Journal of Cancer Therapy
Vol.3 No.6(2012), Article ID:25417,6 pages DOI:10.4236/jct.2012.36142
CXCR4-Expressing Glial Precursor Cells Demonstrate Enhanced Migratory Tropism for Glioma
![]()
Department of Neurological Surgery, Vanderbilt University Medical Center, Nashville, USA.
Email: *moneeb.ehtesham@vanderbilt.edu
Received July 6th, 2012; revised August 8th, 2012; accepted August 17th, 2012
Keywords: Glioma; CXCR4; Neural Precursor Cells; Brain Tumor(s)
ABSTRACT
Malignant glioma remains one of the most intractable of human cancers principally due to the highly infiltrative nature of these neoplasms. The use of neural precursor cells (NPC) has received considerable attention based on their ability to selectively migrate towards disseminated areas of tumor in vivo and their described ability to deliver tumor-directed therapies specifically to infiltrating tumor cells. Fundamental to optimizing the use of these cells for potential clinical translation is the development of an understanding regarding the biologic cues that govern their ability to migrate towards infiltrative glioma foci. To this end, in this paper we detail that NPC selected for double-expression of the glial-precursor marker A2B5 and the cell-surface chemokine receptor, CXCR4, demonstrate enhanced in vitro gliomadirected tropism. These findings demonstrate the relevance of these markers for the phenotypic segregation of an optimally tumor-tropic NPC sub-population as a means of enhancing NPC-based therapeutic strategies for the treatment of glioma.
1. Introduction
Despite advances in surgical techniques and adjuvant therapies, the prognosis for patients with malignant glial tumors remains poor with a median survival following a new diagnosis of glioblastoma multiforme of under one year, and a two year survival rate approaching zero [1]. The therapeutic challenge posed by these neoplasms centers on their highly infiltrative nature. At the microscopic level solitary tumor cells or clusters of neoplastic cells migrate throughout the brain, often to significant distance, resulting in near universal recurrence of disease despite aggressive surgical resection, radiation and chemotherapy. One promising means of specifically directing treatment to disseminated areas of tumor involves the use of neural precursor cells (NPC). These are mutlipotent progenitor cells that can be harvested from either fetal, neonatal or adult tissue sources and are capable of long-term sustained self-renewal in vitro. When subjected to appropriate differentiation-inducing conditions, these cells are capable of terminal differentiation into either neurons or glia. There is an extensive body of literature from our group as well as others that has described the use of these cells to deliver tumoricidal gene products to disseminated tumor foci with significant therapeutic success in pre-clinical models of glioma [2-8]. Notable, however, has been our consistent finding that only a sub-population of NPC appears to display this glioma-tropic migratory activity in vivo [9]. This led us to speculate that the identification of specific phenotypic differences between tumor-tropic versus non-migratory NPC populations may allow us to better characterize and, ultimately, refine for a selected population of NPC optimally primed for glioma therapy. To this end, we have earlier described that NPC that migrate towards disseminated foci of intracranial glioma demonstrate a pheno- -type consistent with that of glial precursor cells (as characterized by expression of A2B5) that also expressed the cell surface chemokine receptor, CXCR4 [9]. The CXCR4 chemokine pathway is known to regulate glial precursor migration in the developing brain [10,11], and glioma cells are known to express significant levels of CXCL12 (the soluble ligand for CXCR4) [12-14] which likely explains why this phenotypic combination is also associated with glioma-tropic NPC populations within our treatment models. In this study, we now detail the use of non-invasive bioluminescence-based imaging to confirm that, within unfractionated NPC, only a portion of cells demonstrate glioma-directed migratory behavior. Furthermore, we describe the use of a magnetic and flow-cytometry based cell-sorting approach to refine for A2B5+ CXCR4+ double-positive NPC and demonstrate that this population displays significantly enhanced tropism for glioma-conditioned medium in vitro. These findings point to the relevance of the use of this refined population of NPC as an optimally tumor-tropic tool for enhanced delivery of tumoricidal gene products to glioma in vivo.
