American Journal of Plant Sciences
Vol.5 No.1(2014), Article ID:41946,7 pages DOI:10.4236/ajps.2014.51015
Identification of F1 Cassava (Manihot esculenta Crantz) Progeny Using Microsatellite Markers and Capillary Electrophoresis
1National Crops Resources Research Institute, Kampala, Uganda; 2Makerere University, Kampala, Uganda; 3International Institute of Tropical Agriculture, Nairobi, Kenya.
Email: kyalivincent@gmail.com
Copyright © 2014 Kyaligonza Vincent et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Kyaligonza Vincent et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received August 27th, 2013; revised December 16th, 2013; accepted January 2nd, 2014
KEYWORDS
Cassava Crosses; Authenticity of F1 Progeny; Molecular Markers
ABSTRACT
Generation of genetic diversity is necessary in improving on the potential of cassava when faced with various biotic and abiotic challenges. Presently, cassava breeders are breeding for a number of traits, such as drought tolerance, early root bulking, yield, starch, beta-carotene, protein, dry matter, pest and disease resistance, by relying on genetic diversity that exists in manihot esculenta germplasm. Controlled pollination is one of the main methods used to generate genetic diversity in cassava. However, the process of controlled pollination especially in an open field is prone to contamination by illegitimate pollen right from the time of pollination, seed collection, nursery bed establishment to planting of the trials. Therefore, authentication of the progeny obtained from cassava crosses is very important for genetic studies. Twelve informative microsatellite markers were used to verify the authenticity of 364 F1 progeny thought to come from four controlled parental crosses. The transmission of each allele at nine microsatellite loci was tracked from parents to progeny in each of the four Namikonga-derived F1 cassava families. Out of the 364 F1 progeny, 317 (87.1%) were true-to-type, 44 (12.1%) were a product of self-pollination and 3 (0.8%) were a product of open pollination. The consistency of the results obtained using microsatellite markers makes this technique a reliable tool for assessing the purity of progeny generated from cassava crosses.
1. Introduction
Cassava (Manihot esculenta Crantz) is one of the most important and widely grown root crops in Africa, Asia and South America with a total production of over 250 million tonnes [1]. Cassava, once regarded as “food for the poor” has become a multipurpose crop that responds to the priorities of developing countries, to trends in the global economy and to the challenges of climate change [2].
Cassava is mainly grown for its starchy storage roots and thus plays a key role in the livelihood of resourcelimited farmers in tropical Africa where it serves both as a food security crop and a source of income generation [3].
Unfortunately, this important crop faces both biotic and abiotic threats which cause yield losses and hinder optimal cassava productivity. Cassava breeders have taken up the challenge to breed for improved varieties for traits such as yield, starch, proteins, early root bulking, beta-carotene, and pest and disease resistance by utilizing the available genetic variation in cassava germplasm [4,5].
Cassava breeding mostly focuses on phenotypic selection of the best performing clones as parents. Such selection can only be effective if the pedigree of clones is correctly identified. Progeny testing, in which individual parents are evaluated on the basis of the performance of their offspring, will not be valid if some of the offspring are illegitimate [6]. These phenotypic methods used in conventional breeding may not be reliable in verifying the authenticity of the progeny generated from cassava crosses due to the possibility of pollen contamination during crossing and human errors during seed collection, nursery bed establishment and planting in the field. If not checked early, this may become an inherent problem in breeding leading to false pedigree information and genotype mix-ups [7]. Thus, accurate identification of progeny from crosses is critical to cassava breeders for the integrity of a durable breeding program.
The use of genomic tools is one of effective method to overcome this problem of progeny identity. Quite a good number of genomic resources have been created to facilitate progress in plant breeding through the application of advanced molecular technologies for crop improvement [8]. Genomic tools such as molecular markers have been used to trace errors in progeny established for plant breeding [9]. In progeny testing, true progeny are detected by the presence of DNA sequences corresponding to both alleles contributed by the two parents [10]. Among several types of molecular markers, microsatellites or simple sequence repeats (SSRs) markers have been utilized in many applications in plant genetic and breeding such as characterization and clarification of parentage-offspring relationship as well as validation of genotype identity [11,12].
In cassava breeding, efforts have been made to verify the progeny generated from crosses using gel electrophoresis to separate the DNA fragments after running a PCR for SSR analysis [13,14]. Here the progeny are identified by comparing their banding pattern of the alleles with that of the parents. However, results from gel electrophoresis are prone to higher percentage errors due to the low resolution power which leads to detection of only a few alleles [15]. Of recent, capillary electrophoresis has been identified as a powerful analytical technique to overcome the challenges of gel electrophoresis of nucleic acids [16]. Capillary electrophoresis detects more alleles and provides higher discriminatory power than gel electrophoresis [15]. In this study, SSR markers from cassava [17,18] and capillary electrophoresis were used to confirm the identity of 364 F1 progeny generated from four cassava parental crosses.
