Case Reports in Clinical Medicine
Vol.2 No.2(2013), Article ID:31765,5 pages DOI:10.4236/crcm.2013.22040
Cranial neuropathies, confusion, and ataxia— Challenges for diagnosis and therapy
![]()
1Department of Neurology, State University of New York, Upstate Medical Center, Syracuse, USA
2Department of Neurology, Edward Hines, Jr. VA Hospital, Hines, USA
3Department of Neurology, Loyola University Chicago Stritch School of Medicine, Maywood, USA; *Corresponding Author: morris.fisher@va.gov
Copyright © 2013 Ajay Sood et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 7 March 2013; revised 20 April 2013; accepted 4 May 2013
Keywords: Bickerstaff’s Brainstem Encephalitis; Botulism; Miller Fisher Syndrome; Opthalmpoplegia; Wernicke’s Encephalopathy
ABSTRACT
Introduction: The differential diagnosis of patients presenting with multiple cranial neuropathies, ataxia, and altered mentation is broad and includes immunologic, infectious, vasculitic and metabolic conditions. Primary considerations are Bickerstaff’s brainstem encephalitis (BBE), the Miller Fisher syndrome (MFS), Wernicke’s encephalopathy and botulism. The initial workup may be unrevealing. Timely treatment is imperative and unnecessary treatment can be associated with serious adverse reactions. Sensitivity to the decisions needed in such patients is therefore important. Case report: A 58-year-old male presented with symptoms of altered mental status, blurred vision, dysphagia and dysarthria, impaired pupillary responses to light, facial diplegia, ataxia, and decreased tendon reflexes after an episode of a self resolving diarrheal disease. Primary initial diagnostic concerns were Bickerstaff’s brainstem encephalitis (BBE), Miller Fisher syndrome (MFS), Wernicke’s encephalopathy and botulism. Initial work-up including cerebrospinal fluid analyses, imaging studies, and an electrodiagnostic examination did not provide information helpful for narrowing this differential. The patient was treated with botulinum antitoxin, thiamine and intravenous immune globulin (IVIG) before the results of specialized tests were available. The patient’s clinical condition improved. Retrospectively, the patient was diagnosed as BBE. Conclusion: This case emphasizes the difficulties in distinguishing between BBE, MFS, and botulism as well as demonstrating the complexities of treating such patients.
1. INTRODUCTION
The differential diagnosis of patients presenting with multiple cranial neuropathies, ataxia, and altered mentation is broad. This differential includes immunologically mediated, infectious, vasculitic and metabolic disorders. An accurate diagnosis requires use of multiple diagnostic modalities. Primary considerations for such patients would be Bickerstaff’s brainstem encephalitis (BBE), Miller Fisher syndrome (MFS), Wernicke’s encephalopathy, and botulism. BBE and MFS are thought to be autoimmune processes possibly due to antecedent viral infections. The clinical distinction between BBE and MFS can be challenging. A recent study of a large number of such patients has emphasized that these patients may be part of a continuous spectrum. Botulism is usually a food borne disease caused by ingestion of food contaminated with Clostridium botulinum producing toxins that block the release of acetylcholine (Ach) from motor nerve terminals. This can result in, severe muscle weakness commonly preceded by GI symptoms and with prominent bulbar signs. This report presents a case of a patient who presented with bulbar signs and ataxia that emphasizes the difficulties in distinguishing between BBE, MFS, and botulism as well as highlighting the complexities of treating such patients.
2. CASE REPORT
2.1. Patient History
A 58 years old right handed man presented with three days of blurring of vision, two days of nasal and slurred speech, one day of diplopia and falls. He had a past history of bipolar disorder, liver disease due to hepatitis C infection, and hyperlipidemia. At presentation, his gait was described as similar to that of someone with alcohol intoxication. He reported that one week prior to presentation he had an episode of self limiting diarrhea. He denied any flu like symptoms. He was taking lithium for a bipolar disease, benzodiazepine for anxiety, and methadone for a past history of substance abuse as well as opiates for chronic low back pain. He had no allergies and no family history of neurological disease. When first seen, he was afebrile with unremarkable vital signs and a normal general physical examination. His neurological exam showed poor attention, nasal dysarthria, dilated sluggishly reactive pupils, bilateral lateral rectus palsy with diplopia, decreased vibration and strength in the legs, depressed reflexes in the right arm and absent reflexes in the legs, absent Babinski responses, and a positive Romberg sign. He was ataxic in the arms and legs with a broad based, unsteady gait. The following day, the patient became increasingly drowsy and his dysarthria worsened. He also exhibited bifacial weakness as well as mild weakness of neck flexion. The patient was admitted to the intensive care unit. Swallowing evaluation showed prominent dysphagia and he was started on a dysphagia diet. He had trouble breathing. Serial negative inspiratory force and vital capacities were reduced but he did not require intubation.
