student’s t-test was used.

Figure 1. Relationship of growth rate (f) and 5-methylcytosine for various fibroblast cell lines.
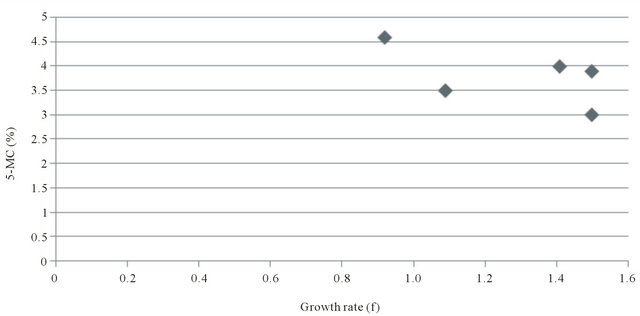
Figure 2. Correlation of growth rate (f = 1/GT) and 5-methylcytosine content in fibroblast cell lines.
Table 2. Genomic DNA methylation between various growth arrest states of the mouse Balb/c 3T3 T fibroblast cell line.
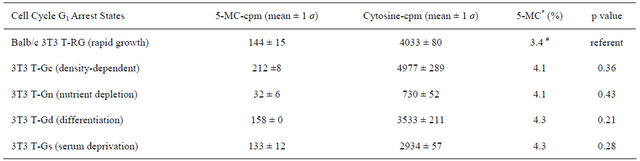
Student’s t-test was used. *3T3-RG is marginally significant statistically from all the combined average of four of the above arrest states (p = 0.06).
Table 3. Comparison of DNA methylation between cultured normal keratinocytes and tumor cell lines.
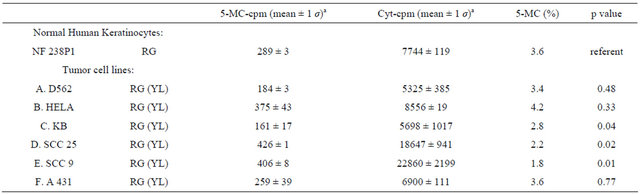
aMean values are given for duplicate TLC plates except KB cell lines, which has triplicate TLC.
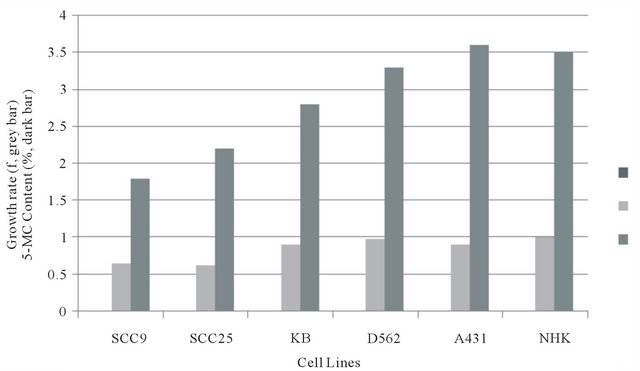
Figure 3. Relationship of Growth rate to 5-methylcytosine content of NHK and ECC lines. aSuspend 2.3 × 105 rapidly dividing K B cells in 4.0 ml of 1.3% methylcellulose for 24 hours. bSuspend 4.3 × 105 rapidly dividing A 431 cells in 4.0 of 1.3% methylcellulose for 24 hours.
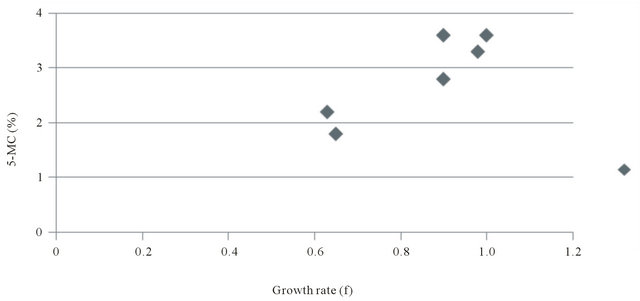
Figure 4. Correlation of growth rate (f) and 5-methylcytosine content of NHK and ECC cell lines.