2. Materials and Methods
2.1. Plant Materials
Five cassava parental genotypes were used to generate the progeny population (Table 1). Namikonga was used as the sole source of pollen and crossed to four genotypes. Hand pollination was done as described by Kawano, [19] at a cassava crossing block established at the National Crops Resources Research Institute, Uganda. The verification of F1 progeny using SSR markers was carried out at the Bioscience Eastern and Central African (BeCA) hub, Kenya.
2.2. Genomic DNA Extraction, Purification and Quantification
Genomic DNA was extracted from five parental genotypes and 364 F1 progeny following the CTAB procedure [20]. Briefly, fresh leaf samples were collected from young leaf lobes weighing about 0.2 g and ground to fine powder in liquid nitrogen. The powder was then transferred into a 1.5 ml sterile Eppendorf tube and DNA extracted using the CTAB protocol. The extracted DNA was purified by phenol extraction [21]. The quality and concentration of the extracted DNA of each sample was determined by taking the readings directly from a Nanodrop spectrophotometer ND-1000-v3.7 (Thermo Scientific) at absorbance ratios of 260/280 and 260/230. Gel electrophoresis was used to determine the quality of the DNA on 0.8% agarose gel in 1 × TAE (i.e. 40 mM Trisacetate buffer in 1 mM EDTA, pH adjusted to 8.3 with acetic acid). Electrophoresis was run at 80V for an hour. The gel was stained with gel red 3× (Biotium) and visualised under UV light (Syngene Bioimaging System). The DNA samples were diluted to a working concentration of 50 ng/µl by addition of appropriate amount of sterile water.
2.3. Genotyping the Parents and the Progeny
In this study, 30 SSR markers were used to screen the parents for polymorphism. These primer pairs were chosen based on their ability to form clear PCR products visualised on an agarose gel and the same optimized PCR conditions. Only markers which were identified to be polymorphic for the parents were selected and used to screen the entire F1 population to confirm genotype identity of each seedling. The forward primer of each of the SSR markers was labeled at the 5’ end of the oligonucleotide using fluorescent dyes (6-FAM, VIC, PET
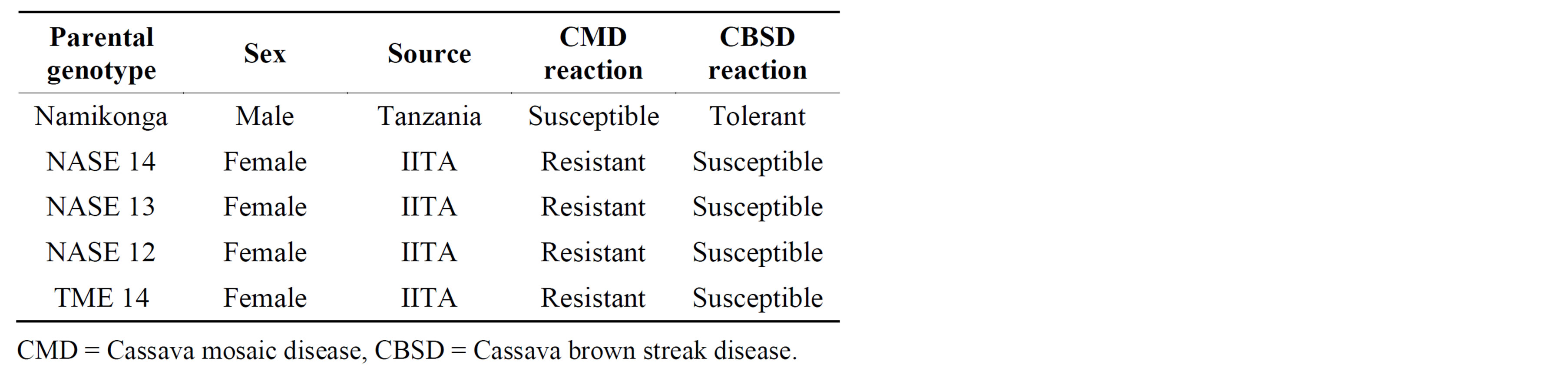
Table 1. Parental lines used to generate four full-sib F1 families.
and NED) to enable detection on the ABI 3730 capillary DNA Genetic Analyzer (Applied Biosystem).
