Neuroscience & Medicine
Vol.2 No.4(2011), Article ID:8873,13 pages DOI:10.4236/nm.2011.24053
The Functions of the Amyloid Precursor Protein Gene and Its Derivative Peptides:III Pharmacological Studies*
![]()
Neurodegenerative Disorders Research Pty Ltd., Subiaco, Australia.
E-mail: #research@ndr.org.au
Received September 27th, 2011; revised November 6th, 2011; accepted November 20th, 2011.
Keywords: Amyloid Precursor Protein, APP, Genes, Dementia, Alzheimer’s Disease
ABSTRACT
Pharmacological studies reveal APP and Aβ have interactions with glutamate and calcium, cytokines, copper/zinc chelators, secretases and presenilins, nicotinic receptors, acetycholinesterase, neurotrophins, non-steroidal anti-inflamematory drugs, monoclonal antibodies to Aβ, protease inhibitors, oestrogen, homocysteine, immediate early genes such as c-fos or c-jun and cholesterol. These functional and pharmacological observations highlight the need for greater understanding of APP and Aβ in brain function and have implications for clinical trials.
1. Pharmacological Studies
1.1. Glutamate and Calcium
Glutamate activates ion channel family receptors and G protein coupled receptors which modulate excitatory synaptic transmission through transduction pathways [1]. The ion channel family may be subclassified depending on their selective agonists: N-methyl-D-aspartate (NMDA), α-Amino-3-hydroxy-S-methylisoxazale-4-proprionic acid (AMPA) and kainic acid.
The glutamatergic system has been implicated in the pathogenesis of AD through an interaction which enhances the neurotoxicity of the amyloidogenic fragment Aβ of the APP gene [2] and the mechanisms of excitotoxic neuronal death involving increases in intracellular Ca2+ and neuronal depolarization [3]. An important functional relationship may exist between glutamate, the APP gene and calcium. The interaction between these three factors may be important in the development of neuro degenerative disorders. The embryogenesis of the nervous system signal transduction pathways are involved in controlling growth cone function, synaptogenesis and natural cell death in the so-called neurodevelopmental triad. Glutamate and its receptor systems and the APP gene, particularly in its Aβ form interact [4]. Calcium is the second messenger mediating rapid (e.g., modelling of microtubular and cytoskeletal proteins) and delayed reactions (e.g., transcriptional regulation of neurotrophins). Soluble forms of APP, sAPPα and sAPPβ probably act through cyclic GMP which encourages potassium channels and reduced Ca2+ in the acute phase [4,5]. Some of the delayed actions of soluble APPs might involve regulation of gene expression through transcription factor NF-kappa-B. That is, soluble APPs counteract the effects of glutamate. The balance between these two opposing forces is critical in the formation of the nervous system and in neurodegeneration where there might also be a role for cyclic GMP in modulating glucose and glutamate transporter mechanisms in synaptosomes [6].
The amyloidogenic Aβ 1-42, the amyloidogenic peptide product of the amyloid precursor protein damages and kills neurons possibly through an effect on the membrane lipid peroxidation, impaired ion-motive ATPases, glucose and glutamate transporters making nerve cells vulnerable to the excitotoxic effects of glutamate [7]. The effects of calcium on mitochondrial function might be critical in apoptosis and necrosis [8]. Calbindin-D28K might stabilise the effects of intracellular calcium and protect against apoptosis in neurotoxicity [9].
Soluble forms of the amyloid precursor protein may inhibit the damaging effects of presenilin 1 mutations by influencing the effects of NF-kappa-B and calcium homeostasis induced by these mutations [10]. This too might work through a mechanism involving mitochondrial toxicity. TNF α expression increases NF-kappa-B which might protect neurons from NMDA and AMPA kainate induced currents [11]. Soluble APPs might stimulate astroglial excitatory amino acid transport to protect the brain against glutamate induced excitotoxicity [12]. sAPPα has been shown to decrease NMDA currents in hippocampal neurons through a mechanism probably involving cGMP [11]. Aβ can damage neurons and render them vulnerable to excitotoxicity and the soluble APP derivatives can protect nerve cells from this process [13,14].
Apolipoprotein E, a risk factor for AD, might increase intracellular calcium and through the NMDA mechanism might damage neurons and encourage calcium influx [14]. The changes in Ca2+ in the endoplasmic reticulum may be critical in apoptosis and neuronal death secondary to excitoxicity [15].
Two studies have reported that NMDA receptors are absent or decreased from cortical and hippocampal regions in AD [16,17]. Other investigations suggest that the degree of cell loss is the reason for this receptor reduction [18,19]. The binding of L-3H-glutamate to the NMDA receptor on synaptic membranes from the hippocampus, fronto-temporal cortex and parietal cortex was unchanged in comparison to the reduction achieved in D-3H-aspartate binding [20]. An assessment of 3HMK801 binding, which is a non-competitive antagonist for the glutamate site of NMDA receptor, was reduced with marked intersubject variability [21].
Studies have investigated the expression of glutamate receptor genes using in situ hybridisation in human disorders and experimental animal models. In a transgenic mouse model carrying the mutated APP gene (the Swedish double mutation) there was no significant change in NR1 mRNA or autoradiographic receptor binding in the hippocampus and other brain regions [22].
There is intriguing molecular diversity within the NMDA receptor system which implies that therapy for neurological disorders might be directed towards specific receptors in localized brain regions in cells involving specific heteromers of the NMDA receptor [23]. Given that NR1 subunit is the fundamental subunit of NMDA receptor channels insight into its functional changes might yield information as to its role in the pathogenesis of AD. The hypothesis was tested that the NMDA receptor is involved in the pathogenesis of AD using in situ hybridization as a means of measuring the expression of the mRNA of the universal subunit of the NMDA receptor NR1. This approach has the advantage of assessing gene function irrespective of changes in protein degradation and catabolism and that anatomical differences in gene expression may be readily quantified [24]. There were trends to a reduction in NR1 in the hippocampus and increased NR1 within the frontal and superficial temporal gyrus which were not significant. There was variation within and between all patients with and without AD in the magnitude of NR1 expression in all anatomical regions studied. The findings suggest heterogeneity in the involvement of the post-synaptic glutamatergic system in AD [24].
Amyloid precursor-like protein 2 (APLP2) belongs to the same family of proteins as APP. APLP2 expression was observed in rat cortical neurones after treatment with glutamate. Lactate dehydrogenase was present in the medium which is indicative of neuronal damage but APLP2 expression was diminished. These observations were not seen with N-methyl-D-aspartate receptor antagonist pre-treatment (MK-801) [25].
In vitro, glutamate receptors promote the nonamyloidogenic APP processing pathway (sAPP). In vivo, intrahippocampal injection of guinea pigs with mGluR agonist 1S, 3R-ACPD resulted in CA1 neurodegeneration. Intraneuronal granules were found in degenerating neurons of the hippocampus after immunocytochemistry with Aβ antibodies. Guinea pigs injected with NMDA displayed neurodegeneration with immunoreactivity to Aβ [26].
The possibility that modification of the glutametegic system might be useful in AD has been supported by the beneficial effects of memantine, a non-competitive NMDA antagonist which leads to functional improvement, reduces dependence and clinical deterioration in patients with moderate to severe AD [27,28].
1.2. Cytokines
A number of in vitro studies have suggested an interaction between interleukin 1 (IL-1) and APP. IL-1 has been shown to stimulate the APP promoter [29]; to elevate APP mRNA in human endothelial cells [30,31]; and to increase neuronal APP mRNA in synergistic interaction with IL-6 [32]. Systemic injections of lipopolysaccharide have also been shown to increase IL-1 and IL-6 mRNAs in the cerebellum of mice, associated with elevation of Kunitz Protease Inhibitor isoforms of APP (KPI+) [33]. This study also found that mice with the stagger mutation, which have no Purkinje cells, have increased APP KPI+. Elevation of both IL-1 and APP KPI+ have been demonstrated in glial cells from human temporal lobectomy specimens removed from patients with refractory complex partial seizures [34]. The effects of rhIL-1ra on neuronal survival after kainic acid and its effect on expression of mRNAs for KPI– and KPI+ isoforms of APP and the glial fibrillary acid protein (GFAP)—a measure of astrocyte activation—have been investigated [35]. The cytokine rhIL-1ra prevented neuronal death after kainic acid and secondary changes in APP and GFAP mRNAs in some brain regions by mechanisms independent of inhibition seizure activity and modification of physiological variables [35]. Cytokine manipulation might yield treatments in AD.
1.3. Copper/Zinc Chelators: Transition Metals
The transition metal ions Cu2+, Zn2+ and Fe3+, are found at elevated levels in the neocortex of AD brains and in higher concentrations within the amyloid plaques [36]. In vitro, metals such as Cu2+, Zn2+ and to a lesser extent Fe3+, elevate synthetic Aβ aggregation [37]. Aβ binds to these metals in vitro which exist in higher levels in the AD brain [37-39].
Treatment with metal chelators induced Aβ aggregates in vitro resulting in the solubilization of Aβ [39,40]. Aβ was also solubilized by metal chelators in post-mortem brain tissue [41]. Therapeutic agents that specifically chelate the Cu2+ and Zn2+ in the neocortex might be valuable in the treatment of AD. Clioquinol (a Cu2+/Zn2+ chelator) was administered to transgenic mice resulting in an increase in soluble Aβ whereas APP, and synaptophysin were unaffected [42].