Extent of genomic DNA methylation of cycling and suspension-arrested NHK and ECC cell lines.
Table 4 presents data showing the extent of genomic DNA methylation between cycling and suspension-arrested NHK, and three different ECC cell lines. For NHK (NF 284P1) there was a dramatic decrease (59%) in overall DNA base labeling (C + 5-MC) between cycling (RG) and suspension-arrested (SUS) cultured cells, indicative of suspension-induced growth arrest, There also was 7% increase in 5-MC labeling, indicating a relative increase in methylation due to suspension arrest. For KB cells, there was a 3% decline to total DNA base labeling upon suspension in semi-solid medium, indicating that suspension did not significantly induce growth arrest. Nevertheless, there was a 41% increase in 5-MC base labeling, indicating that suspension perse of KB results in DNA hypermethylation. Flow cytometric analysis of DNA contents (Table 5) confirmed that suspension of KB in semi-solid medium actually led to a decrease in cells in G1 (39%) relative to cycling KB cells (56%), and a relative increase in combined S-phase plus G2/M (61%) compared to cycling cells (44%). Table 4 present data showing that SCC25 displayed a 73% decrease in total DNA base labeling upon culture in semi-solid medium, suggesting that suspension and loss of anchorage arrests SCC25 tumor cells. An even greater decrease in 5-MC base labeling was observed upon suspension of SCC25 cells, resulting in hypomethylation of SCC25 genomic DNA relative to cycling cells. Finally, for A431 tumor cells suspension culture induced a 33% increase in overall DNA labeling over cycling cells, indicating that suspension in semi-solid medium not only failed to induce growth arrest, but actually enhanced overall DNA synthesis. Further, hypomethylation was observed for suspension of A431 without growth arrest. Table 5 present flow cytometric analysis of DNA contents A431 cycling and suspension cultures. It confirms cell cycling occurred in A431 suspension cultures resulting in a decreased proportion of G1 cells (19%) relative to cycling cells (35%).
Table 4. Comparison of genomic DNA methylation of dividing cultures versus suspension-arrested cultures of normal keratinocytes and several epithelioid tumor cells.
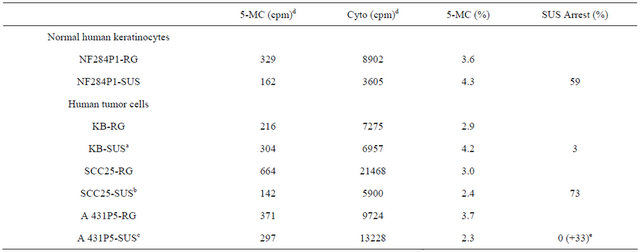
Table 5. Cell cycle analysis of human tumor cell lines suspended in semisolid medium.
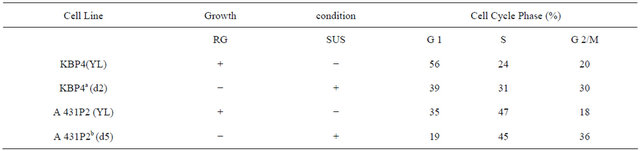
aSuspend 2.3 × 105 rapidly dividing K B cells in 4.0 ml of 1.3% methylcellulose for 24 hours. bSuspend 4.0 × 105 rapidly dividing SCC25 cells in 4.0 ml of 1.3% methylcellulose for 24 hours. cSuspend 4.3 × 105 rapidly dividing A 431 cells in 4.0 of 1.3% methylcellulose for 24 hours. dValues given are for results of one TLC plate for each cell line. eSuspension enhanced cytosine labeling.