Amplification of DNA samples was carried out in a GeneAmp®PCR System 9700 (Applied Biosystems Inc.). PCR reactions consisted of 10 μl volumes containing 1 µl of 50 ng genomic DNA, 1 µl of 1× reaction buffer (10 mM Tris-HCl at pH 8.3 and 50 mM KCl), 0.8 µl of 0.2 μM of each forward and reverse primer, 0.8 µl of 2.0 mM MgCl2, 0.8 µl of 0.2 mM of each deoxynucleotide triphosphates (dNTP), 2.3 µl of sterile water and 0.1 U Taq DNA polymerase. The thermocycling profile consisted of an initial denaturation step for 3 minutes at 95˚C, followed by 30 cycles of denaturation at 94˚C for 30 s, annealing at 57˚C for 1 minute and primer extension at 72˚C for 1 minute and a final extension cycle of 10 minutes at 72˚C. The success of amplification was checked on 2% agarose gel run in 1 × TBE buffer. Band sizes were determined by comparing with a 1 Kb ladder (Bioneer).
Evaluation of polymorphism and segregation of parental alleles was conducted using the ABI 3730 capillary DNA Genetic Analyzer (Applied Biosystem). The PCR products from each sample were co-loaded in three groups on the basis of dye colour and fragment size to ensure separation of each fragment in comparison with others. For fragment analysis, 1 µl of pooled PCR product was added to 9 µl of HIDI formamide/Liz500 size standard mix (ABI) prepared by adding 20 µl of Liz 500 to 900 µl of HIDI. Thereafter, the pooled PCR products together with standard mix were denatured at 95˚C for 3 minutes followed by cooling on ice for 5 minutes before capillary electrophoresis. The run conditions for fragment analysis were; run voltage of 15 Kv, sample injection voltage of 2 Kv for 10 s, run temperature of 66˚C and laser power of 25 mW.
3. Data Analysis
The amplified fragments were viewed, sized and binned using the Gene mapper v.3.7 software (Applied Biosystems). This software performs allele calls which include peak detection and fragment size matching. The data from the size standard (LIZ) is used to determine a standard curve plotting mobility of the fragments against the known size. Fragments arising from the PCR products were compared with the standard curve and their sizes determined.
For all the SSR markers used in the parent-offspring test, their respective alleles were scored for each progeny. If only maternal alleles were present, the progeny was considered to be a product of self-fertilization. If a non-parental allele was recorded together with a maternal allele, the progeny was considered to be a product of contamination by foreign pollen. If maternal and paternal alleles were present, then the progeny was considered to be a true progeny for the respective cross.
4. Results and Discussion
This study was aimed at identifying false progeny among four Namikonga-derived F1cassava families using SSR markers and capillary electrophoresis.
4.1. DNA Extraction and Quality
The CTAB method (Doyle and Doyle, 1987) was satisfactory for DNA extraction from all the five parents and the 364 F1 progeny. The quality of DNA was good (Figure 1), with A260/A280values ranging between 1.77 and 2.01. The concentration of DNA ranged between 152 and 5818 ng/µl.
4.2. Genotyping the Parents and the Progeny Using SSR Markers
Genomic DNA from the parents was amplified with 30 SSR primer pairs from the cassava genome. These primer pairs were chosen based on their ability to form clear PCR products on gel (Figure 2) and worked at the same optimized PCR conditions. Results based on gel electrophoresis could not clearly reveal polymorphism between the parents due to low resolution (Figure 2). When the PCR products were subjected to capillary electrophoresis, clear polymorphism between the parents was revealed.
Out of the 30 SSR primer pairs screened on the capillary ABI 3730 DNA Genetic Analyzer with parents of the four families, 18 SSR primer pairs (60%) were monomorphic (Figure 3) while 12 (40%) primer pairs were polymorphic between the parents (Table 2) and segregated in the respective F1 progeny (Figures 4 and 5).
Molecular marker analysis showed that from a total of 364 progenies tested, 317 (87.1%) were true-to-type F1s, 44 (12.1%) were selfs and three (0.8%) were a product of open pollination. The highest percentage of true-to-type F1 progeny (88.8%) was obtained from the family NASE 14 × Namikonga, while the lowest (77.3%) was obtained in the family NASE 13 × Namikonga (Table 3). All progeny considered true-to-type had both paternal and maternal alleles and existed as heterozygotes at all marker loci (Figures 4 and 5). The presence of a Namikonga allele in the form of the heterozygote in the progeny of any of the four Namikonga-derived families indicated that the original bi-parental cross succeeded. Since the progeny were derived from bi-parental crosses, all the selfs came from the female parents. However, the presence of 1.2% off-to-type F1 progeny which had foreign alleles in addition to the maternal allele at all the tested maker loci in the family NASE 14 × Namikonga-

Figure 1. Gel photo showing the quality of nine DNA samples. (M = 1Kb ladder, 1 - 5 = parents, 6 - 9 = progeny).