2.2. Laboratory Studies
The results of his laboratory studies are shown in Table 1. His elevated gamma gluteryl transminase and ammonia were attributed to his chronic hepatitis C infection. In addition, tests were negative for HIV, syphilis, Lyme’s disease, West Nile virus, arboviruses, entero viruses, herpes simplex virus, cytomegalovirus, Epstein Barr virus B12, folate, and thiamine levels were within normal limits. A CT of the head, an MRI of the brain, and an MRA of the head and neck were unremarkable. Electrodiagnostic studies showed normal compound motor action potentials (CMAP) in the distribution of the left tibial, peroneal, median and ulnar nerves. Left tibial F wave latencies were mildly prolonged. Left ulnar F wave latencies were within normal limits. Sensory nerve conduction studies of the left sural, median and ulnar nerves were unremarkable in both conduction and amplitude. Needle EMG examination of selected muscles of the right arm and leg including the trapezius and cervical parapsinal did not reveal meaningful abnormalities. Slow (2 Hz) and rapid (20 Hz) repetitive stimulation studies recording from the abductor pollicis brevis and trapezius muscles did not reveal meaningful incremental or decreemental response.

Table 1. Laboratory studies.
2.3. Treatment and Course
The patient was treated with thiamine. He was also empirically treated with 5 days of intravenous immunoglobulin (IVIG) treatment before the results of stool cultures or anti-GQ1b testing were available. Even after five days of treatment of IVIG, the patient continued to deteriorate and he was closely monitored in the intensive care unit. The Center for Disease Control (CDC) was contacted for possible botulism. Based on discussions with the CDC, it was thought that the patient should be treated for botulism prior to the results of diagnostic tests. Anti-botulinum heptavalent horse serum was given in a single infusion. Two days after IVIG therapy and completion of botulinum antitoxin treatment, the patient started to improve. His confusion began to resolve. His speech became less dysarthric, ophthalmoplegia less prominent, and reflexes in the legs could be elicited. He was subsequently transferred to a rehabilitation facility primarily for therapy of his gait ataxia. Results of stool and serum studies from the CDC proved negative for botulinum toxin. Anti-GQ1b antibody titers were positive.
3. DISCUSSION
Given the constellation of the patient’s symptoms and findings including altered mental status, he was diagnosed retrospectively with BBE.
This patient emphasizes that accurate diagnoses of patients presenting with multiple cranial neuropathies can be challenging due to overlapping features. In our patient, primary considerations were BBE, MFS, Wernicke’s encephalopathy and botulism as they share common features. Clinical distinction of BBE from MFS is challenging. Some now consider both of these entities a part of the same spectrum [1]. IgG autoantibodies to GQ1b are strongly associated with the MFS as well as its central nervous variant BBE. BBE includes acute ataxia and opthalmoplegia as well as impaired consciousness. Common features are variable involvement of cranial nerves and ataxia. BBE has been associated with mental status changes whereas MFS has not [2].
In 1951 Bickerstaff and Cloak [3] described three patients who presented with drowsiness, ophthalmoplegia and ataxia thought due to “mesencephalitis and rhombencephalitis”. Further studies with similar patients included extensor plantar responses and hemisensory loss [4]. These patients have subsequently been referred to as BBE. In a series of 62 such patients [5], the most common symptoms for BBE included symmetrical external ophthalmoplegia (100%), ataxia (100%), alteration of consciousness (74%), a flaccid symmetrical tertaparesis (60%), a facial diplegia (45%), a Babinski sign (40%), internal opthalmoplegia (34%), and brisk tendon reflexes (34%). 30% of patients were reported to have an abnormal MRI although specific abnormalities were not described.