The specific reduction of APP-bound copper (II) to copper (I) by APP [43] results in lipid and protein damage [44] and oxidative stress in a cell-free system [45,46]. The effect of copper on APP knock-out (APP–/–) and wild-type (WT) mouse neurons was compared as to whether APP and copper interact to create oxidative stress in neurons [47]. WT neurons were specifically affected by copper concentrations equivalent to physiological conditions more than APP–/–. Oxidative stress was observed through increased levels of peroxidation products. These findings suggest that there is a specific copper-APP interaction which has important applications in AD. Treatment with Clioquinol, a metal protein binding substance that inhibits zinc and copper ions, has been shown to slow cognitive decline in a small sample of AD patients [48]. APP might function as an iron export ferroxidase whose function is inhibited by zinc [49].
1.4. Presenilins, Notch, Secretases
Presenilin has homology to Notch genes. Notch genes are involved in intracellular signalling and development, and may have important roles in the physiological regulation of differentiation within the haemopoietic system— functional properties which may limit the development of compounds which antagonize the actions of presenilin proteins. Mutagenesis experiments of two transmembrane aspartates in PS1 and PS2 abrogate γ-secretase activity and the production of Aβ, suggesting that aspartate sites are critical in the proteolytic cleavage of APP [50]. PS1 mRNA is found in the same neurons as APP [51], PS1 has been identified in the endoplasmic reticulum and Golgi apparatus, and N-terminal fragments are found in the synaptic organelles [52].
There may be a stoichometric interaction between APP and presenilin as both of these proteins form complexes with each other in living cells [53,54]. There may also be an interaction between these proteins at the cell surface which may be important in cell-cell adhesion and signalling which might activate tyrosine kinase [55]. Compounds which modulate the function of presenilins and the γ-secretase are potential treatments for AD.
β-secretase activity could be targeted therapeutically in AD, such that its inhibition would lead to decreased levels of Aβ. A study of 61 AD patients and 33 controls measured the cleavage of APP β-amyloid fragment by β-secretase (BACE) in frontal temporal and cerebellar regions [56]. BACE activity was increased in the temporal and frontal cortex but not in the cerebellum where Aβ plaques do not aggregate; the duration of disease was proportional to the activity of BACE in the temporal cortex.
1.5. Nicotinic Receptors and Acetylcholinesterase in AD
Cholinergic activity is known to be hypoactive in most regions of an AD brain and substances which inhibit acetylcholinesterase (AChE) are now used as a treatment for mild to moderate AD [57-63]. AchE expression is higher in and around neuritic plaques of the AD brain. Beta-actin promoter was used in transgenic mouse brain to increase the level of the APP C-terminal 100 amino fragment (APP CT100). APP CT100 and Aβ levels were increased in the brain along with AchE isoforms. AchE was shown to increase with increasing Aβ [64]. Choline acetyltransferase levels were measured in patients with no cognitive impairment, mild cognitive impairment, mild AD, and late AD. Acetylcholine activity was lower in late AD patients but higher in the superior frontal cortex of mild cognitive impaired patients [65]. This finding might explain some of the variable results obtained with cholinesterase inhibitors.
Cleavage of APP can result in either soluble APP or insoluble APP (Aβ-component of neuritic plaques). It is thought that the pathogenesis of AD results from low levels of soluble APP and protein kinase C (PKC). One study with primary cultures rat basal forebrain found that AChE inhibitors (ambenonium and metrifonate) increased PKC levels and cell-associated APP levels in cells and in the medium. The increase in PKC levels and cell-associated APP levels results in an increase in α-secretase activity resulting in an elevation of N-terminal APP, reducing Aβ [57,58,66-68].
1.6. Neurotrophins
Nerve growth factor (NGF) has been shown to be influential in APP processing in vitro. NGF drug treatments could limit cholinergic hypoactivity in the cortex of AD affected brains. TrkA (tyrosine kinase A NGF receptor) has been shown to increase APP processing and p75NTR (neurotrophin receptor) affects APP transcription [69]. This non-amyloidogenic APP processing pathway involving TrkA and p75 NTR might stimulate cholinergic activity.
1.7. Non Steroidal Anti Inflammatory Drugs and Related Substances
Ibuprofen is a non-steroidal anti-inflammatory drug (NSAID) that delays amyloid deposition in transgenic mice [70]. Reduced IL-1β levels and a reduction in the total area of Aβ deposition after 6 months of continuous administration of ibuprofen were observed.
Nonsteroidal anti-inflammatory drugs influence the development of AD [71]. An inverse proportional relationship between the duration of NSAID use and the onset of AD has been observed.
APP processing and formation of amyloidogenic APP holoprotein is enhanced by neurotransmitters such as prostaglandins and norepinephrine by elevating cellular cAMP levels. The conversion of APP to its soluble form is enhanced by neurotransmitters that stimulate phosphatidylinositol hydrolysis by activating muscarinic, serotoninergic or metabotropic glutamate receptors. A study has found that some neuroimmunophilin ligands (cyclosporin A and FK506) inhibited the over expression of APP by prostaglandin E2. This in turn reduced the synthesis of amyloidogenic APP holoprotein. Nonsteroidal inflammatory agents and cyclooxygenase-2 (COX- 2) inhibitors might inhibit the accumulation of amyloid plaques in AD by reducing the levels of amyloidogenic APP holoprotein as observed in cultured neurons or astrocytes and promote neuronal regeneration [72].
The immune system may contribute an important role in AD. Inflammatory proteins (such as eicosanoids, cytokines and complement components) are released by microglia and astrocytes of the immune system and are known to be associated with neuritic plaques. Inflammatory proteins are thought to be stimulated by Aβ production and deposition. Aβ is thought to stimulate microglia and astrocytes to release the inflammatory proteins and stimulate a neurotoxic response causing cognitive impairment. Under normal conditions microglia stimulate Aβ-specific T cell production to degrade Aβ.
In cortical astrocyte cultures it was shown that PGE2 receptor activation promotes cAMP and induces APP mRNA and amyloidogenic APP holoprotein production [73]. Agents that prevent APP over expression such as phospholipase A2 inhibitors (like dexamethasone), antiinflammatory treatments (aspirin, indomethacin), or prostaglandin synthase inhibitors may be useful in the treatment of AD.
Complement proteins are found in amyloid plaques in AD brains. Aβ plays a role in complement activation and perhaps chronic inflammation in AD. Pharmacotherapies that inhibit Aβ complement activation might be useful in AD treatment.
1.8. Immunization and Monoclonal Antibodies
Studies of PDAPP mice (which over express mutant human APP) suggest that Aβ immunisation might treat and prevent the neuro pathological changes of AD [74]. Immunization of PDAPP mice prior to the onset of AD neuropathology reduced the formation of β-amyloid plaques.
A monoclonal antibody 22C11 that binds to the extracellular domain of APP was used to determine a possible role of APP in neurons. DNA cleavage and condensation within the nucleus were observed along with neuron degeneration when cortical neurons were exposed to 22C11. The effects of 22C11 were blocked by pretreating the neurons with the general caspase inhibitor N-benzyloxycarbonyl-Val-Ala-Asp(O-methyl)-fluromethyl ketone. GSH ethyl ester (GEE) penetration of the cortical neurons also resulted in the prevention of the effects of 22CC11. 22C11 toxicity was enhanced by incubating the neurons with buthionine sulfoximine (gamma-glutamylcysteinyl synthetase). Neuritic degeneration was observed followed by caspase dependent apoptosis when the monoclonal antibody binds to APP. This is suggestive of the involvement of APP in neuronal cell death in AD [75].
Trials immunizing patients to moieties of Aβ had to be aborted because of deaths from a T-lymphocyte encephalitis, however, neuritic plaques were reduced at autopsy [76,77].
1.9. Protease Inhibitors
Aβ is the main constituent of plaques in AD and this peptide is formed by the enzymatic cleavage of the transmembrane protein APP by β- and γ-secretases respectively.
A Swedish pedigree of familial AD identifies a double mutation at the β-secretase cleavage site at the amino terminal of APP (APPSw) [78]. A protease inhibitor prevents the cleavage of APPSw by β-secretase thereby decreasing Aβ secretion [79]. Protease inhibitors show promise in AD pharmacotherapy as they block β-secretase cleavage of APP.
Cysteine protease inhibitors increased the amount of the APP extracellular domain by twofold. Protease inhibitors increased the appearance of incompletely glycosylated APP and increasing the amount of APP entering the secretory pathway. Cysteine proteases quickly degrade APP molecules [80].
Metalloendopeptidase EC 3.4.24.15 (MP24.15) promotes the degradation of Aβ [81]. SKNMC human neuroblastoma cell lines were transfected with MP24.15 cDNA in the sense and antisense directions. There was increased Aβ degradation in the sense-transfected cells and decreased Aβ degradation in antisense-transfected cells. Pretreatment with serine proteinase inhibitors (4-2-aminoethylbenzenesulfonyl fluoride) completely blocked Aβ degradation. Similarly α1-antichymotrypsin (a serpin family inhibitor) also blocked Aβ degradation. MP24.15 was found to be vital for the proteolytic cascade for the activation of the serine proteinases. The development of pharmacotherapeutics promoting Aβ degradation requires a better understanding of the cascade including activation of serine proteinase by MP24.15.
The proteasome is a multicatalytic complex involved in the degradation of polyubiquitinated proteins. The proteasome modulates the intracellular concentration of presenilins 1 and 2. These two proteins, when mutated, appear responsible for most of early onset forms of AD which is thought to be an effect favouring the deposition of long forms of Aβ leading to amyloidogenesis. Controlling presenilins concentrations could have drastic repercussions on cell physiology as suggested by the observation that proteasome inhibitors drastically potentiate the “pathogenic” presenilin function. The possibility of considering the proteasome as a potential target for therapeutic intervention in AD is important [82].