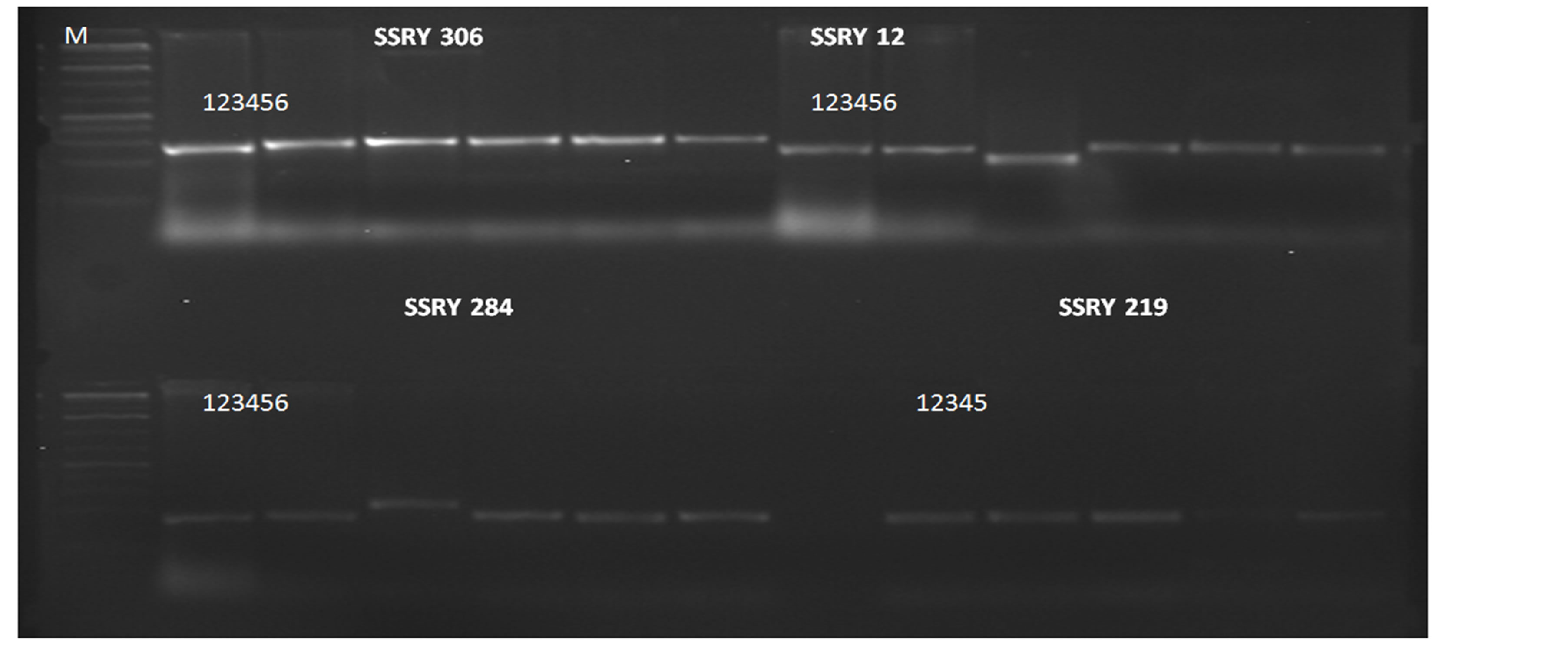
Figure 2. Banding pattern of SSRY 306, SSRY 12, SSRY 284, SSRY 219. Sample 1 = Namikonga, 2 = NASE 14, 3 = TME 14, 4 = NASE 12, 5 = NASE 13 and 6 = F1 progeny.

Figure 3. Electropherogram showing marker SSRY 64 monomorphic for all parents.