2. Materials and Methods
Isolation of NPC, generation of intracranial tumor models, and in vivo bioluminescence imaging. Murine fetal NPC were harvested from the frontoparietal regions of day 15 mouse fetuses as described earlier [6]. NPC were cultured and maintained as previously detailed [5, 6]. The 9L and GL26 glioblastoma cell lines were obtained from ATCC (Manassas, VA) and Dr. John Yu (Los Angeles, CA), respectively, and maintained in vitro per established protocol. NPC were engineered to express firefly luciferase by means of infection with the firefly-luciferase gene bearing replication-defective adenovirus using methodology described by us earlier [5,6]. Intracranial tumors were established as follows: Six to eight week old athymic nude mice were obtained from Charles River Laboratories (Wilmington, MA) and anesthetized using intraperitoneal ketamine and xylazine. They then underwent stereotactic implantation of 5 × 104 glioma cells in 3 µl of 1.2% methylcellulose/MEM in the left corpus striatum as reported previously [5,6]. Control animals received injections of the identical volume of cell-free methylcellulose/MEM. Five days later, the animals underwent implantation of 2 × 105 NPC expressing firelfly luciferase in the contralateral hemisphere using similar implantation methodology. Six days following this second implantation, animals were anesthetized using inhaled isoflurane and images in the Xenogen IVIS 200 imaging system, allowing for quantitation of light emission following intraperitoneal administration of Dluciferin (150 µg/g body weight). For immunohistochemical studies, athymic nude mice underwent stereotactic implantation of GL26 glioma cells in the left cerebral hemisphere followed by inoculation of β-galactosidase expressing NPC (NPC-LacZ) [6,13] into the contralateral hemisphere one week later. After an additional seven days, brains from these mice were harvested and stained using standard immunofluorescence techniques using antibodies against β-galactosidase, A2B5, GFAP, CNPase, E-NCAM, beta III tubulin, PDGFRα (Chemicon; Temecula, CA).
In vitro selection of A2B5+ CXCR4+ NPC. Fetal murine NPC cultures were supplemented with 10 ng/ml recombinant ciliary neurotrophic factor (CNTF) and 20 ng/ml bone-morphogenic protein-2 (BMP-2) (Peprotech, Rocky Hill, NJ). This methodology has been previously described as promoting induction of a glial-precursor cell fate-commitment from within undifferentiated NPC [15]. 24 hours following supplementation of cultures with these factors, cells were isolated and then incubated with a PE-conjugated anti-A2B5 primary antibody (Chemicon) for 30 - 45 minutes, followed by a microbead tagged secondary antibody (Miltenyi Biotec; Auburn, CA). A positive selection strategy was then applied using a magnetic cell sorting (MACS) separation column (Miltenyi Biotec) wherein microbead-tagged A2B5+ cells were eluted as a separate enriched population using methodology as previously detailed [16]. Further enrichment was performed by labeling cells with a FITC-conjugated anti-CXCR4 antibody (BD Pharmingen) and utilizing fluorescence activated cell sorting to separate a population of A2B5+ CXCR4+ double-positive cells.
In vitro NPC migration assay. All chemotaxis experiments were performed using a chemotaxis chamber (Neuro Probe, Gathersburg, MD) consisting of pairs of culture wells separated by a 5 um porous polycarbonate membrane, utilizing methodology detailed by us earlier [9]. Lower wells were filled with GL-26 conditioned medium harvested as described [9]. Fresh DMEM supplemented with 10% FBS and 1% penicillin/streptomycin was used as a control (unconditioned media. Following placement of the porous membrane, approximately 1.5 × 105 disaggregated NPC were added to the top chambers. The chamber system was incubated at 37˚C for four hours, after which media from lower wells were collected and quantitatively analyzed for cell content using flow cytometry against a defined number of fluorescent beads (BD Pharmingen) utilizing methodology detailed by us previously [9]. This allowed for quantification of the percentage of cells added to each top chamber that had migrated to the bottom chamber. For neutralization assays, anti-CXCR4 (40 μg/ml) (R&D Systems, Minneapolis, MN) monoclonal antibody was incubated with NPC for 30 minutes at room temperature prior to the assay. Control samples were incubated with identical concentrations of isotype-matched nonspecific antibody (BD Pharmingen). All experiments were performed in triplicate.