4. Discussion
Our results provide strong evidence that genomic DNA methylation patterns of cultured mammalian cell lines are affected by growth rate-effecting culture conditions. First, we examined the effect of four different culture manipulations that arrest murine embryonic fibroblasts (Balb/c 3T3 T proadipocytes) in the G1 phase of the cell cycle (see Table 2). This included growth arrest due to nutriaent-depletion (Gn), confluent-arrest (Gc), serum-deprivation (Gs) and adipocyte-induced differentiation coupled growth arrest (GD) .This result suggests that the enzymes involved in de novo DNA methylation (DNMT3a and DNMT3b) may be activated by arrest in the G1 phase of the cell cycle. In each case G1 phase cell cycle growth arrest was accompanied by significant genomic DNA hypermethylation. This is the first report that global DNA hypermethylation occurs during the transition from rapid cycling to growth arrest in normal embryonic murine fibroblasts. Transition to reversible G1 phase arrest is known to require protein synthesis and gene expression as well as the silencing of many growth promoting genes. Hypermethylation in G1 phase cells suggests that the enzymes involved in de novo DNA methylation (DNMT3a and DNMT3b) may be activated by arrest in the G1 phase of the cell cycle. We also report and confirm that chemically-(3T3-MCA) and virally (3T3-SV)-transformed murine embryonic fibroblast cells are hypermethylated relative to their normal counterparts (Swiss 3T3). Hypermethylation in cancer cells is usually interpreted to mean that gene silencing has occurred to suppress tumor suppressor genes.
We next asked the question whether there is direct relationship between growth rate of cells and the extent of DNA methylation. This was accomplished by determined for each cell culture its average population generation time (GT) and it reciprocal (f) with 5-MC content for that growth rate. Linear regression analysis established that three different murine embryonic fibroblast cell lines tested (Swiss 3T3, Balb/c 3T3 T, and C3H 10T1/2) neatly demonstrate a strong positive correlation. By contrast, the two tumorigenic and transformed derivatives of the Swiss 3T3 line when included in the plot with the three normal fibroblasts are clearly outliers that reduce the correlation to non-significance. This appears to be caused by their slower than predicted growth rates.
Earlier studies reported that cancer cells display either hypomethylation or hypermethylation relative to untransformed cells [5]. When the methylated DNA of NHK was compared to six human ECC tumor cell lines, there was no change for Hela and A431cells, while the four other ECC cell lines tested (D562, KB, SCC25 and SCC9) were all hypomethylated. Hypomethylation in tumor cells is generally associated with gene derepression occurring in the promoter regions of island CpG DNA sequences [5]. Again, we examined the relationship of growth rate of NHK and ECC cell lines to their pair-wise 5-MC contents. Linear regression analysis of the data revealed a strong negative correlation. Tumor cells appear to link lower extents of DNA methylation with correspondingly longer growth rates. It should be noted that each of the ECC cell lines was established in culture from an epithelial type cancer tissue, and are thus the consequence of human cancer carcinogenesis not the result of a direct chemical or viral oncogenesis as was the case for the murine embryonic fibroblasts cells.
Finally, we examined growth arrest of cycling epithetlial cells induced by loss of anchorage to a solid surface (anoikis). All of the ECC cell lines were grown on a solid surface before suspension in semi-solid medium. The consequence of anoikis on DNA methylation patterns for NHK cell strain was typical G1 phase growth arrest confirming our previous studies [32]. Anokis and G1 phase growth arrest was accompanied by genomic DNA hypermethylation. Based on these results we would expect that cessation of cell cycling and arrest in the G1 phase may be implemented by de novo DNA methylation likely in promoter regions of cell cycle-related genes. Although, a recent report [34] indicated that DNMT3a can methyllate DNA in non-promoter region.
SCC25 tumor derived epithelial cell underwent suspension-induced growth arrest and a drastic decrease in DNA base labeling. Yet, they were even more hypomethylated relative to cycling SCC25 cells. Neither KB nor A431 tumor-derived epitheliod cell lines were arrested by suspension culture and both showed a further 5-MC hypomethylation relative to their cycling counterparts. Growth arrest of SCC25 cells by suspension culture is somewhat surprising as we previously reported that it was defective in response G1 arrest by exposure to TGF-β [35] and defective in coupling growth arrest with terminal squamous differentiation [36]. It would appear that another primary defect in SCC cells is the uncoupling of DNA hypermethylation of cell cycle-related genes from growth arrest [12]. Recently, a higher frequency of aberrant hypermethylation of the FOXE1 gene was reported in cutaneous squamous cell carcinoma [37]. KB and A431 cells appear to be more defective in responding to growth controlling signals such as suspendsion than SCC25 cells. Overall, we have not seen any clear correlation whether hypermethylation or hypomethylation is more likely to occur during growth arrest in tumor-derived cells.