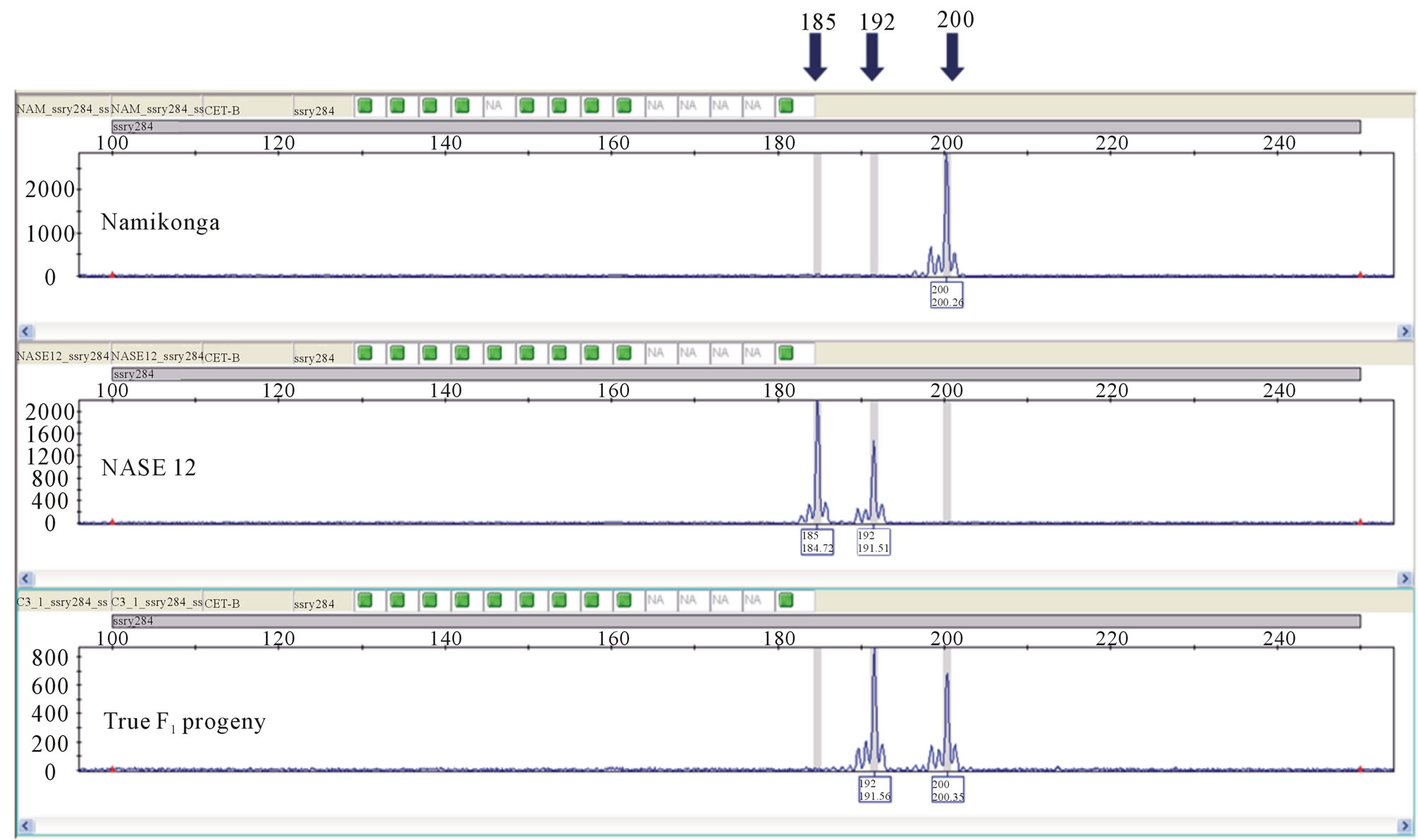
Figure 4. Electropherogram showing the segregation of parental alleles in an F1 progeny using marker SSRY 284. Namikonga (200, 200), NASE 12 (185, 192) and progeny (192, 200).