To understand the mechanisms of APP degradation, it has been established that in the presence of proteasome inhibitors, the cystolic molecular chaperon Hsc73, interacts with the cytoplasmic domain of APP (carboxy-terminal) which signals lysosomal proteolysis. Hsc73 binds to the various mutated isoforms of APP (as found in the Swedish or Dutch mutations) in equal amounts and this is suggestive of an Hsc73 attachment mechanism dependent on the conformation of the APP secretory cleavage site [83].
N-acetyl-leucyl-leucyl-norleucinal (ALLN or LLnL) is a calpain inhibitor [84-88] inhibits proteasome activities at high concentrations [89]. Two studies suggest that Aβ 40 and Aβ 42 are produced by different γ-secretases as ALLN inhibits Aβ 40 but enhances Aβ 42 production [90,91]. Another study showed that both Aβ 40 and Aβ 42 are inhibited by high ALLN concentrations and increased at low ALLN concentrations [92]. One group examined the effects of ALLN on 293 cells expressing APP (or carboxy terminal 100 amino acids; C100) and found that low concentrations of ALLN increased Aβ 40 and Aβ 42 (to a greater extent than Aβ 40) secretion and high ALLN concentrations reduced Aβ 40 and Aβ 42 secretion. C100 seems to be either processed by γ-secretase or by a degradation pathway (inhibited by low concentrations of ALLN) [93]. This gives evidence supporting that γ-secretase is inhibited by high levels of ALLN.
In studies using canine kidney cells (MDCK) and human embryonic kidney cells (HEK) the calpain 1 inhibitor LLnL (ALLN) and lysosomotropic agent ammonium chloride (NH4Cl) were used to inhibit the degradation of PS1 and APP-c100 (containing the Aβ fragment) [94-98]. It was observed that APP-C100 formed a higher molecular mass complex with PS1 fragments. When PS1 was immunoprecipitated, a large amount of APP-C100 followed suit. This is suggestive that PS1 may directly interact with APP-C100 to control Aβ deposition [99].
In APP processing α-secretase seems to be a Ca2+ dependent protease, as does calpain. In AD protease abnormalities seem to occur in the processing of APP when it is cleaved excessively by β or γ-secretases and α-secretase activity is inactivated. Intracellular Ca2+ imbalance seems to be prevalent in AD [100,101].
One study suggests that cysteine aspartate-specific proteases (caspase) directly contribute to AD pathogenesis. Caspases cleave APP encouraging the amyloidogenic processing pathway of the protein. Caspases also cleave presenilins thus promoting apoptosis. Presenilin C-terminal fragments are known to have antiapoptotic functions [102]. Caspase inhibitors could have therapeutic benefits in AD.
The presenilin 2 mutation (N141T-PS2) inhibits secretion of the α-secretase cleaved product of APP in human HEK293 cells [103]. Wild type (wt-) PS2 increases APPα secretion in human HEK293 cells. These effects are enhanced by two proteasome inhibitors Z-IE(Ot-Bu)A-Leucinal and lactacystin.
1.10. Oestrogen
A study using ovarectomized guinea-pigs suggests that the absence of ovarian oestrogen in postmenopausal women might increase Aβ 40 and Aβ 42 concentrations in the brain and 17b-estradiol (E2) treatment decreased Aβ deposition [104]. Postmenopausal women taking oestrogen replacement therapy showed significant delays in the development of AD [105,106]. Some studies reveal that oestrogen therapy is not useful in treating existing AD neuro pathology [107,108]. An experimental study investigated the effects of oestrogen (E2) on ovarectomized female rats following focal ischemia. Over expression of APP mRNA was reduced after E2 treatment [109].
1.11. Homocysteine and Oxidative Stress
Homocysteine is an amino acid which is neurotoxic [110-114] and is known to accumulate in many neurodegenerative disorders including Alzheimer’s disease [115- 117].
Homocysteine is known to augment Aβ neurotoxicity [118]. Homocysteine is a known ligand for the NMDA receptor [119]. Previous studies have shown that NMDA receptor agonists also increase Aβ toxicity [120,121]. Apoptosis was blocked by addition of vitamin E, an antioxidant, and N-acetyl-L-cytoseine (a glutathione precursor) following treatment with homocysteine and Aβ in cultural cells. This suggests that oxidative stress plays a role in apoptosis and might be important in AD.
1.12. Immediate Early Genes
The protooncogenes c-fos and c-jun are members of a set of genes known as cellular immediate early genes, and are believed to play an important role in stimulus-transcripttion coupling [122]. Many stimuli induce c-fos mRNA and protein including long-term potentiation, seizures, ischaemia and electrical stimulation [122,123]. These stimuli require the influx of Ca2+ through the N-methylD-aspartate (NMDA) ionophore and L-type Ca2+ channels [124]. The induction of c-fos by ischemia, seizures and other stimuli can be blocked by NMDA receptor antagonists such as the non-competitive antagonist dizocilpine (MK-801) [125-128]. Immediate early genes bind to AP-1 sites in the promoter regions of genes, and given that APP has AP-1 sites suggests potential therapies by modulation of immediate early gene activation of APP.
1.13. Antisense Approaches
Autosomal dominant mutations in the presenilin 1 gene
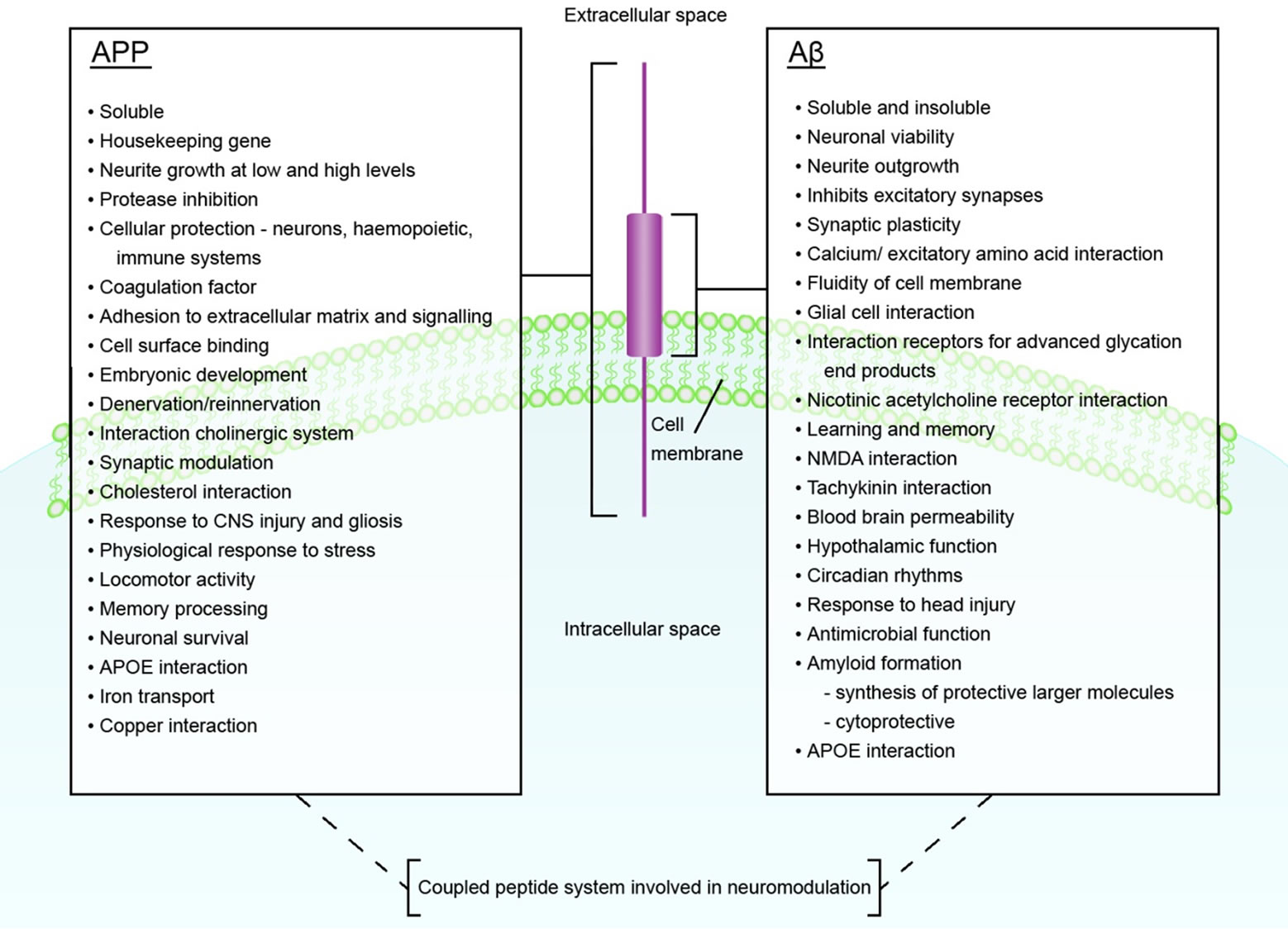
Figure 1. The Functions of APP and Aβ.
(PS1) increased concentrations of Aβ (1-42) in earlyonset AD patients. In transfected cell medium and transgenic mouse brain with PS1 mutations the level of Aβ 11-42 was increased. A human cell line expressing inducible antisense PS1 RNA found that Aβ (42) increased five-fold after 14 days of treatment whilst PS1 holoprotein decreased by 90% [129]. Antisense oligonucleotides given intracerebroventricularly into SAMP8 mice which over express APP decreased the expression of APP and improved abnormalities in learning [130].