3. Results
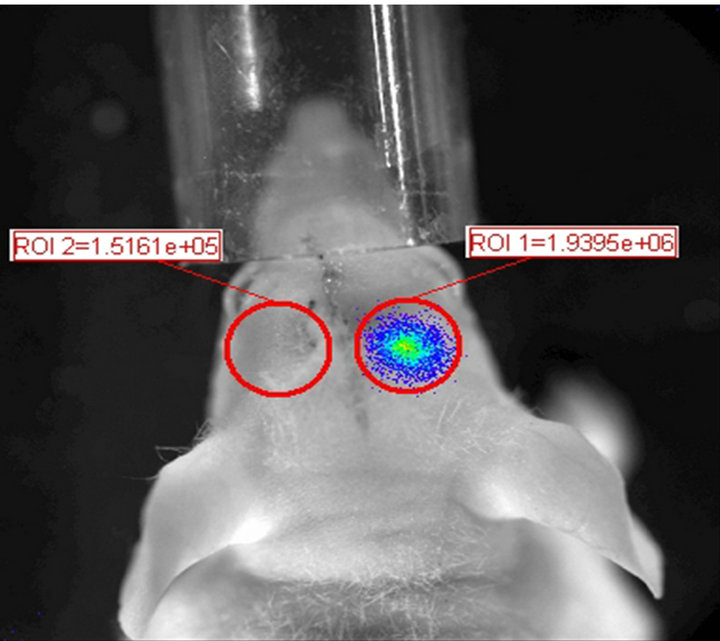
Non-invasive in vivo bioluminescence imaging demonstrates the presence of a sub-population within NPC that exhibits glioma tropic migratory behavior. With the goal of developing a non-invasive modality for the tracking of NPC migration within the brain, we labeled fetal murine NPC with firefly-luciferase and injected these cells into the contralateral cerebral hemisphere within the brains of nude mice bearing pre-established intracranial gliomas. As depicted in Figure 1(a), six days following implantation NPC injected into the brains of control mice without pre-existing intracranial tumors remained largely localized to the ipsilateral cerebral hemisphere. In contrast, NPC injected into tumor bearing brains displayed marked migratory activity across the midline and redistribution into the vicinity of the already established tumor. These findings provide an elegant non-invasive imaging modality and confirm our earlier post-mortem histological analyses which demonstrated similar results [5,6,9]. Also, of note, despite the migration of a marked number of implanted NPC into the contralateral tumorbearing hemisphere, we again noted that a significant portion of the implanted cells did not demonstrate this tumor tropic property and remained within the ipsilateral hemisphere. This finding again supports our earlier data indicating that NPC contain differential populations with differing tumor-tropic migratory potential [9].
Tumor-tropic NPC demonstrate expression of the glial precursor markers A2B5 and GFAP but do not express markers of neuronal or oligodendroglial fatecommitment or differentiation. With the goal of identifying certain phenotypic characteristics that could help delineate the sub-population of NPC that demonstrate an enhanced in vivo tumor tropic migratory capacity, we utilized immunofluorescence histochemistry to analyze β-galactosidase expressing NPC that had migrated towards contralaterally implanted GL26 gliomas in mouse brains. As represented in Figure 1(b), we found that β- galactosidase expressing NPC that had migrated across midline into the opposite tumor-bearing hemisphere demonstrated expression of the glial precursor markers A2B5 and GFAP. Conversely, they did not express the neuronal precursor marker E-NCAM nor the neuronal differentiation marker beta-III tubulin and were also negative for markers of oligodendroglial commitment (PDGFRα) and oligodendroglial differentiation (CNPase) (not shown). Consistent with our prior studies, these findings again demonstrate that the tumor-tropic migratory capacity of NPC appears to be limited to those cells that bear phenotypic characteristics indicative of a likely astrocytic glial fate-commitment.