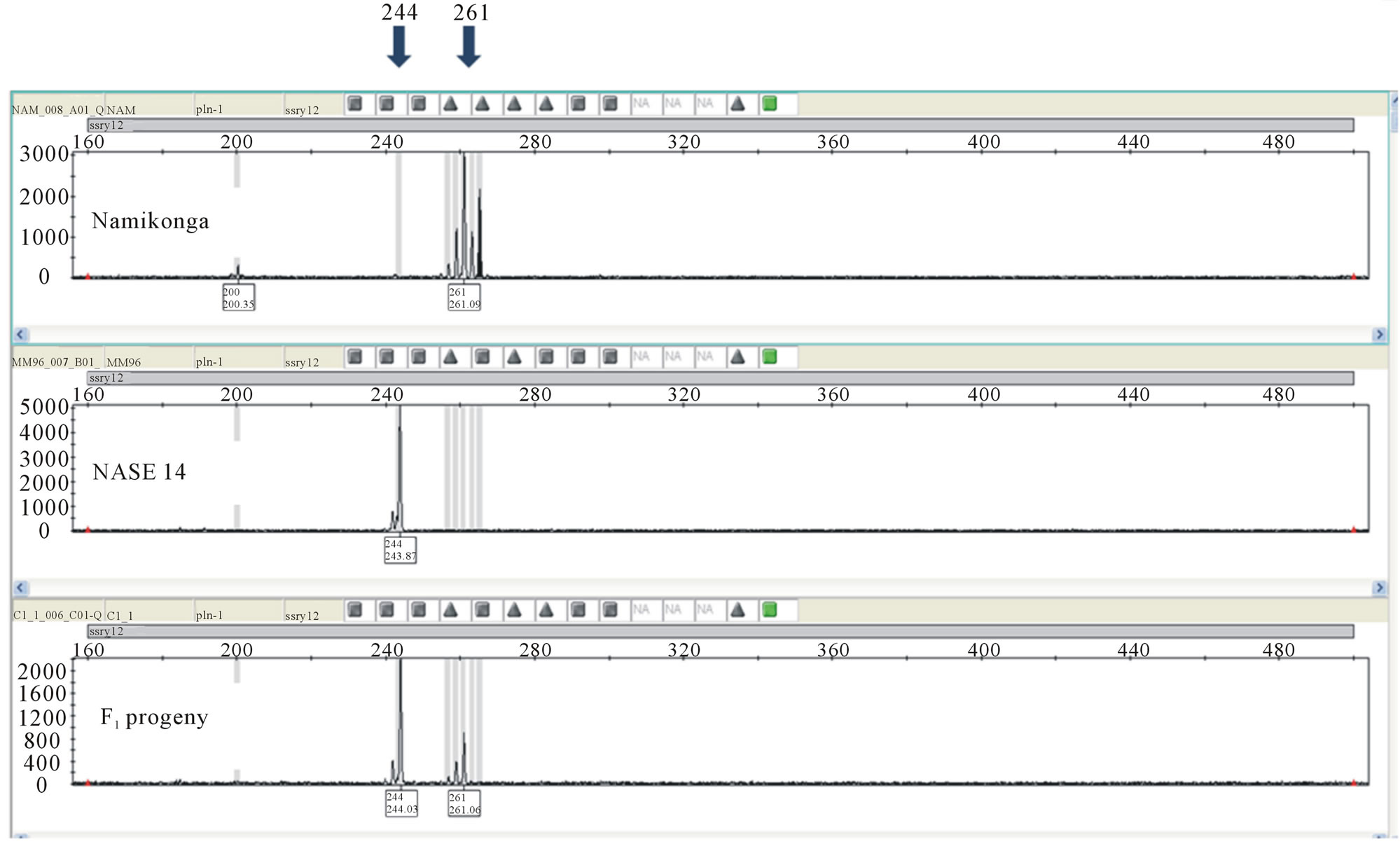
Figure 5. Electropherogram showing the segregation of parental alleles in an F1 progeny using marker SSRY 12. Namikonga (261, 261), NASE 14 (244, 244) and progeny (244, 261).
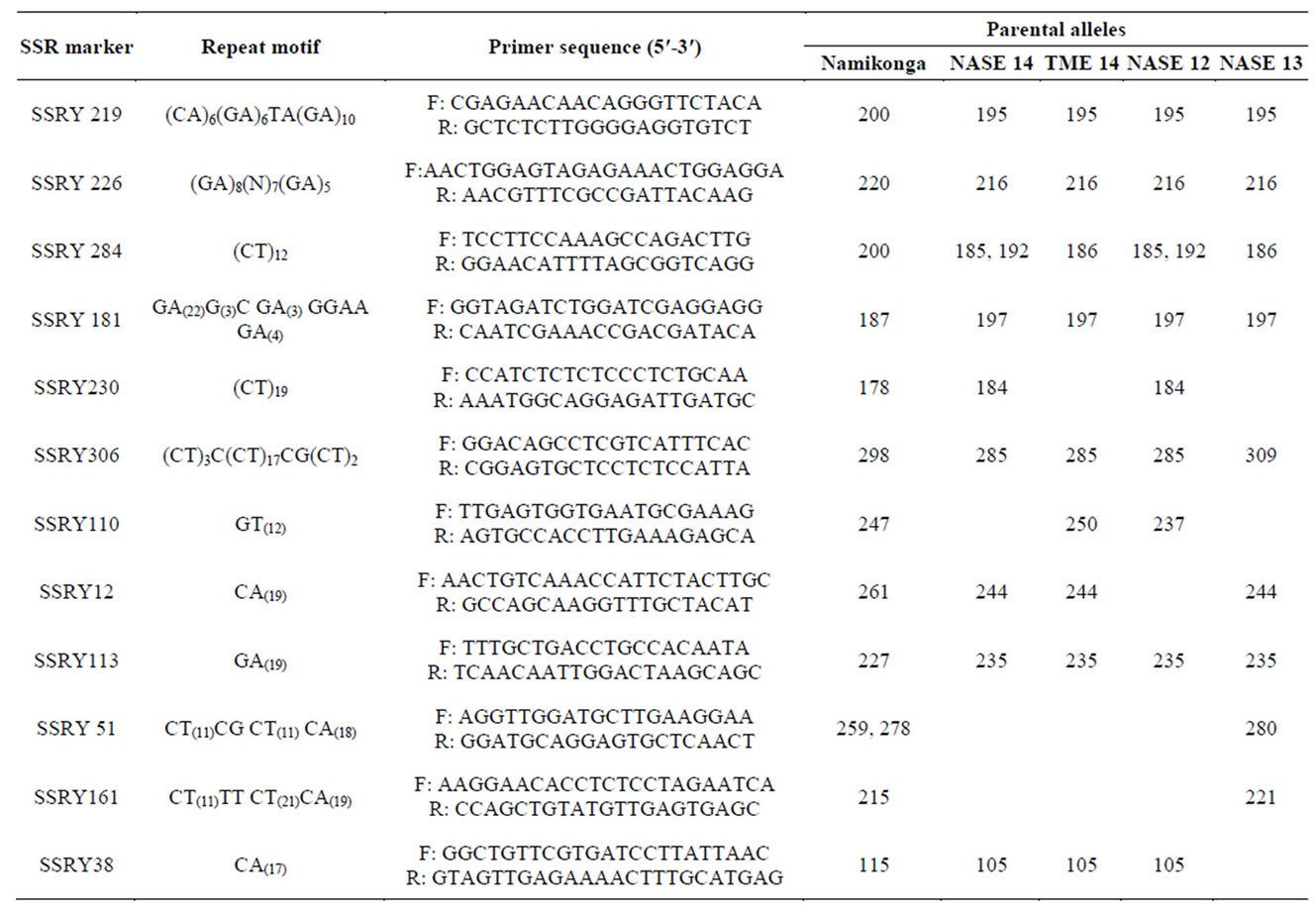
Table 2. Polymorphic SSR markers used in progeny test.

Table 3. Results for progeny test using SSR markers.
(Table 3) demonstrates that there were other genotypes that caused pollen contamination besides the female parent.
5. Conclusion
To verify the authenticity of the obtained progeny, all the F1 seedlings were genotyped using nine polymorphic SSR markers from the cassava genome. Results from the progeny test indicated that there were some genotype mix-ups due to pollen contamination during genetic crosses. Twelve point nine percent of the progeny were off-types. This contamination was caused by selfing (pollen from female parents) and out-crossing (pollen from unintended parents). The polymorphism generated by the SSR primers was useful for authenticating the F1 genotypes. This also highlights the obstacles of cassava breeding i.e. errors in pollinations. This is one of the few studies that has confirmed the true-to-type identity of progeny prior to genetic analysis. It is desirable