1.14. Cholesterol
Manin-Darby canine kidney (MDCK) cells were transfected with cDNA APP with a 42 amino acid truncation at the C-terminus (DeltaC). A unique Aβ sequence was found and immunoprecipitated with an Aβ 17-24-specific monoclonal antibody (4G8) but not with Aβ 1-16-specific monoclonal antibody (BAN50). Treatment of DeltaC MDCK cells with the cholesterol synthesis inhibitor compactin, or the cholesterol binding drug filipin, resulted in the immunoprecipitation of Aβ by BAN50 but not 4 GB. These results suggest that Aβ production is cholesterol-dependent [131].
Clinical studies have revealed that cholesterol-lowering statin drugs might reduce the risk of AD [132].
2. Conclusions
APP and Aβ have many proposed actions in the central nervous system and together probably function as a coupled peptide system with a fundamental role in neuromodulation (Figure 1). Improved understanding of the functions of these neuropeptides will help in the interpretation of data collected from clinical trials, such as those currently in progress assessing the contribution of Aβ immunization in AD.
REFERENCES
- Y. Takumi, A. Matsubara, E. Rinvik and O. P. Otterson, “The Arrangement of Glutamate Receptors at Excitatory Synapses,” Annals of the New York Academy of Sciences, Vol. 868, 1999, pp. 474-482. doi:10.1111/j.1749-6632.1999.tb11316.x
- J.-Y. Koh, L. L. Yang and C. W. Cottman., “Beta Amyloid Protein Increases the Vulnerability of Cultured Cortical Neurons to Excitotoxic Damages,” Brain Research, Vol. 433, 1990, pp. 315-320. doi:10.1016/0006-8993(90)91355-K
- J. I. M. Dubinsky, “Examination of the Role of Calcium in Neuronal Death,” Annals of the New York Academy of Sciences, Vol. 679, No. 1, 1993, pp. 34-42. doi:10.1111/j.1749-6632.1993.tb18287.x
- M. P. Mattson and K. Furukawa, “Signalling Events Regulating the Neurodeveolpmental Triad. Glutamate and Secreted Forms of Beta Amyloid Precursor Protein as Examples,” Perspectives on Developmental Neurobiology, Vol. 5, No. 1998, pp. 337-352.
- P. K. Panegyres, “The Functions of the Amyloid Precursor Protein Gene,” Reviews in Neuroscience, Vol. 12, 2001, pp. 1-39. doi:10.1515/REVNEURO.2001.12.1.1
- M. P. Mattson, Z. H. Guo and J. D. Geiger, “Secreted Form of Amyloid Precursor Protein Enhances Basal Glucose and Glutamate Transport and Protects against Oxidative Impairment of Glucose and Glutamate Transport in Synaptosomes by a Cyclic GMP Mediated Mechanism,” Journal of Neurochemistry, Vol. 73, 1999, pp. 532-537. doi:10.1046/j.1471-4159.1999.0730532.x
- M. P. Mattson and W. A. Pederson, “The Effects of APP Derivatives and Oxidative Stress on Basal Forebrain Cholinergic Systems in Alzheimer’s Disease,” International Journal of Developmental Neuroscience, Vol. 16, No. 7-8, 1998, pp. 737-753. doi:10.1016/S0736-5748(98)00082-3
- I. I. Kruman and M. P. Mattson, “Pivotal Role of Mitochondrial Calcium Uptake in Neural Cell Apoptosis and Necrosis,” Journal ofNeurochemistry, Vol. 72, No. 2, 1999, pp. 529-540. doi:10.1046/j.1471-4159.1999.0720529.x
- R. P. Wernyj, M. P. Mattson and S. Christakos, “Expression of Calbindin-D28k in C6 Glial Cells Stabilizes Intracellular Calcium Levels and Protects against Apoptosis Induced by Calcium Ionophore and Amyloid B Peptide,” Brain Research: Molecular Brain Research, Vol. 64, 1999, pp. 69-79. doi:10.1016/S0169-328X(98)00307-6
- Q. Guo, N. Robinson and M. P. Mattson, “Secreted P-Amyloid Precursor Protein Counteracts the Pro-Apoptotic Action of Mutant Presenilin 1 Bioactivation of NF-KB and Stabilizattion of Calcium Homeostasis,” Journal of Biological Chemistry, Vol. 273, No. 20, 1998, pp. 12341-12351. doi:10.1074/jbc.273.20.12341
- K. Furukawa and M. P. Mattson, “The Transcription Factor NF-KB Mediates Increases in Calcium Currents and Decreases in NMDA—and AMPA I Kainate—Induced Currents Induced by Tumor Necrosis Factor-A in Hippocampal Neurons,” Journal of Neurochemistry, Vol. 70, No. 5, 1998, pp. 1876-1886. doi:10.1046/j.1471-4159.1998.70051876.x
- E. Masliah, J. Raber, M. Afford, N. Mallory, N. P. Mattson, D. Yang, D. Wong and L. Nucke, “Amyloid Protein Precursor Stimulates Excitatory Amino Acid Transport. Implications for Roles in Neuroprotection and Pathogenesis,” Journal of Biological Chemistry, Vol. 273, No. 20, 1998, pp. 12548-12554. doi:10.1074/jbc.273.20.12548
- M. P. Mattson, “Cellular Actions of Beta-Amyloid Precursor Protein and Its Soluble and Fibrilogenic Derivatives,” Physiological Reviews, Vol. 77, No. 4, 1997, pp. 1081-1132.
- M. Tolar, J. N. Keller, S. Chan, M. P. Mattson, N. A. Marques and N. K. Crutcher, “Truncated Apolipoprotein E (APOE) Causes Increased Intracellular Calcium and May Mediate APOE Neurotoxicity,” Journal of Neuroscience, Vol. 19, No. 16, 1999, pp. 7100-7110.
- M. P. Mattson, F. N. LaFerla, S. L. Chang, M. A. Leissring, P. N. Shepel and J. D. Geiger, “Calcium Signalling in the ER, Its Role in Neuronal Plasticity and Degenerative Disorders,” Trends in Neuroscience, Vol. 23, No. 5, 2000, pp. 222-229. doi:10.1016/S0166-2236(00)01548-4
- J. T. Greenamyre, A. B. Penny, A. B. Young, C. J. D’Amato, S. P. Hicks and I. Sholson, “Alterations in L-glutamate Binding in Alzheimer’s and Huntington’s Diseases,” Science, Vol. 227, No. 4693, 1985, pp. 1496- 1499. doi:10.1126/science.2858129
- J. T. Greenamyre, A. B. Penny, A. B. Young and C. J. D’Amato, “Dementia of the Alzheimer’s Type, Changes in Hippocampal L-[3H] Glutamate Binding,” Journal of Neurochemistry, Vol. 48, 1987, pp. 543-55l. doi:10.1111/j.1471-4159.1987.tb04127.x
- J. W. Geddes, C. Chang, S. N. Cooper, I. T. Loft and C. W. Cotman, “Density and Distribution of NMDA Receptors in Human Hippocampus in Alzheimer’s Disease,” Brain Research, Vol. 399, 1986, pp. 156-161. doi:10.1016/0006-8993(86)90611-6
- D. T. Monaghan, J. W. Geddes, D. Yao, C. Chung and C. W. Cotman, “[3H] TCP Binding Site in A1zheimer’s Disease,” Neuroscience Letters, Vol. 73, No. 2, 1987, pp. 197-200. doi:10.1016/0304-3940(87)90017-6
- R. H. P. Porter, P. J. Robert and R. S. J. Briggs, “NMDA Receptor Status in Elderly Normal Individuals and Those with AD,” Annals of the New York Academy of Sciences, Vol. 695, No. 1, 1993, pp. 50-53.
- J. Ulas, L. C. Brunner, L. W. Geddes, W. Choe and C. W. Cotman, “N-methyl-D-aspartate Receptor Complex in the Hippocampus of Elderly Normal Individuals and Those with AD,” Neuroscience, Vol. 49, No. 1, 1992, pp. 45-61. doi:10.1016/0306-4522(92)90075-D
- J.J. Cha, L. A. Farrell, S. F. Ashmed, A. Frey, K.K. Hsaio-Ashe, A. B. Young, J. B. Penney, J. J. Locascio, B. T. Hyman and M. C. Irizarry, “Glutamate Receptor Dysregulation in the Hippocampus of Transgenic Carrying Mutated Human Amyloid Precursor Protein,” Neurobiological Disorders, Vol. 81, 2000, pp. 90-102.