In 1956 [6], C. Miller Fisher described three cases of ophthalmoplegia, ataxia and arefexia. He postulated that this disorder was a variant of GBS due to the presence of arefexia and CSF albuminocytological dissociation. This condition has subsequently been called MFS. This is considered a variant of GBS since some of these patients may progress to this syndrome. Although not necessarily diagnostic, the presence of anti-GQ1b antibodies is characteristic. EMG abnormalities are usually relatively limited but abnormal sensory nerve conductions studies are consistent findings [7].
In a report of 18 patients, Al-Din et al. [8] argued that BBE is a distinct clinical entity with pathology originating in the CNS. Roper et al. [1], however, felt that 6 of these 18 patients fit the classical description of MFS and therefore BBE and MFS were a spectrum of the same clinical entity. A series of 581 cases of BBE and MFS has been described by Ito et al. [2]. In this study, BBE was diagnosed if the patients had altered consciousness; those with normal consciousness and areflexia were diagnosed as MFS. In both groups, greater than 50% of patients were positive for antiGQ1b with a somewhat higher percentage in the MFS group. Approximately onefifth of the patients in both groups were found to be positive for Campylobacter jejuni infection. There were a higher percentage of patients positive for CSF albuminocytologic dissociation in the BBE group as compared to the MFS group. MRI abnormalities were more common in BBE group but the incidence was only 10%. Ito et al. [2] thought that MFS and BBE should be considered as part of a continuum which should include patients difficult to definitely place in either of these categories.
Treatment for both MFS and BBE has included intravenous immune globulin (IVIG) and plasma exchange although there are no randomized control trials to evaluate the efficacy of such therapies. Multiple retrospective studies have suggested that IVIG and plasma exchange should be started as early as possible for BBE because of poor clinical prognosis; however these therapies do not alter the course of disease in MFS probably due to a generally good clinical outcome [9].
Botulism is usually food borne and caused by ingestion of inadequately sterilized contaminated foods. There are eight known types of toxins (A, B, Ca, Cb, D, E, F, and G) produced by the anaerobic bacterium Clostridium botulinum. The toxin blocks the release of Ach from motor nerve terminals [10]. This produces long-lasting, severe muscle paralysis symptoms usually starting 12 - 36 hours after ingestion. Muscle paralysis is usually preceded by GI symptoms. Clinical findings include diplopia, blurred vision, dysphagia and dysarthria, dysphonia, impaired pupillary responses to light, descending symmetrical flaccid paralysis, and variably reduced tendon reflex responses. Management involves supportive care and heptavalent antitoxin. Electrophysiological abnormalities in botulism tend to evolve with time and may not be present early in the disease. The electrodiagnostic findings are those associated with a presynaptic neuromuscular junction disorder. These abnormalities typically include reduced compound muscle action potential (CMAP) amplitudes, at least 20% facilitation of CMAP amplitude during tetanic stimulation or following sustained contraction, persistence of facilitation for at least two minutes after activation, and no post-activation exhaustion. Single fiber EMG demonstrates markedly increased jitter and blocking. Jitter and blocking may decrease as the firing rate increases [10]. Patients are generally treated with botulinium antitoxin which is derived from horse serum.
Ophthalmoplegia, ataxia, and a confusional state are the classic clinical triad of Wernicke’s encephalopathy due to thiamine deficiency usually from a poor nutritional state [11]. Treatment is thiamine replacement.
An accurate diagnosis of such conditions requires use of multiple diagnostic modalities including electrodiagnostic studies: brain imaging; x-ray studies; cerebrospinal fluid analysis; and, evaluation of serologic markers, body fluid and stool cultures. The laboratory tests from cerebrospinal fluid (CSF) studies may not be diagnostic for up to several weeks, and electrophysiological testing can be unrevealing for up to 4 weeks. Some of the studies require samples sent to specialized laboratories and can take up to several weeks. Our patient posed difficulties as to accurate and timely diagnosis and treatment. There are case reports of patients with overlapping presentations where the diagnoses and treatments posed a clinical challenge (Table 2). Although all of these patients improved clinically, some patients developed complications, some patients ended with multiple treatments, and some with treatment delays. A similar patient has been considered of such complexity and interest to warrant a recent discussion in the New York Times Magazine [12].