5. Acknowledgements
One author (JJW) extends his sincere appreciation for financial assistance provided by grants to the laboratory of Dr. Robert E. Scott, then Professor of Cell Biology at the Mayo Medical School, Rochester, MN, during this author’s prior tenure there.
REFERENCES
- A. K. Maunakea, I. Chepelev and K. Zhao, “Epigenome Mapping in Normal and Disease States,” Circulation Research, Vol. 107, No. 3, 2010, pp. 327-339. doi:10.1161/CIRCRESAHA.110.222463
- K. D. Robertson and A. P. Wolffe, “DNA Methylation in Health and Disease,” Nature Reviews, Vol. 1, No. 1, 2002, pp. 11-19.
- H. Denis, M. N. Ndulova and F. Fuks, “Regulation of Mammalian DNA Methyltransferases: A Route to New Mechanisms,” EMBO Reports, Vol. 12, No. 7, 2011, pp. 647-656. doi:10.1038/embor.2011.110
- T. Mikeska, C. Bock, H. Do and A. Dobrovic, “DNA Methylation Biomarkers in Cancer: Progress towards Clinical Implementation,” Expert Review of Molecular Diagnostics, Vol. 12, No. 5, 2012, pp. 473-487. doi:10.1586/erm.12.45
- R. A. Weinberg, “The Biology of Cancer,” Garland Science, Taylor & Francis Group, LLC, New York, 2007.
- P. A. Jones and S. B. Baylin, “The Fundamental Role of Epigenetic Events in Cancer,” Nature Reviews Genetics, Vol. 3, No. 6, 2002, pp. 415-428.
- R. Jaenisch and A. Bird, “Epigenetic Regulation of Gene Expression: How the Genome Integrates Intrinsic and Environmental Signals,” Nature Genetics, Vol. 33, 2003, pp. 245-254. doi:10.1038/ng1089
- S. J. Clark and J. Melki, “DNA Methylation and Gene Silencing in Cancer, Which Is the Guilty Party,” Oncogene, Vol. 21, No. 35, 2002, pp. 5380-5387. doi:10.1038/sj.onc.1205598
- P. M. Das and R. Singai, “DNA Methylation and Cancer,” Journal of Clinical Oncology, Vol. 22, No. 22, 2004, pp. 4632-4642. doi:10.1200/JCO.2004.07.151
- C. Stirzaker, J. Z. Song, B. Davidson and S. J. Clark, “Transcriptional Gene Silencing Promotes DNA Hypermethylation through a Sequential Change in Chromatin Modification in Cancer Cells,” Cancer Research, Vol. 64, No. 11, 2004, p. 3871. doi:10.1158/0008-5472.CAN-03-3690
- V. Mutskov and G. Felsenfeld, “Silencing of Transgene Transcription Precedes Methylation of Promoter DNA and Histone H3 Lysine 9,” The EMBO Journal, Vol. 23, No. 1, 2004, pp. 138-149. doi:10.1038/sj.emboj.7600013
- S. K. Patra, “Ras Regulation of DNA Methylation,” Experimental Cell Research, Vol. 314, No. 6, 2008, pp. 1193-1201. doi:10.1016/j.yexcr.2008.01.012
- E. G. Toraño, S. Petrus, A. F. Fernandez and M. F. Fraga, “Global DNA Hypomethylation in Cancer: Review of Validated Methods and Clinical Significance,” Clinical Chemistry and Laboratory Medicine, Vol. 50, No. 10, 2012, pp. 1733-1742. doi:10.1515/cclm-2011-0902
- R. G. Liteplo and R. S. Kerbel, “Reduced Levels of DNA 5-Methylcytosine in Metastatic Variants of Human Melanoma Cell Line MeWo,” Cancer Research, Vol. 47, No. 9, 1987, pp. 2264-2267.