A sub-population of glial precursor cells further enriched for expression of the cell surface chemokine receptor CXCR4 demonstrate even greater tropism for glioma in vitro. We have previously demonstrated that activity of the CXCR4 cell surface chemokine receptor
 (a)
(a)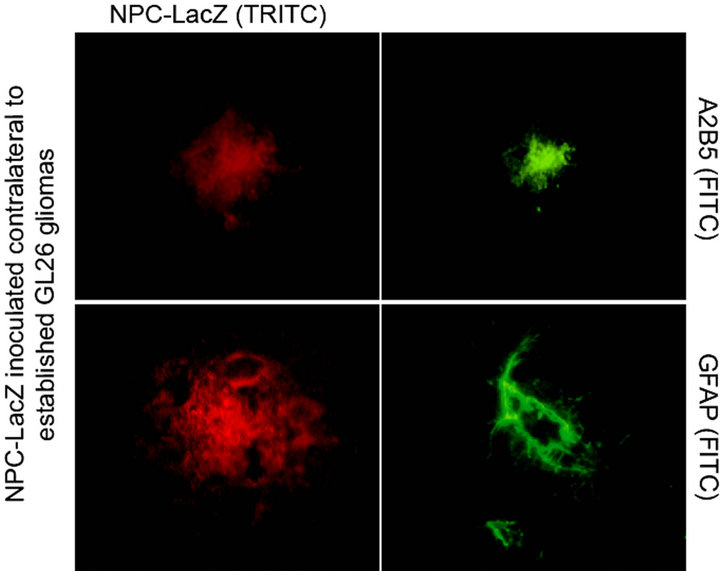 (b)
(b)
Figure 1. (a) NPC demonstrate significant, albeit non-uniform, intracranial migratory tropism for contralateral glioma. NPC engineered to express firefly luciferase (NPC-luc) when inoculated into the right corpus striatum in a non-tumor bearing athymic nude mouse (left panel) do not demonstrate any significant migratory activity after 6 days, as determined by in vivo bioluminescence imaging (BLI). However, when NPC-luc are inoculated into the right corpus striatum of a nude mouse that has a previously implanted 9L glioma growing in the left cerebral hemisphere, a significant number of cells traversed into the opposite hemisphere by day 6 following NPC inoculation (right panel). BLI allows for the quantitation of NPC derived luminescence from a demarcated area located over each cerebral hemisphere. A quantitative assessment of NPC migration efficiency towards contralateral tumor can be calculated by dividing NPC derived photon flux from the tumor bearing hemisphere by the total photon flux from both sides of the head. This gives a value for Percent total photon redistribution into contralateral hemisphere. For the control non-tumor bearing animal (left) this value is 7.2% versus 44.1% for the tumor bearing animal (right). Even though photon flux values cannot be equilibrated to cell counts, these results still indicate that despite the significant tumor-tropic capacity of NPC, a significant portion of transplanted cells do not migrate towards contralateral tumor. (b) NPC migrating towards contralateral tumor in vivo demonstrate phenotypic markers of glial precursors. Brains from mice inoculated with 9L glioma cells in the left cerebral hemisphere followed by NPC injection into the right hemisphere were harvested and immunocytochemically stained for the glial precursor markers A2B5 and GFAP. As depicted, NPC found to have migrated into the contralateral tumor bearing hemisphere demonstrated expression of these markers indicative of a glial precursor phenotype whereas the NPC that remained within the ipsilateral hemisphere and failed to migrate towards contralateral tumor were negative for A2B5 (not shown). Of note, tumor-tropic NPC were also negative for the neuronal precursor marker E-NCAM and also did not express markers of neuronal differentiation (beta-III tubulin), oligodendroglial precursors (PDGFRα) or oligodendroglial differentiation (CNPase) [not shown] indicative of that fact that glioma-tropic NPC populations most likely consist of astrocytic precursors.