- H. Monyer, R. Sprengel, R. Schoepfer, A. Herb, M. Higuchi, H. Lomeli, N. Burnashev, B. Sakmann and P. H. Seeburg, “Heteromeric NMDA Receptors, Molecular and Functional Distinction of Subtypes,” Science, Vol. 256, No. 5060, 1992, pp. 1217-1221. doi:10.1126/science.256.5060.1217
- P. K. Panegyres, K. Zafiris-Toufexis and B. A. Kakulas, “The mRNA of the NR1 Subtype of Glutamate Receptor in Alzheimer’s Disease,” Journal of Neural Transmission, Vol. 109, 2002, pp. 77-89. doi:10.1007/s702-002-8238-9
- M. T. Webster, N. Amin, B. Pearce and P. T. Francis, “Glutamate Toxicity in Rat Cultured Neurones, Effects on Amyloid Precursor-Like Protein 2,” Neuroscience Letters, Vol. 276, No. 2, 1999, pp. 107-110. doi:10.1016/S0304-3940(99)00800-9
- D. T. Stephenson and J. A. Clemens, “Metabotropic Glutamate Activation in Vivo Induces Intraneuronal Amyloid Immunoreactivity in Guinea Pig Hippocampus,” Neurochemistry International, Vol. 33, No. 1, 1998, pp. 83-93. doi:10.1016/S0197-0186(05)80012-9
- B. Winblad and N. Poritis, “Memantine in Severe Dementia, Results of the 9M-Best Study (Benefit and Efficacy in Severely Demented Patients during Treatment with Memantine),” International Journal of Geriatric Psychiatry, Vol. 14, No. 2, 1999, pp. 135-146. doi:10.1002/(SICI)1099-1166(199902)14:2<135::AID-GPS906>3.0.CO;2-0
- B. Reisberg, R. Doody, A. Stoffler, F. Schmitt, S. Ferris, H. J. Mobius and Memantine Study Group, “Memantine in Moderate-to-Severe Alzheimer’s Disease,” New England Journal of Medicine, Vol. 348, 2003, pp. 1333-1341. doi:10.1056/NEJMoa013128
- R. J. Donnelly, A. J. Friedhoff, B. Beer, A. J. Blume and M. P. Vite, “Interleukin-1 (IL-1) Stimulates the BetaAmyloid Precursor Protein Promotor,” Cell Molecular Neurobiology, Vol. 10, 1990, pp. 485-495. doi:10.1007/BF00712843
- D. Goldgaber, H. W. Harris and T. Hla, “Interleukin 1 Regulates Synthesis of Amyloid B-Protein Precursor mRNA in Human Endothelial Cells,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 86, 1989, pp. 7606-7610. doi:10.1073/pnas.86.19.7606
- G. Forloni, F. Demicheli, S. Giorgi, C. Bendotti and N. Ageretti, “Expression of APP mRNAs in Endothelial, Neuronal and Glial Cells, Modulation by Interleukin-1,” Molecular Brain Research, Vol. 16, No. 1-2, 1992, pp. 128-134. doi:10.1016/0169-328X(92)90202-M
- G. E. Maestre, B. A. Tate, R. E. Majocha and C. A. Marotta, “Membrane Surface Ruffling in Cells that OverExpress Alzheimer Amyloid β/A4 C-Terminal Peptide,” Brain Research, Vol. 62, 1993, pp. 145-149. doi:10.1016/0006-8993(93)90311-A
- B. Brugg, Y. L. Dubreuil, G. Huber, E. E. Wollman, N. Delhaye-Bouchaud and J. Mariani, “Inflammatory Processes Induce B-Amyloid Precursor Protein Changes in Mouse Brain,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 92, 1995, pp. 3032-3035. doi:10.1073/pnas.92.7.3032
- J. G. Sheng, F. A. Boop, R. E. Mrak and W. S. T. Griffin, “Increased Neuronal B-Amyloid Precursor Protein Expression in Human Temporal Lobe Epilepsy, Association with Interleukin-1a Immunoreactivity,” Journal of Neurochemistry, Vol. 62, 1994, pp. 1872-1879.
- P. K. Panegyres and J. Hughes, “The Neuroprotective Effects of the Recombinant Interleukin-1 Receptor Antagonist Rhil-1ra after Excitotoxic Stimulation with Kainic Acid and Its Relationship to the Amyloid Precursor Protein Gene,” Journal of Neurological Sciences, Vol. 154, No. 2, 1998, pp. 123-132. doi:10.1016/S0022-510X(97)00214-1
- M. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, “Copper, Iron and Zinc in Alzheimer’s Disease Senile Plaques,” Journal of Neurological Sciences, Vol. 158, No. 1, 1998, pp. 47-52. doi:10.1016/S0022-510X(98)00092-6
- A. I. Bush, W. H. Pettingal, G. Multhaup, M. D. Paradis, J. P. Vonsattel, J. F. Gusella, K. Beyreuther, C. L. Masters and R. E. Tanzi, “Rapid induction of Alzheimer Aβ Amyloid Formation by Zinc,” Science, Vol. 265, No. 5177, 1994, pp. 1464-1467. doi:10.1126/science.8073293
- A. I. Bush, W. H. Pettingell, M. D. Paradis and R. E. Tanzi, “Modulation of A beta Adhesiveness and Secretase Site Cleavage By Zinc,” Journal of Biological Chemistry, Vol. 269, No. 16, 1994, pp. 12152-12158.
- X Huang, C. S. Atwood, R. D. Moir, M. A. Hartshorn, J. P. Vonsattel, R. E. Tanzi and A. I. Bush, “Zinc-Induced Alzheimer’s Abeta1-40 Aggregation Is Mediated by Conformational Factors”, Journal of Biological Chemistry, Vol. 272, No. 42, 1997, pp. 26464-26470. doi:10.1074/jbc.272.42.26464
- C. S. Atwood, R. D. Moir, X. Huang, R. C. Scarpa, N. M. E. Bacarra, D. M. Romano, M. K. Hartshorn, R. E. Tanzi and A. I. Bush, “Dramatic Aggregation of Alzheimer Abeta by Cu(II) is Induced by Conditions Representing Physiological Acidosis,” Journal of Biological Chemistry, Vol. 273, No. 21, 1998, pp. 12817-12826. doi:10.1074/jbc.273.21.12817
- R. A. Cherny, J. T. Legg, C. A. Mclean, D. P. Fairlie, X. Huang, C. S. Atwood, K. Beyreuther, R. E. Tanzi, C. L. Masters and A. I. Bush, “Aqueous Dissolution of Alzheimer’s Disease Aβ Amyloid Deposits by Biometal Depletion”, Journal of Biological Chemistry, Vol. 274, No. 33, 1999, pp. 23223-23228. doi:10.1074/jbc.274.33.23223
- R. A. Cherny, C. S. Atwood, M. E. Xilinas, D. N. Gray, W. D. Jones, C. A. Mclean, K. J. Barnham, I. Volitakis, F. W. Fraser, Y. S. Kim, X. Huang, L. E. Goldstein, R. D. Moir, J. T. Lim, K. Beyreuther, H. Zheng, R. E. Tanzi, C. L. Masters and A. I. Bush, “Treatment with Copper-Zinc Chelator Markedly and Rapidly Inhibits B-Amyloid Accumulation in Alzheimer’s Disease Transgenic Mice,” Neurology, Vol. 30, No. 3, 2001, pp. 665-676.
- G. Multhaup, A. Schlicksupp, L. Hesse, D. Beher, T. Rupper, C. L. Masters and K. Beyreuther, “The Amyloid Precursor Protein of Alzheimer’s Disease in the Reduction of Copper (II) to Copper (I),” Science, Vol. 271, No. 5254, 1996, pp. 1406-1409. doi:10.1126/science.271.5254.1406
- M. R. Gunther, P. M. Hanna, R. P. Mason and M. S. Cohen, “Hydroxyl Radical Formation from Cuprous Ion and Hydrogen Peroxide, A Spin-Trapping Study,” Archives of Biochemistry and Biophysics, Vol. 316, No. 1, 1995, pp. 515-522. doi:10.1006/abbi.1995.1068
- M. A. Deibel, W. D. Ehmann and W. R. Markesbery, “Copper, Iron and Zinc Imbalances in Severely Degenerated Brain Regions in Alzheimer’s Disease, Possible Relation to Oxidative Stress,” Journal of Neurological Sciences, Vol. 143, No. 1, 1996, pp. 137-142. doi:10.1016/S0022-510X(96)00203-1
- M. A. Lovell, J. D. Robertson, W. J. Teesdale, J. L. Campbell and W. R. Markesbery, “Copper, Iron and Zinc in Alzheimer’s Disease Senile Plaques,” Journal of Neurological Sciences, Vol. 158, No. 1, 1998, pp. 47-52. doi:10.1016/S0022-510X(98)00092-6
- G. Multhaup, T. Ruppert, A. Schlicksupp, L. Hesse, E. Bill, R. Pipkor, C. L. Masters and K. Beyreuther, “Copper-Binding Amyloid Precursor Protein Undergoes a Site-Specific Fragmentation in the Reduction of Hydrogen Peroxide,” Biochemistry, Vol. 37, No. 20, 1998, pp. 7224-7230. doi:10.1021/bi980022m
- C. W. Ritchie, A. I. Bush, A. Mackinnon, S. Macfarlane, M. Mastwyk, L. Macgregor, L. Kiers, R. Chern, Q. X. Li, A. Tammer, D. Carrington, C. Mavros, I. Volitakis, M. Xilinas, D. Ames, S. Davis, K. Beyreuther, R. E. Tanzi and C. L. Masters, “Metal-Protein Attenuation with Iodochlorhydroxyquin (Clioquinol) Targeting Abeta Amyloid Deposition and Toxicity in Alzheimer Disease, A Pilot Phase 2 Clinical Trial,” Archives of Neurology, Vol. 60, No. 12, 2003, pp. 1685-1691. doi:10.1001/archneur.60.12.1685
- J. A. Duce, A. Tsatsanis, M. A. Cater, S. A. James, E. Robb, K. Wikhe, S. L. Leong, K. Perez, T. Johanssen, M. A. Greenough, H.-H. Cho, D. Galatis, R. D. Moir, C. L. Masters, C. Mclean, R. E. Tanzi, R. Cappai, K. J. Barnham, G. D. Ciccotosto, J. T. Rogers and A. I. Bush, “Iron-Export Ferroxidase Activity of β-Amyloid Precursor Protein is Inhibited by Zinc in Alzheimer’s Disease,” Cell, Vol. 142, No. 6, 2010, pp. 857-867. doi:10.1016/j.cell.2010.08.014
- M. S. Wolfe, W. Xia, B. L. Ostaszewski, T. S. Diehl, W. T. Kimberly and D. J. Selkoe, “Two Transmembrane Aspartates in Presenilin-1 Required for Presenilin Endoproteolysis and Gamma-Secretase Activity,” Nature, Vol. 398, No. 6727, 1999, pp. 513-517. doi:10.1038/19077
- Y. Shen and R. Li, “Expressing Mrnas for Presenilin-1 and Amyloid Precursor Protein (APP-695) from Same Neuronal Populations in Rat Hippocampus,” Brain Research Bulletin, Vol. 46, No. 3, 1998, pp. 233-236. doi:10.1016/S0361-9230(97)00455-3
- D. Beher, C. Elle, J. Underwood, J. B. Davis, R. Ward, E. Karran, C. L. Masters, K. Beyreuther and G. Multhaup, “Proteolytic Fragments of Alzheimer’s Disease-Associated Presenilin 1 Are Present in Synaptic Organelles and Growth Cone Membranes of Rat Brain,” Journal of Neurochemistry, Vol. 72, No. 4, 1999, pp. 1564-1573. doi:10.1046/j.1471-4159.1999.721564.x
- A. Weidemann, K. Paliga, U. Kurrwang, C. Czech, G. Evin, C. L. Masters and K Beyreuther, “Formation of Stable Complexes between Two Alzheimer’s Disease Gene Products, Presenilin-2 and Beta-Amyloid Precursor Protein,” Nature Medicine, Vol. 3, 1997, pp. 328-332. doi:10.1038/nm0397-328
- W. Xia, J. Zhang, R. Perez, E. H. Koo and D. J. Selkoe, “Interaction Between Amyloid Precursor Protein and Presenilins in Mammalian Cells, Implications for the Pathogenesis of Alzheimer Disease,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 94, 1997, pp. 8208-8213. doi:10.1073/pnas.94.15.8208
- N. N. Dewji and S. J. Singer, “Specific Intercellular Binding of the Beta-Amyloid Precursor Protein to the Presenilins Induces Intercellular Signaling, Its Significance for Alzheimer’s Disease,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 95, 1998, pp. 15055-15060. doi:10.1073/pnas.95.25.15055
- R. N. Rosenberg, “Explaining the Cause of the Amyloid Burden in Alzheimer Disease,” Archives of Neurology, Vol. 59, No. 9, 2002, pp. 1367-1368. doi:10.1001/archneur.59.9.1367
- S. L. Rogers and L. T. Friedhoff, “The Efficacy and Safety of Donepezil in Patients with Alzheimer’s Disease, Results of a US Multicenter, Randomized, Double-Blind, Placebo-Controlled Trial,” Dementia, Vol. 7, No. 6, 1996, pp. 293-303.