- A. P. Feinberg, C. W. Gehrke, K. C. Kuo and M. Ehrlich, “Reduced Genomic 5-Methylcytosine Content in Human Colonic Neoplasia,” Cancer Research, Vol. 48, No. 5, 1988, pp. 1159-1161.
- A. Eden, F. Gaudet, A. Waghmare and R. Jaenisch, “Chromosomal Instability and Tumors Promoted by DNA Hypomethylation,” Science, Vol. 300, No. 5618, 2003, p. 455. doi:10.1126/science.1083557
- F. Gaudet, J. G. Hodgson, A. Eden, L. Jackson-Grusby, J. Dausman, J. W. Gray, H. Leonhardt and R. Jaenisch, “Induction of Tumors in Mice by Genomic Hypomethylation,” Science, Vol. 300, No. 5618, 2003, pp. 489-492. doi:10.1126/science.1083558
- T. M. Holm, L. Jackson-Grusby, T. Brambrink, Y. Yamada, W. M. 3rd Rideout and R. Jaenisch, “Global Loss of Imprinting Leads to Widespread Tumorigenesis in Adult Mice,” Cancer Cell, Vol. 8, No. 4, 2005, pp. 275- 285. doi:10.1016/j.ccr.2005.09.007
- H. Cui, M. Cruz-Correa, F. M. Giardiello, D. F. Hutcheson, D. R. Kafonek, S. Brandenburg, Y. Wu, X. He, N. R. Powe and A. P. Feinberg, “Loss of IGF2 Imprinting: A Potential Marker of Colorectal Cancer Risk,” Science, Vol. 299, No. 5613, 2003, pp. 1753-1755. doi:10.1126/science.1080902
- F. Tian, F. Z.Tang, G. Song, Y. Pan, B. He, Q. Bao and S. Wang, “Loss of Imprinting of IGF2 Correlates with Hypomethylation of the H19 Differentially Methylated Region in the Tumor Tissue of Colorectal Cancer Patients,” Vol. 5, No. 6, 2012, pp. 1536-1540.
- M. J. Hoffmann and W. A. Schulz, “Causes and Consequences of DNA Hypomethylation in Human Cancer,” Biochemistry and Cell Biology, Vol. 83, No. 3, 2005, pp. 296-321. doi:10.1139/o05-036
- E. Daura-Oller, M. Cabre, M. Montero, J. L. Paternain, and A. Romeu, “Single Gene Hypomethylation and Cancer: New Insights into Coding Region Feature Trends,” Bioinformation, Vol. 3, No. 8, 2009, pp. 340-343. doi:10.6026/97320630003340
- R. Lister, M. Pelizzola, R. H. Dowen, R. D. Hawkins, G. Hon, J. Tonti-Filippini, J. R. Nery. L. Lee, Z. Ye, Q. M. Ngo, L. Edsall, J. Antosiewicz-Bourget, R. Stewart, V. Ruotti, A. H. Millar, J. A. Thomson, B. Ren and J. R. Ecker, “Human DNA Methylomes at Base Resolution Show Widespread Epigenetic Differences,” Nature, Vol. 462, No. 7271, 2009, pp. 315-322. doi:10.1038/nature08514
- J. F. Costello, M. Krywinski and M. A. Marra, “A First Look at Entire Human Methylomes,” Nature Biotechnology, Vol. 27, No. 12, 2009, pp. 1130-1132. doi:10.1038/nbt1209-1130
- T. G. Jenkins and D. T. Carrell, “Dynamic Alterations in the Paternal Epigenetic Landscape Following Fertilization,” Front Genet, Vol. 3, 2012, p. 143.