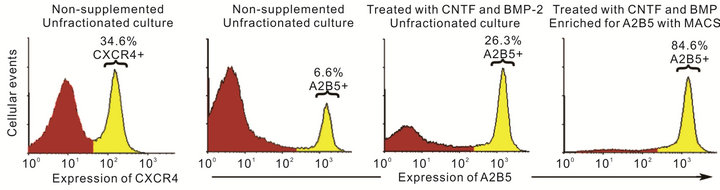
mediates the migratory potential of NPC in vitro [9]. Having identified glioma tropic precursor cells as being mainly glial precursor in phenotype (Figure 1(b) and [9]), we now wished to determine whether the enrichment of a double-selected population of NPC based on both of these characteristics may demonstrate superior tumor-directed migratory capacity. To this end, we utilized a magnetic and flow-cytometry based cell sorting techniques to enrich for subpopulations of NPC based on expression of A2B5 alone or a combination of A2B5 and CXCR4. As depicted in Figure 2, this methodology allowed us to successfully segregate these cells with good efficacy. We next investigated the tumor-directed migratory capacity of these cells by performing an in vitro migration assay quantitating their ability to migrate towards GL26 glioma-conditioned media using methodology similar to that described by us previously [9]. As demonstrated in Figure 3, we again noted that unfractionated NPC demonstrated enhanced tumor-tropic activity by displaying significantly greater migration towards tumorconditioned medium as opposed to non-neoplastic fibroblast conditioned-media. Most notably, however, we found that cells double-selected for A2B5 and CXCR4 expression demonstrated the most marked migration towards glioma medium (P < 0.01; Tukey HSD). Importantly, when A2B5+ CXCR4+ NPC were pre-treated with a CXCR4 neutralizing antibody, this enhanced migratory capability could be reduced to levels similar to that for A2B5+ single-sorted NPC but still remained superior to that of unfractionated NPC alone (P < 0.05; Tukey HSD). These findings confirm that it is possible to fractionate a selected subpopulation of A2B5+ CXCR4+ NPC and that these cells demonstrate an enhanced tumor-directed migratory capability as compared to that of unfractionated NPC.
4. Discussion
Malignant glioma remains one of the most intractable
 (a)
(a)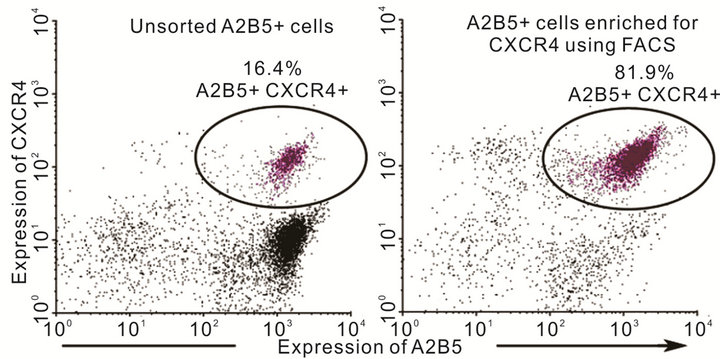 (b)
(b)
Figure 2. Enrichment of chemokine receptor expressing glial precursors from within fetal NPC. With the objective of enriching for a select population of glial precursors (identified on the basis of A2B5 expression) that were positive for the chemokine receptor CXCR4, we induced glial differentiation in NPC by supplementing cultures with recombinant CNTF and BMP-2. After 24 hours, cells were collected and stained with A2B5-specific primary and PE-conjugated secondary antibodies. The cells were then sorted using PE-targeted magnetic bead-based separation (MACS). As illustrated in (a), treatment of NPC cultures with CNTF and BMP-2 significantly increased the number of A2B5+ glial precursors. Following magnetic sorting, a highly enriched population of A2B5+ cells was obtained. Enriched glial precursors were then stained with a FITC-conjugated anti-CXCR4 antibody and the A2B5+ CXCR4+ fraction was separated using FACS. As demonstrated in (b). A significantly enriched population of A2B5+ CXCR4+ cells was thereby obtained.