that such steps become a common practice. The consistency of the data obtained using these SSRs in cassava shows that this technique is efficient for assessing the purity of progeny generated from crosses.
Acknowledgements
The authors acknowledge the International Institute of Tropical Agriculture (IITA) that provided the financial support through the National Agricultural Research Organization (NARO). We are grateful to the team at Biosciences eastern and central Africa (BeCA) hub and at the National Crops Resources Research Institute (NaCRRI) for their supports.
REFERENCES
- FAOSTAT, “Cassava Production,” 2013. http://faostat3.fao.org/home/index.html#SEARCH_DATA
- FAO, “Cassava, a 21st Century Crop,” Save and Grow Cassava; A Guide to Sustainable Production Intensification, Food and Agriculture Organization of the United Nations, Rome, 2013, pp. 1-18.
- FAO, “Why Cassava?” 2008. http://www.fao.org/ag/agp/agpc/gcds/
- New Agriculturist, “Green Light for Yellow Cassava,” 2012. http://www.new-ag.info/en/developments/devItem.php?a=2402
- N. Nagib and O. Rodomiro, “Breeding Cassava to Feed the Poor,” Scientific American, 2010, pp. 78-84.
- R. Corley, “Illegitimacy in Oil Palm Breeding,” Journal of Oil Palm Research, Vol. 17, 2005, pp. 64-69.