- S. L. Rogers, R. A. Doody, R. C. Mohs and L. T. Friedhoff, “Donepezil Improves Cognition and Global Function in Alzheimer’s Disease, A 15-Week, Double-Blind, Placebo-Controlled Study,” Archives of Internal Medicine, Vol. 158, No. 9, 1998, pp. 1021-1031. doi:10.1001/archinte.158.9.1021
- S. L. Rogers, M. R. Farlow, R. S. Doody, R. Mohs and L. T. Friedhoff, “A 24-Week, Double-Blind, Placebo-Controlled Trial of Donepezil in Patients with Alzheimer’s Disease,” Neurology, Vol. 50, No. 1, 1998, pp. 136-145.
- A. Burns, M. Rossor, J. Hecker, S. Gauthier, H. Petit, H. J. Moller, S. L. Rogers and L. T. Friedhoff, “The Effects of Donepezil in Alzheimer’s Disease—Results from a Multinational Trial,” Dementia and Geriatric Cognitive Disorders, Vol. 10, No. 3, 1999, pp. 237-244.
- P. N. Tariot, P. R. Solomon, J. C. Morris, P. Kershaw, S. Lilienfeld and C. Ding, “A 5-Month, Randomized, Placebo-Controlled Trial of Galantamine in AD,” Neurology, Vol. 54, 2000, pp. 2269-2276.
- M. A. Raskind, P. A. Cyrus, B. B. Ruzicka and B. I. Gulanski, “The Effects of Metrifonate on the Cognitive, Behavioural and Functional Performance of Alzheimer’s Disease Patients,” Journal of Clinical Psychiatry, Vol. 60, No. 5, 1999, pp. 318-325. doi:10.4088/JCP.v60n0510
- M. Rösler, R. Anand, A. Cicin-Sain, S. Gauthier, Y. Agid, P. Dal-Bianco, H. B. Stahelin, R. Hartman and M. Gharabawi, “Efficacy and Safety of Rivastigmine in Patients with Alzheimer’s Disease, International Randomised Controlled Trial,” British Medical Journal, Vol. 318, No. 7184, 1999, pp. 633-640.
- G. Sberna, J. Saez-Valero, Q. X. Li, C. Czech, K. Beyreuther, C. L. Masters, C. A. Mclean and D. H. Small, “Acetylcholinesterase Is Increased in the Brains of Transgenic Mice Expressing the C-Terminal Fragment (CT100) of the Beta-Amyloid Precursor of Alzheimer’s Disease,” Journal of Neurochemistry, Vol. 71, No. 2, 1998, pp. 723-731. doi:10.1046/j.1471-4159.1998.71020723.x
- P. Tiraboschi, L. A. Hansen, M. Alford, A. Merdes, E. Masliah, L. J. Thal and J. Corey-Bloom, “Early and Widespread Cholinergic Losses Differentiate Dementia with Lewy Bodies from Alzheimer Disease,” Archives of General Psychiatry, Vol. 59, No. 10, 2002, pp. 946-951. doi:10.1001/archpsyc.59.10.946
- P. J. Whitehouse, D. L. Price, R. G. Struble, A. W. Clark, J. T. Coyle and M. R. Delon, “Alzheimer’s Disease and Senile Dementia, Loss of Neurons in the Basal Forbrain,” Science, Vol. 215, No. 4537, 1982, pp. 1237-1239. doi:10.1126/science.7058341
- M. Weinstock, “The Pharmocotherapy of Alzheimer’s Disease Based on Cholinergic Hypothesis, An Update,” Neurodegeneration, Vol. 4, No. 4, 1995, pp. 349-356. doi:10.1006/neur.1995.0042
- [69] E. Giacobini, “The Cholinergic System in Alzheimer’s Disease,” Progressive Brain Research, Vol. 84, 1990, pp. 321-332. doi:10.1016/S0079-6123(08)60916-4
- S. Rossner, U. Ueberham, R. Schliebs, J. R. Perez-Polo and V. Bigl, “P75 and TRKA Receptor Signalling Independently Regulate Amyloid Precursor Protein mRNA Expression, Isoform Composition, and Protein Secretion in PC12 Cells,” Journal of Neurochemistry, Vol. 71, 1998, pp. 757-766. doi:10.1046/j.1471-4159.1998.71020757.x
- G. P. Lim, F. Yang, T. Chu, P. Chen, W. Beech, B. Teter, T. Tran, O. Ubeda, K. Hsiao Ashe, S. A. Frautschy and G. M. Cole, “Ibuprofen Suppresses Plaque Pathology and Inflammation in a Mouse Model for Alzheimer’s Disease,” Journal of Neuroscience, Vol. 20, No. 15, 2000, pp. 5709- 5714.
- C. M. Beard, S. C. Waring, P. C. O’Brien, L. T. Kurland and E. Kokmen, “Nonsteroidal Anti-Inflammatory Drug Use and Alzheimer’s Disease, A Case-Control Study in Rochester, Minnesota, 1980 Through 1984,” Mayo Clinic Procedures, Vol. 73, 1998, pp. 951-955. doi:10.4065/73.10.951
- R. K. Lee and R. J. Wurtman, “Regulation of APP Synthesis and Secretion by Neuroimmunophilin Ligands and Cyclooxygenase Inhibitors,” Annals of the New York Academy of Sciences, Vol. 920, No. 1, 2000, pp. 261-268. doi:10.1111/j.1749-6632.2000.tb06934.x
- R. K. Lee, S. Knapp and R. J. Wurtman, “Prostaglandin E2 Stimulates Amyloid Precursor Protein Gene Expression, Inhibition by Immunosuppressants,” Journal of Neuorsciences, Vol. 19, No. 3, 1999, pp. 940-947.