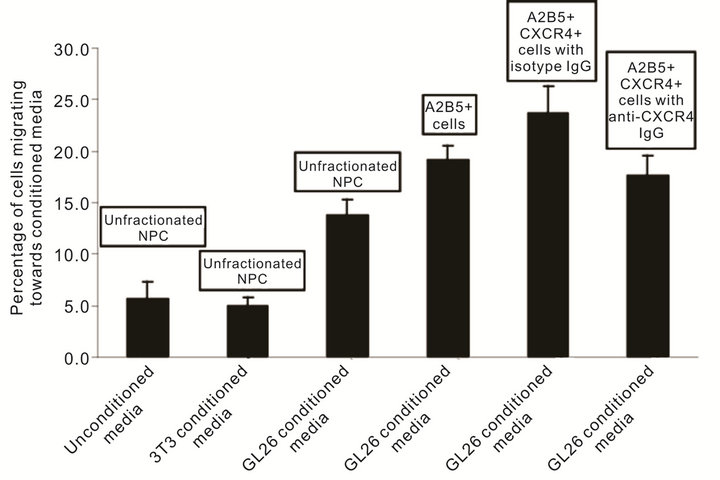
Figure 3. Chemokine receptor expressing glial precursors demonstrate enhanced glioma-tropic migration in vitro. On the basis of our earlier retrospective observation indicating that in vivo glioma-tropic NPC largely consisted of A2B5+ and CXCR4+ cells, we wished to prospectively assess whether NPC populations, that had been enriched for on the basis of these markers, demonstrated increased glioma-tropic migration. Using a two-chamber chemotaxis system, we assessed the migratory capacity of heterogenous unfractionated fetal murine NPC as well as fractionated populations consisting of either A2B5+ cells or a further enriched fraction of A2B5+ CXCR4+ double-positive cells. We observed that unfractionated NPC demonstrated negligible migration towards either unconditioned medium or media that had been conditioned by control cultured 3T3 fibroblasts. As expected, however, unfractionated NPC demonstrated significantly enhanced migration towards GL26 glioma cell cultured medium (P < 0.01; Tukey HSD). Of importance was our further finding that both the A2B5+ as well as A2B5+ CXCR4+ fractions demonstrated enhanced in vitro glioma-tropic migration compared to unfractionated NPC (P < 0.01 for both; Tukey HSD). The migration seen with A2B5+ CXCR4+ double-positive cells was greater than for A2B5+ alone (P < 0.05; Tukey HSD). The pre-treatment of A2B5+ CXCR4+ cells with an inhibitory CXCR4 specific antibody brought the migratory rate of these cells down to a level similar to that for A2B5+ single-positive cells. However, despite the inhibition of cell-surface CXCR4, the migratory potential of A2B5+ CXCR4+ double positive cells still remained higher than that for unfractionated NPC (P < 0.05; Tukey HSD). This indicates that the enhanced glioma-tropic potential of A2B5+ cells is likely to be dependent on additional chemotactic signaling mechanisms, apart from CXCR4. Furthermore, in NPC, the expression of CXCR4 and the presence of a glial precursor differentiation state (i.e. A2B5+) may act synergistically to enhance the patho-tropic migratory capacity of these cells.