- D. P. Khasa, S. Nadeem, B. Thomas, A. Robertson and J. Bousquet, “Application of SSR Markers for Parentage Analysis of Populus Clones,” Forest Genetics, Vol. 10, No. 4, 2003, pp. 73-281.
- M. Ferguson, J. Sarah, J. Timothy, W. Steve, D. Christopher, Y. Joe, P. Marri, R. Ismail and P. Etienne, “Identification, Validation and High-Throughput Genotyping of Transcribed Gene SNPs in Cassava,” Theoretical and Applied Genetics, Vol. 124, 2011, pp. 685-695. http://dx.doi.org/10.1007/s00122-011-1739-9
- S. Riaz, S. Vezzulli, E. Harbertson and M. A. Walker, “Use of Molecular Markers to Correct Grape Breeding Errors and Determine the Identity of Novel Sources of Resistance to Xiphinemaindex and Pierceûs Disease,” American Journal of Enology and Viticulture, Vol. 58, 2007, pp. 494-498.
- N. Manigbas and L. Villegas, “Microsatellite Markers in Hybridity Tests to Identify True Hybrids of Sugarcane,” Philippine Journal of Crop Science, Vol. 29, 2004, pp. 23-32.
- S. Gomez, N. Denwar, T. Ramasubramanian, C. Simpson, G. Burow, J. Burke, N. Puppala and M. Burow, “Identification of Peanut Hybrids Using Microsatellite Markers and Horizontal Polyacrylamide Gel Electrophoresi,” Peanut Science, Vol. 35, No. 2, 2008, pp. 123-129. http://dx.doi.org/10.3146/PS07-109.1
- J.-F. Li, G.-B. Ma and L. Xu, “SSR Markers for Identification of Purity of Melon Hybrids,” Chinese Journal of Agricultural Biotechnology, Vol. 5, No. 3, 2008, pp. 223- 229. http://dx.doi.org/10.1017/S147923620800243X
- C. Mohan, P. Shanmugasundaram and N. Senthil, “Identification of True Hybrid Progenies in Cassava Using Simple Sequence Repeat (SSR) Markers,” Bangladesh Journal of Botany, Vol. 42, No. 1, 2013, pp. 155-159. http://dx.doi.org/10.3329/bjb.v42i1.15906
- G. Otti, F. Ayodele, A. Ikpan and G. Melaku, “Development of Genomic Tools for Verification of Hybrids and Selfed Progenies in Cassava (Manihot Esculenta),” African Journal of Biotechnology, Vol. 10, 2011, pp. 17400- 17408.
- G. Vinay, D. Grant, E. Alan, J. Philip and G. Bryan, “Gel versus Capillary Electrophoresis Genotyping for Categorizing Treatment Outcomes in Two Anti-Malarial Trials in Uganda,” Malaria Journal, 2010, pp. 9-19.
- J. Butler, E. Buel, C. Federica and R. Bruce, “Forensic DNA Typing by Capillary Electrophoresis Using the ABI Prism 310 and 3100 Genetic Analyzers for STR Analysis,” Electrophoresis, Vol. 25, 2004, pp. 1397-1412. http://dx.doi.org/10.1002/elps.200305822
- P. Chavarriaga-Aguirre, M. M. Maya, M. W. Bonierbale, S. Kresovich, M. A. Fregene, J. Tohme and G. Kochert, “Microsatellites in Cassava (Manihot esculenta Crantz): Discovery, Inheritance and Variability,” Theoretical and Applied Genetics, Vol. 97, 1998, pp. 493-501. http://dx.doi.org/10.1007/s001220050922
- R. E. C. Mba, P. Stephenson, K. Edwards, S. Melzer, J. Mkumbira, U. Gullberg, K. Apel, M. Gale, J. Tohme and M. Fregene, “Simple Sequence Repeat (SSR) Markers Survey of the Cassava (Manihot esculenta Crantz) Genome: Towards an SSR-Based Molecular Genetic Map of Cassava,” Theoretical and Applied Genetics, Vol. 102, 2001, pp. 21-31. http://dx.doi.org/10.1007/s001220051614
- K. Kawano, “Cassava,” In: W. R. Fehr and H. H. Hadley, Eds., Hybridization of Crop Plants, American Society of Agronomy, Madison, 1980, pp. 225-233.
- J. J. Doyle and J. L. Doyle, “A Rapid DNA Isolation Procedure for Small Quantities of Fresh Leaf Tissue,” Phytochemical Bulletin, Vol. 19, 1987, pp. 11-15.
- R. Powell and F. Gannon, “Purification of DNA by Phenol Extraction and Ethanol Precipitation,” Oxford Practical Approach Series, Oxford University Press, Oxford, 2002.