- D. Schenk, R. Barbour, W. Dunn, G. Gordon, H. Grajeda, T. Guido, K. Hu, J. Huang, K. Johnson-Wood, K. Khan, D. Kholodenko, M. Lee, Z. Liao, I. Lieberburg, R. Motter, L. Mutter, F. Soriano, G. Shopp, N. Vasquez, C. Vandevert, S. Walker, M. Wogulis, T. Yednock, D. Games and P. Seubert, “Immunization with Amyloid-β Attenuates Alzheimer-Disease-Like Pathology in PDAPP Mouse,” Nature, Vol. 400, No. 6740, 1999, pp. 173-177. doi:10.1038/22124
- T. T. Rohn, K. J. Ivins, B. A. Bahr, C. W. Cotman and D. H. Cribbs, “A Monoclonal Antibody to Amyloid Precursor Protein Induces Neuronal Apopotosis,” Journal of Neurochemistry, Vol. 74, No. 6, 2000, pp. 2331-2342. doi:10.1046/j.1471-4159.2000.0742331.x
- J. A. Nicoll, D. Wilkinson, C. Holmes, P. Steart, H. Markham and R. O. Weller, “Neuropathology of Human Alzheimer Disease after Immunization with AmyloidBeta Peptide, A Case Report,” Nature Medicine, Vol. 9, No. 4, 2003, pp. 448-452. doi:10.1038/nm840
- I. Ferrer, M. Boada Rovira, M. L. Sanchez Guerra, M. J. Rey and F. Costa-Jussa, “Neuropathology and Pathogenesis of Encephalitis Following Amyloid-Beta Immunization in Alzheimer’s Disease,” Brain Pathology, Vol. 14, No. 1, 2004, pp. 11-20. doi:10.1111/j.1750-3639.2004.tb00493.x
- M. Mullan, F. Crawford, K. Axelman, H. Houlden, L. Lilius, B. Winbald and L. Lannefelt, “A Pathogenic Mutation for Probable Alzheimer’s Disease in the APP Gene at the N-Terminus of Beta-Amyloid,” Nature Genetics, Vol. 1, 1992, pp. 345-347. doi:10.1038/ng0892-345
- M. L. Steinhilb, R. S. Turner and J. R. Gaut, “The Protease Inhibitor, MG132, Blocks Maturation of the Amyloid Precursor Protein Swedish Mutant Preventing Cleavage By β-Secretase,” Journal of Biological Chemistry, Vol. 276, No. 21, 2001, pp. 4476-4484. doi:10.1074/jbc.M008793200
- J. F. Hare, “Protease Inhibitors Divert Amyloid Precursor Protein to the Secretory Pathway,” Biochemical and Biophysical Research Communications, Vol. 281, No. 5, 2001, pp. 1298-1303. doi:10.1006/bbrc.2001.4507
- R. Yamin, E. G. Malgeri, J. A. Sloane, W. T. Mcgraw and C. R. Abraham, “Metalloendopeptidase EC 3.4.24.15 Is Necessary for Alzheimer’s Amyloid-β Peptide Degradation,” Journal of Biological Chemistry, Vol. 274, 1999, pp. 1877-18784.
- F. Checler, C. A. Da Costa, K. Ancolio, N. Chevallier, E. Lopez-Perez and F. Marambaud, “Role of the Proteasome in Alzheimer’s Disease,” Biochemical and Biophysical ACTA, Vol. 1502, No. 1, 2000, pp. 133-138.
- Z. Kouchi, H. Sorimachi, K. Suzuki and S. Ishiura, “Proteasome Inhibitors Induce the Association of Alzheimer’s Amyloid Precursor Protein with Hsc73,” Biochemical and Biophysical Research Communications, Vol. 254, No. 3, 1999, pp. 804-810. doi:10.1006/bbrc.1998.9977
- M. Citron, T. Diehl, G. Gordon, A. Biere, P. Seubert and D. J. Selkoe, “Evidence That the 42- And 40-Amino Acid Forms of Amyloid Beta Protein Are Generated from the Beta-Amyloid Precursor Protein by Different Protease Activities,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 93, 1996, pp. 13170-13175. doi:10.1073/pnas.93.23.13170
- J. Higaki, D. Quon, Z. Zhong and B. Cordell, “Inhibition of Beta-Amyloid Formation Identifies Proteolytic Precursors and Subcellular Site of Catabolism,” Neuron, Vol. 14, No. 3, 1995, pp. 651-659. doi:10.1016/0896-6273(95)90322-4
- H.-W. Klafki, D. Abramowski, R. Swoboda, P. A. Paganetti and M. Staufenbiel, “The Carboxyl Termini of Beta-Amyloid Peptides 1-40 and 1-42 Are Generated by Distinct Gamma-Secretase Activities,” Journal of Biological Chemistry, Vol. 271, No. 8, 1996, pp. 28655- 28659. doi:10.1074/jbc.271.45.28655
- T. Yamazaki, C. Haas, T. C. Saido, S. Omura and Y. Ihara, “Specific Increase in Amyloid Beta-Protein 42 Secretion Ratio by Calpain Inhibition,” Biochemistry, Vol. 36, 1997, pp. 8377-8383. doi:10.1021/bi970209y
- H. W. Klafki, P. A. Paganetti, B. Sommer and M. Staufenbiel, “Calpain Inhibitor I Decreases Beta A4 Secretion from Human Embryonal Kidney Cells Expressing Beta-Amyloid Precursor Protein Carrying the APP670/ 671 Double Mutation,” Neuroscience Letters, Vol. 201, No. 1, 1995, pp. 29-32. doi:10.1016/0304-3940(95)12122-K
- A. Viniski, C. Michaud, J. Powers and M. Orlowski, “Inhibition of the Chymotrypsin-Like Activity of the Pituitary Multicatalytic Proteinase Complex,” Biochemistry, Vol. 31, 1992, pp. 9421-9428. doi:10.1021/bi00154a014
- M. Citron, T. Diehl, G. Gordon, A. Biere, P. Seubert and E. Selkoe, “Evidence That the 42- and 40-Amino Acid Forms of Amyloid Beta Protein Are Generated from the Beta-Amyloid Precursor Protein by Different Protease Activities,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 93, 1996, pp. 13170-13175. doi:10.1073/pnas.93.23.13170
- H.-W. Klafki, D. Abramowski, R. Swoboda, P. A. Paganetti and M. Staufenbiel, “The Carboxyl Termini of Beta-Amyloid Peptides 1-40 and 1-42 Are Generated by Distinct Gamma-Secretase Activities,” Journal of Biological Chemistry, Vol. 271, No. 45, 1996, pp. 28655- 28659. doi:10.1074/jbc.271.45.28655
- T. Yamazaki, C. Haas, T. C. Saido, S. Omura and Y. Ihara, “Specific Increase in Amyloid Beta-Protein 42 Secretion Ratio by Calpain Inhibition,” Biochemistry, Vol. 36, 1997, pp. 8377-8383. doi:10.1021/bi970209y
- L. Zhang, L. Song and E. M. Parker, “Calpain Inhibitor I Increases B-Amyloid Peptide Production by Inhibiting the Degradation of the Substrate of -Secretase,” Journal of Biological Chemistry, Vol. 274, No. 13, 1999, pp. 8966-8972. doi:10.1074/jbc.274.13.8966
- C. Haass, M. G. Schlossmacher, A. Y. Hung, C. VigoPelfrey, A. Mellon, B. L. Ostaszewski, I. Lieberburg, E. H. Koo, D. Schenk and D. B. Teplow, “Amyloid B-Peptide Is Produced by Cultured Cells During Normal Metabolism,” Nature, Vol. 359, 1992, pp. 322-325. doi:10.1038/359322a0
- G. Verdile, R. N. Martins, M. Duthie, E. Holmes, P. H. St George-Hyslop and P. E. Fraser, “Inhbiting Amyloid Precursor Protein C-Terminal Cleavage Promotes an Interaction with Presenilin 1,” Journal of Biological Chemistry, Vol. 275, No. 27, 2000, pp. 20794-20798. doi:10.1074/jbc.C000208200
- G. L. Caporaso, S. E. Gandy, J. D. Buxbaum, T. V. Ramabhadran and P. Greengard, Protein Phosphorylation Regulates Secretion of Alzheimer Beta/A4 Amyloid Precursor Protein,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 89, 1992, pp. 3055-3059. doi:10.1073/pnas.89.7.3055
- H. Steiner, A. Capell, B. Pesold, M. Citron, P. M. Kloetzel, D. J. Selkoe, H. Romig, K. Mendla and C. Haass, “Expression of Alzheimer’s Disease-Associated Presenilin-1 Is Controlled by Proteolytic Degradation and Complex Formation,” Journal of Biological Chemistry, Vol. 273, No. 48, 1998, pp. 32322-32331. doi:10.1074/jbc.273.48.32322
- L. L. Zhang, L. X. Song and E. M. Parker, “Calpain Inhibitor I Increases Beta-Amyloid Peptide Production by Inhibiting the Degradation of the Substrate of GammaSecretase. Evidence That Substrate Availability Limits Beta-Amyloid Peptide Production,” Journal of Biological Chemistry, Vol. 274, No. 13, 1999, pp. 8966-8972. doi:10.1074/jbc.274.13.8966
- G. Verdile, R. N. Martins, M. Duthie, E. Holmes, P. H. St George-Hyslop and P. E. Fraser, “Inhibiting Amyloid Precursor Protein C-Terminal Cleavage Promotes an Interaction with Presenilin 1,” Journal of Biological Chemistry, Vol. 274, No. 25, 2000, pp. 20794-20798. doi:10.1074/jbc.C000208200
- J. F. Disterhoft, W. E. Gispen, J. Traber and Z. S. Khachaturian (Eds.), “Calcium Hypothesis of Aging and Dementia,” Annals of the New York Academy of Sciences, 1994, p. 747.
- M. Chen, “Is Alzheimer’s Disease Associated with a Decreased Intracellular Level of Calcium?” Frontiers in Bioscience, Vol. 3, 1998, Let 1-2.
- C. L. Wellington and M. R. Hayden, “Caspases and Neurodegeneration, on the Cutting Edge of New Therapeutic Approaches,” Clinical Genetics, Vol. 57, No. 1, 2000, pp. 1-10. doi:10.1034/j.1399-0004.2000.570101.x
- P. Marambaud, C. Alves Da Costa, K. Ancolio and F. Checler, “Alzheimer’s Disease-Linked Mutation of Presenilin 2 (N141 I-PS2) Drastically Lowers Appalpha Secretion, Control by Proteasome,” Biochemical and Biophysical Research Communications, Vol. 252, No. 1, 1998, pp. 134-138. doi:10.1006/bbrc.1998.9619
- S. S. Petanceska, V. Nagy, D. Frail and S. Gandy, “Ovariectomy and 17beta-Estradiol Modulate the Levels of Alzheimer’s Amyloid Beta Peptides in Brain,” Neurology, Vol. 54, No. 12, 2000, pp. 2212-2217.