human cancers. Current state-of-the art treatment consists of surgical resection in combination with radiation and chemotherapy. Despite notable advances in surgical navigation, imaging technology and adjuvant therapies, median survival for patients with this disease remains extremely poor with a five-year survival rate approaching zero [1]. The treatment-refractory nature of these tumors stems, to a significant extent, from their highly invasive nature, resulting in near universal recurrence of disease after therapy. As such, it is imperative to develop novel treatment strategies that focus specifically on targeting and elimination of the disseminated neoplastic burden within the brain. The use of tumor-tropic NPC offers such a potential approach and the glioma-directed migratory capacity of these cells has been described extensively in the literature by us as well as others [2-7,17]. Notable, however, has been our repeated finding that tumor-tropic activity appears to be limited to a sub-population of cells within NPC [9]. Certainly, from a translational perspective, where the distances that infiltrative tumor cells disseminate over within the brain, are logarithmically greater than the scale encountered within the rodent brain, it will be imperative to identify the biologic mechanisms that govern the ability of certain NPC populations to migrate towards distant tumor foci. With the goal of defining the phenotypic characteristics of the NPC that display glioma-directed migration, we have previously described using immunohistochemical analyses that tumor-tropic NPC populations express markers reflective of cells that have adopted an astroglial precursor fate-commitment. In addition, we separately detailed that NPC migrating towards glioma cells in vivo also express increased levels of the cell surface chemokine receptor CXCR4, and in vitro neutralization of CXCR4 correspondingly impairs the tumor-tropic activity of NPC. In this study, we now detail a methodology to enrich NPC for the phenotypic characteristics that we have previously identified as being associated with an enhanced glioma-tropic migratory activity. We describe how use ofa combination sorting strategy utilizing magnetic-bead and flow-based cell sorting techniques can allow for the separation of a double-selected CXCR4-expressing glial precursor cell population from within NPC and that these cells demonstrate increased migration towards gliomaconditioned medium in vitro. Taken together, these findings argue strongly towards further studies involving the selected use of a A2B5+ CXCR4+ subpopulation for in vivo glioma-directed therapies. Based on our findings, the use of this sub-population should enhance the targeting of disseminated glioma foci and improve upon our earlier reported therapeutic results using unfractionated NPC in pre-clinical glioma models.
5. Acknowledgements
The studies described in this paper were supported, in part, by NIH grant NS051557 (to M.E.).
REFERENCES
- T. S. Surawicz, F. Davis, S. Freels, E. R. Laws Jr. and H. R. Menck, “Brain Tumor Survival: Results from the National Cancer Data Base,” Journal of Neuro-Oncology, Vol. 40, No. 2, 1998, pp. 151-160. doi:10.1023/A:1006091608586
- K. S. Aboody, A. Brown, N. G. Rainov, et al., “Neural Stem Cells Display Extensive Tropism for Pathology in Adult Brain: Evidence from Intracranial Gliomas,” Proceedings of the National Academy of Sciences, Vol. 97, 2000, pp. 12846-12851. doi:10.1073/pnas.97.23.12846
- S. Benedetti, B. Pirola, B. Pollo, et al., “Gene Therapy of Experimental Brain Tumors Using Neural Progenitor Cells,” Nature Medicine, Vol. 6, 2000, pp. 447-450. doi:10.1038/74710
- M. Ehtesham, P. Kabos, K. L. Black and J. S. Yu, “Use of Neural Stem Cells as Therapeutic Vehicles for the Treatment of Malignant Glioma,” Expert Review of Neurotherapeutics, Vol. 3, No. 6, 2003, pp. 883-895. doi:10.1586/14737175.3.6.883
- M. Ehtesham, P. Kabos, M. A. Gutierrez, et al., “Induction of Glioblastoma Apoptosis Using Neural Stem CellMediated Delivery of Tumor Necrosis Factor-Related Apoptosis-Inducing Ligand,” Cancer Research, Vol. 62, No. 24, 2002, pp. 7170-7174.