In addition, Critchley et al. [13] reported clinical features of 27 patients (age range 14 months to 74 years) identified in an outbreak of botulism due to the consumption of contaminated hazelnut yogurt in Lancashire, England and North Wales in 1989. The incubation period between ingestion of yogurt and the onset of symptoms ranged from few hours to 2 days. Clinical symptoms were consistent with botulism but included drowsiness in 16 of the 27 patients. All serological tests for botulism in the affected patients were negative; however the diagnosis of botulism was made by identification of Clostridium botulinum type B toxin in one tin of hazelnut puree, in the stool specimen of 1 patient, and in two cartons of hazelnut yogurt obtained from patients’ homes. As soon as the diagnosis had been made, all new patients received antitoxin. This case series emphasizes that drowsiness (16/27 patients) may be frequent in botulism. As in our patient, this could be confounding for an accurate diagnosis. This would be especially true when the serological studies for botulism are falsely negative.
This manuscript discuses the first patient reported with
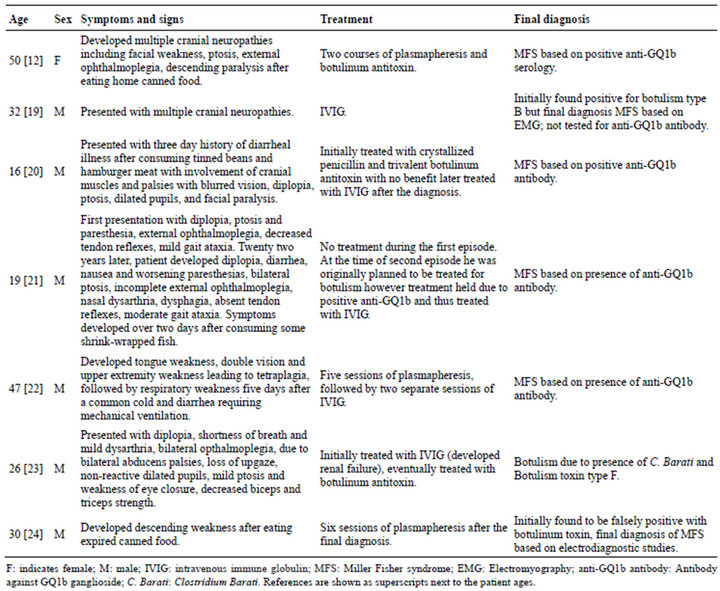
Table 2. Patients with overlapping findings for BBE, MFS, and botulism.
acute cranial neuropathies diagnosed in retrospect with BBE who was treated with both botulinum antitoxin and IVIG. Such treatment was arguably appropriate and reinforces the complexity of decision making in such patients. This issue is important since the early treatment can be critical but the treatments themselves not without potential complications. The response to botulism is best if given within first 24 hours of exposure [14], and the sooner the treatment, the better the outcome. There is, however, risk of anaphylaxis, thermal reactions, and serum sickness with botulinum antitoxin [15-17]. Treatment risks with IVIG particularly relevant for BBE include anaphylactic reactions, acute renal failure and thrombo-embolic events [18].
4. CONCLUSION
As is clear from the above case report, patients with acute multiple cranial neuropathies can pose a diagnostic challenge. Sensitivity to these issues is important since appropriate treatment can be life saving but such treatment itself has its own complexities.
REFERENCES
- Ropper, A.H. (1983). The CNS in Guillain-Barre syndrome. Archives of Neurology, 40, 397-398. doi:10.1001/archneur.1983.04050070027003
- Ito, M., et al. (2008) Bickerstaff’s brainstem encephalitis and Fisher syndrome form a continuous spectrum: Clinical analysis of 581 cases. Journal of Neurology, 25, 674- 682. doi:10.1007/s00415-008-0775-0
- Bickerstaff, E.R. and Cloake, P.C. (1951) Mesencephalitis and rhombencephalitis. British Medical Journal, 2, 77-81. doi:10.1136/bmj.2.4723.77
- Bickerstaff, E.R. (1957) Brain-stem encephalitis; further observations on a grave syndrome with benign prognosis. British Medical Journal, 1, 1384-1387. doi:10.1136/bmj.1.5032.1384
- Odaka, M., et al. (2003) Bickerstaff’s brainstem encephalitis: Clinical features of 62 cases and a subgroup associated with Guillain-Barre syndrome. Brain, 126, 2279- 2290. doi:10.1093/brain/awg233
- Fisher, M. (1956) An unusual variant of acute idiopathic polyneuritis (syndrome of ophthalmoplegia, ataxia and areflexia). New England Journal of Medicine, 25, 57-65. doi:10.1056/NEJM195607122550201
- Aranyi, Z., Kovacs, T., Sipos, I. and Bereczki, D. (2013) Miller Fisher syndrome: Brief overview and update with a focus on electrophysiological findings. European Journal of Neurology, 19, 15-20.