- A. Razin, C. Webb, M. Szyf, J. Yisraeli, A. Rosenthal, T. Naveh-Many, N. Sciaky-Gallili and H. Cedar, “Variations in DNA Methylation during Mouse Cell Differentiation in Vivo and in Vitro,” Proceedings of National Academy Sciences of the USA, Vol. 81, No. 8, 1984, pp. 2275-2279. doi:10.1073/pnas.81.8.2275
- W. Reik, W. Dean, and J. Walter, “Epigenetic Reprogramming in Mammalian Development,” Science, Vol. 293, No. 5532, 2001, pp. 1089-1093. doi:10.1126/science.1063443
- M. R. Choi, Y. H. In, J. Park, T. Park, K. H. Jung, J. C. Chai, M. K. Chung, Y. S. Lee and Y. G. Chai, “GenomeScale DNA Methylation Pattern Profiling of Human Bone Marrow Mesenchymal Stem Cells in Long-Term Culture,” Experimental & Molecular Medicine, Vol. 44, No. 8, 2012, pp. 503-512. doi:10.3858/emm.2012.44.8.057
- J. J. Wille and R. E. Scott, “Topography of the Predifferentiation GD Growth Arrest State Relative to Other Growth Arrest States in the G1 Phase of the Cell Cycle,” Journal of Cellular Physiology, Vol. 112, No. 1, 1982, pp. 115-122. doi:10.1002/jcp.1041120117
- R. E. Scott, D. L. Florine, J. J. Wille Jr. and K. Yun, “Coupling of Growth Arrest and Differentiation at a Distinct State in G1 Phase of the Cell Cycle, GD,” Proceedings of National Academy Sciences of the USA, Vol. 79, No. 3, 1982, pp. 845-849. doi:10.1073/pnas.79.3.845
- S. T. Boyce and R. G. Ham, “Calcium Regulated Differentiation of Normal Human Epidermal Keratinocytes in Chemically Defined Clonal Cultures and Serum-Free Serial Cultures,” Journal of Investigative Dermatology, Vol. 81, Suppl. 1, 1983, pp. 33-40. doi:10.1111/1523-1747.ep12540422
- M. R. Pittelkow, J. J. Wille Jr. and R. E. Scott, “Two Functionally Distinct Classes of Growth Arrest States in Human Prokeratinocytes that Regulate Clonogenic Potential,” Journal of Investigative Dermatology, Vol. 86, No. 4, 1986, pp. 410-417. doi:10.1111/1523-1747.ep12285684
- V. L. Wilson and P. A. Jones, “The Effect of Tissue Culture Cell Aging on DNA Methylation,” Science, Vol. 220, No. 4601, 1983, pp. 1055-1057. doi:10.1126/science.6844925
- H. Wu, V. Coskin, J. Tao, W. Xie, W. Ge, K. Yoshikawa, K. E. Lee. Y. Zhang and Y. Sun, “Dnmt3a-dependent Non-Promoter Methylation Facilitates Transcription of Neurogenic Genes,” Science, Vol. 329, No. 5990, 2010, pp. 444-448. doi:10.1126/science.1190485
- G. D. Shipley, M. R. Pittelkow, J. J. Wille Jr., R. E. Scott and H. L. Moses, “Reversible Inhibition of Normal Human Prokeratinocytes by Type-Beta Transforming Growth Factor/Growth Inhibitor in Serum-Free Medium,” Cancer Research, Vol. 46, No. 4, 1986, pp. 2068-2071.
- D. Breitkreutz, H. J. Stark, P. Plein, M. Baur and N. E. Fusenig, “Differential Modulation of Epidermal Keratinization in Immortalized (HaCat) and Tumorigenic Human Skin Keratinocytes (Hacat-Ras) by Retinoic Acid and Extracellular Ca2+ Differentiation,” Vol. 54, No. 3, 1993, pp. 201-217. doi:10.1111/j.1432-0436.1993.tb01602.x
- I. Venza, M. Visalli, B. Tripoido, G. De Grazia, S. Loddo, D. Teti and M. Venza, “FOXE1 is a Target for Aberrant Methylation in Cutaneous Squamous Cell Carcinoma,” British Journal of Dermatology, Vol. 162, No. 5, 2009, pp. 1093-1097. doi:10.1111/j.1365-2133.2009.09560.x
NOTES
*Corresponding author.