- M. X. Tang, D. Jacobs, Y. Stern, K. Marder, P. Schofield, B. Gurland, H. Andrews and R. Mayeux, “Effect of Oestrogen During Menopause on Risk and Age at Onset of Alzheimer’s Disease,” Lancet, Vol. 348, No. 9025, 1996, pp. 429-432. doi:10.1016/S0140-6736(96)03356-9
- L. Melton, “Sex Is All in the Brain, Report of a Novartis Foundation Symposium on the Neural and Cognitive Effects of Oestrogens,” Trends Endocrinol Metabolism, Vol. 11, 2000, pp. 69-71. doi:10.1016/S1043-2760(99)00231-3
- V. W. Henderson, A. Paganini-Hill, B. L. Miller, R. J. Elble, P. F. Reyes, D. Shoupe, C. A. Mccleary, R. A. Klein, A. M. Hake and M. R. Farlow, “Estrogen for Alzheimer’s Disease in Women, Randomised Double-Blind, Placebo-Controlled Trial,” Neurology, Vol. 54, 2000, pp. 295-301.
- R. A. Mulnard, C. W. Cotman, C. Kawas, R. J. Elble, P. F. Reyes, D. Shoupe, C.A. McCleary, R. A. Klein, A. M. Hake and M. R. Farlow, “Estrogen Replacement Therapy for Treatment of Mild to Moderate Alzheimer’s Disease, A Randomised Control Trial, Alzheimer’s Disease Corporative Study,” Journal of the American Medical Association, Vol. 283, No. 8, 2000, pp. 1007-1015. doi:10.1001/jama.283.8.1007
- J. Shi, K. S. Panickar, S. H. Yang, O. Rabbani, A. L. Day and J. W. Simpkins, “Estrogen Attenuates Over-Expression of Beta-Amyloid Precursor Protein Messenger RNA in an Animal Model of Focal Ischemia,” Brain Research, Vol. 810, No. 1-2, 1998, pp. 87-92. doi:10.1016/S0006-8993(98)00888-9
- P. I. Ho, S. C. Collins, S. Dhitavat, D. Ortiz, D. Ashline, E. Rogers and T. B. Shea, “Homocysteine Potentiates B-Amyloid Neurotoxicity, Role of Oxidative Stress,” Journal of Neurochemistry, Vol. 78, 2001, pp. 249-253. doi:10.1046/j.1471-4159.2001.00384.x
- S. Sawada, S. Takada and C. Yamamoto, “Excitatory Actions of Homocysteic Acid on Hippocampal Neurons,” Brain Research, Vol. 238, No. 1, 1982, pp. 282-285. doi:10.1016/0006-8993(82)90798-3
- M. Lee, H. K. Strahlendorf and J. C. Strahlendorf, “Differential Effects of N-Methyl-D-Aspartic Acid and LHomocysteine Acid on Cerebellar Purkinje Neurons,” Brain Research, Vol. 456, No. 1, 1988, pp. 104-112. doi:10.1016/0006-8993(88)90351-4
- S. A. Lipton, W. K. Kin, Y. B. Choi, S. Kumar, D. M. D-Emilia, P. V. Rayudu, D. R. Amelle and J. S. Stamler, “Neurotoxicity Associated with Dual Actions of Homocysteine at the N-Methyl-D-Aspartate Receptor,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 94, 1997, pp. 5923-5928. doi:10.1073/pnas.94.11.5923
- I. I. Kruman, C. Culmsee, S. L. Chan, Y. Kruman, Y. Z. Guo, L. Penix and M. P. Mattson, “Homocysteine Elicits a DNA Damage Response in Neurons that Promotes Apoptosis and Hypersensitivity to Excitotoxicity,” Journal of Neuroscience, Vol. 20, No. 18, 2000, pp. 6920- 6926.
- R. Clarke, A. D. Smith, K. A. Jobst, H. Retsum, I. Sutton and P. M. Ueland, “Folate, Vitamin B12, and Serum Total Homocysteine Levels in Confirmed Alzheimer’s Disease,” Archives of Neurology, Vol. 55, No. 11, 1998, pp. 1449-1455. doi:10.1001/archneur.55.11.1449
- C. G. Gottfries, W. Lehmann and B. Reglan, “Early Diagnosis of Cognitive Impairment in the Elderly with the Focus on Alzheimer’s Disease,” Journal of Neural Transmission, Vol. 105, 1998, pp. 773-783. doi:10.1007/s007020050094
- J. Miller, “Homocysteine and Alzheimer’s Disease,” Nutrition Reviews, Vol. 57, 1999, pp. 126-129.
- P. I. Ho, S. C. Collins, S. Dhitavat, D. Ortiz, D. Ashline, E. Rogers and T. B. Shea, “Homocysteine Potentiates Beta-Amyloid Neurotoxicity, Role of Oxidative Stress,” Journal of Neurochemistry, Vol. 78, 2001, pp. 249-253. doi:10.1046/j.1471-4159.2001.00384.x
- M. L. Zeise, T. Knopfel and W. Zieglgansberger, “(+1-)- Beta-Parachlorophenylglutamate Selectivity Enhances the Depolarizing Response to L-Homocysteine Acid in Neocortical Neurons of the Rat Evidence for a Specific Uptake System,” Brain Research, Vol. 359, No. 6393, 1988, pp. 325- 327.
- M. P. Mattson, B. Cheng, D. Davis, K. Bryant, I. Leiberburg and R. E. Rydel, “Beta Amyloid Peptides Destabilized Calcium Homeostasis and Render Human Cortical Neurons Vulnerable to Excitotoxicity,” Journal of Neuroscience, Vol. 12, No. 2, 1992, pp. 376-389.
- C. W. Gray and A. J. Patel, “Neurodegeneration Mediated By Glutamate and Beta-Amyloid Peptide a Comparison and Possible Interaction,” Brain Research, Vol. 691, No. 1, 1995, pp. 169-179. doi:10.1016/0006-8993(95)00669-H
- J. I. Morgan and T. Curran, “Stimulus-Transcription Coupling and Nervous System, Involvement of the Inducible Protooncogenes Fos and Jun,” Annual Reviews in Neuroscience, Vol. 14, 1991, pp. 421-451. doi:10.1146/annurev.ne.14.030191.002225
- M. Sheng and M. E. Greenburg, “The Regulation and Function of c-Fos and Other Immediate Early Genes in the Nervous System,” Neuron, Vol. 4, 1990, pp. 477-485. doi:10.1016/0896-6273(90)90106-P
- H. Bading, D. D. Ginty and M. E. Greenberg, “Regulation of Gene Expression in Hippocampal Neurones by Distinct Signalling Pathways,” Science, Vol. 260, No. 5105, 1993, pp. 181-186. doi:10.1126/science.8097060
- M. Dragunow, W. C. Abraham, M. Goulding, S. E. Mason, H. A. Robertson and R. L. M. Faull, “Long Term Potentiation and the Induction of c-Fos mRNA Proteins in the Dentate Gyrus of Unanaesthetized Rats,” Neuroscience Letters, Vol. 101, No. 3, 1989, pp. 274-280. doi:10.1016/0304-3940(89)90545-4
- D. E. Herrera and H. A. Robertson, “Unilateral Induction of c-Fos Protein in Cortex Following Cortical Devascularization,” Brain Research, Vol. 503, No. 2, 1989, pp. 205-213. doi:10.1016/0006-8993(89)91665-X
- A. M. Szekely, M. I. Barbaccia and E. Costa, “Activation of Specific Glutamate Receptor Subtypes Increases c-Fos Proto-Oncogene Expression in Primary Cultures of Neonatal Rat Cerebellar Granule Cells,” Neuropharmacology, Vol. 26, 1987, pp. 1779-1782. doi:10.1016/0028-3908(87)90132-8
- A. M. Szekely, M. I. Barbaccia, H. Alho and E. Costa, “In Primary Cultures of Cerebellar Granule Cells the Activation of NMDA-Sensitive Glutamate Receptors Induces c-Fos mRNA Expression,” Molecular Pharmacology, Vol. 35, 1989, pp. 401-408.
- L. M. Refolo, C. Eckman, C. M. Prada, D. Yager, K. Sambamurti, N. Mehta, J. Hardy and S. G. Younkin, “Antisense-Induced Reduction of Presenilin 1 Expression Selectively Increases the Production of Amyloid Beta 42 in Transfected Cells,” Journal of Neurochemistry, Vol. 73, No. 6, 1999, pp. 2383-2388. doi:10.1046/j.1471-4159.1999.0732383.x
- V. B. Kumar, S. A. Farr, J. F. Flood, V. Kemalsh, M. Franko, W. Banks and J. Morley, “Site-Directed Antisense Oligonucleotide Decreases the Expression of Amyloid Precursor Protein and Reverses Deficits in Learning and Memory in Aged SAMP8 Mice,” Peptides, Vol. 21, No. 12, 2000, pp. 1769-1775. doi:10.1016/S0196-9781(00)00339-9
- T. Mizuno, C. Haas, M. Michikawa and K. Yanagisawa, “Cholesterol-Dependent Generation of a Unique Amyloid Beta-Protein from Apically Missorted Amyloid Precursor Protein in MDCK Cells,” Biochemistry and Biophysical ACTA, Vol. 1373, No. 1, 1998, pp. 119-130. doi:10.1016/S0005-2736(98)00097-2
- B. Wolozin, W. Kellman, P. Rousseau, G. G. Celesia and G. Siegel, “Decrease Prevalence of Alzheimer’s Disease Associated with 3-Hydroxy-3-Methylglutaryl Coenzyme a Reductase Inhibitors,” Archives of Neurology, Vol. 57, No. 10, 2000, pp. 1439-1443. doi:10.1001/archneur.57.10.1439
NOTES
*Three parts to be published in three consecutive issues.