- Al-Din, A.N., Anderson, M., Bickerstaff, E.R. and Harvey, L. (1982) Brainstem encephalitis and the syndrome of Miller Fisher: A clinical study. Brain, 105, 481-495. doi:10.1093/brain/105.3.481
- Mori, M. and Kuwabara, S. (2011) Fisher syndrome. Current Treatment Options in Neurology, 13, 71-78. doi:10.1007/s11940-010-0103-8
- Sanders, D.B. and Howard Jr., J.F. (2008) Disorders of neuromuscular transmission. In: Bradley, W.G., Ed., Neurology in Clinical Practice, Butterworth-Heinemann/Elsevier, Philadelphia, 2283-2402.
- Aminoff, M.J., Greenberg D.A. and Simon, R.P. (2009) Disorders of cognitive function. In: Simon, R.P., Greenberg, D. and Aminoff, M.J., Eds., Clinical Neurology, McGraw-Hill, New York, 1-68.
- Sanders, L. (2011). The woman with creeping paralysis. The New York Times, 5 June 2011.
- Critchley, E.M., Hayes, P.J. and Isaacs, P.E. (1989) Outbreak of botulism in north west England and Wales. Lancet, 334, 849-853. doi:10.1016/S0140-6736(89)93010-9
- Tacket, C.O., Shandera. W.X., Mann, J.M., Hargrett, N.T. and Blake, P.A. (1984) Equine antitoxin use and other factors that predict outcome in type A foodborne botulism. American Journal of Medicine, 76, 794-798. doi:10.1016/0002-9343(84)90988-4
- Gottlieb, S.L., et al. (2007) Long-term outcomes of 217 botulism cases in the Republic of Georgia. Clinical Infectious Diseases, 45, 174-180. doi:10.1086/518890
- Sobel, J. (2005) Botulism. Clinical Infectious Diseases, 41, 1167-1173. doi:10.1086/444507
- Black, R.E. and Gunn, R.A. (1980) Hypersensitivity reactions associated with botulinal antitoxin. American Journal of Medicine, 69, 567-570. doi:10.1016/0002-9343(80)90469-6
- Katz, U., Achiron, A., Sherer, Y. and Shoenfeld, Y. (2007) Safety of intravenous immunoglobulin (IVIG) therapy. Autoimmunity Reviews, 6, 257-259. doi:10.1016/j.autrev.2006.08.011
- Chang, V.H. and Robinson, L.R. (2000) Serum positive botulism with neuropathic features. Archives of Physical Medicine and Rehabilitation, 8, 122-126.
- Boylu, E.E., Togrol, R.E., Senol, M.G. Ozdaē, M.F. and Saraçoğlu, M. (2010) Role of anti-GQ1B antibody in differential diagnosis of acute ophthalmoparesis. Journal of Neuropsychiatric Disease and Treatment, 6, 119-122. doi:10.2147/NDT.S8219
- Dörr, J., Dieste, F.J., Klaasen van Husen, D., Zipp, F. and Vogel, H.P. (2006) A case of recurrent Miller Fisher syndrome mimicking botulism. Neurological Sciences, 27, 424-425. doi:10.1007/s10072-006-0723-7
- Susuki, K., Takahashi, H., Yuki, N., Ohsawa, H., Hirata, K. and Sakata, I. (2001) Guillain-Barre syndrome mimicking botulism. Journal of Neurology, 248, 720-721. doi:10.1007/s004150170125
- Chadderdon, S.M., Hagg, D.S. and Jacaoby, D.B. (2004) Botulism secodary to Clostirdium baratii type F. Chest, 126, 947S-948S.
- Yeh, Y.-C., Lin, K.-P., Lai, K.-L., Lllang, J.-F. and Liao, K.-K. (2008). Serum false-positive in Guillain-Barré syndrome. Scholarly Research Exchange, 2008, 1-3. doi:10.3814/2008/430531