- M. Ehtesham, P. Kabos, A. Kabosova, T. Neuman, K. L. Black and J. S. Yu, “The Use of Interleukin 12-Secreting Neural Stem Cells for the Treatment of Intracranial Glioma,” Cancer Research, Vol. 62, No. 20, 2002, pp. 5657-5663.
- U. Herrlinger, C. Woiciechowski, M. Sena-Esteves, et al., “Neural Precursor Cells for Delivery of ReplicationConditional HSV-1 Vectors to Intracerebral Gliomas,” Molecular Therapy, Vol. 1, No. 4, 2000, pp. 347-357. doi:10.1006/mthe.2000.0046
- K. Y. Mapara, C. B. Stevenson, R. C. Thompson and M. Ehtesham, “Stem Cells as Vehicles for the Treatment of Brain Cancer,” Neurosurgery Clinics of North America, Vol. 18, No. 1, 2007, pp. 71-80. doi:10.1016/j.nec.2006.10.001
- M. Ehtesham, X. Yuan, P. Kabos, et al., “Glioma Tropic Neural Stem Cells Consist of Astrocytic Precursors and Their Migratory Capacity Is Mediated by CXCR4,” Neoplasia, Vol. 6, No. 3, 2004, pp. 287-293. doi:10.1593/neo.03427
- F. Lazarini, T. N. Tham, P. Casanova, F. ArenzanaSeisdedos and M. Dubois-Dalcq, “Role of the AlphaChemokine Stromal Cell-Derived Factor (SDF-1) in the Developing and Mature Central Nervous System,” Glia, Vol. 42, No. 2, 2003, pp. 139-148. doi:10.1002/glia.10139
- K. Reiss, R. Mentlein, J. Sievers and D. Hartmann, “Stromal Cell-Derived Factor 1 Is Secreted by Meningeal Cells and Acts as Chemotactic Factor on Neuronal Stem Cells of the Cerebellar External Granular Layer,” Neuroscience, Vol. 115, No. 1, 2002, pp. 295-305. doi:10.1016/S0306-4522(02)00307-X
- S. Barbero, R. Bonavia, A. Bajetto, et al., “Stromal Cell-Derived Factor 1alpha Stimulates Human Glioblastoma Cell Growth through the Activation of Both Extracellular Signal-Regulated Kinases 1/2 and Akt,” Cancer Research, Vol. 63, No. 8, 2003, pp. 1969-1974.
- M. Ehtesham, J. A. Winston, P. Kabos and R. C. Thompson, “CXCR4 Expression Mediates Glioma Cell Invasiveness,” Oncogene, Vol. 25, No. 19, 2006, pp. 2801- 2806. doi:10.1038/sj.onc.1209302
- S. A. Rempel, S. Dudas, S. Ge and J. A. Gutierrez, “Identification and Localization of the Cytokine SDF1 and Its Receptor, CXC Chemokine Receptor 4, to Regions of Necrosis and Angiogenesis in Human Glioblastoma,” Clinical Cancer Research, Vol. 6, No. 1, 2000, pp. 102- 111.
- P. Rajan and R. D. McKay, “Multiple Routes to Astrocytic Differentiation in the CNS,” Journal of Neuroscience, Vol. 18, No. 10, 1998, pp. 3620-3629.
- M. C. Nunes, N. S. Roy, H. M. Keyoung, et al., “Identification and Isolation of Multipotential Neural Progenitor Cells from the Subcortical White Matter of the Adult Human Brain,” Nature Medicine, Vol. 9, 2003, pp. 439- 447. doi:10.1038/nm837
- M. Ehtesham, C. B. Stevenson and R. C. Thompson, “Stem Cell Therapies for Malignant Glioma,” Neurosurgical Focus, Vol. 19, No. 3, 2005, p. E5. doi:10.3171/foc.2005.19.3.6
NOTES
*Corresponding author.