Advances in Microbiology
Vol.3 No.1(2013), Article ID:29240,10 pages DOI:10.4236/aim.2013.31015
Characterization of NADase-Inactive NAD+ Glycohydrolase in Streptococcus pyogenes
Department of Bacteriology, Graduate School of Medical Sciences, Nagoya City University, Nagoya, Japan
Email: tadaoh@med.nagoya-cu.ac.jp
Received December 7, 2012; revised January 8, 2013; accepted February 10, 2012
Keywords: Streptococcus pyogenes; NAD+ Glycohydrolase; NADase; SPN; Streptococcal; SF370
ABSTRACT
Background: Streptococcus pyogenes secretes NAD+ glycohydrolase (NADase, also known as SPN or Nga). All S. pyogenes strains examined to date possess the gene that encodes SPN (spn), but some strains produce SPN that lacks detectable NADase activity. Although there is much evidence to support that SPN’s NADase activity contributes to virulence, there is very little evidence that NADase-inactive SPN has detectable functions. Results: In order to characterize the NADase-inactive SPN, we firstly attempted to clone the NADase-inactive spn allele in Escherichia coli. Although we obtained recombinants which were shown to have the correct size insert, all had some mutations in the spn allele. Therefore, we attempted to change the mutated nucleotides back to the original nucleotides. While a nucleotide mutagenesis (inverse PCR method) easily changed a target nucleotide of control genes back to the original nucleotides, the mutations of NADase-inactive spn allele were never successfully converted back to the original nucleotides. Finally the mutant spn alleles were sub-cloned into another vector (pLZ12-Km2), which is maintained in both E. coli and S. pyogenes. The resultant plasmids were subjected to nucleotide mutagenesis using inverse PCR; the resultant mutagenized plasmid DNAs were used to transform both E. coli and S. pyogenes strains. We observed successful nucleotide substitutions back to the original spn nucleotide sequence in S. pyogenes transformants, but not in E. coli transformants. Thus, the NADase-inactive spn allele was successfully cloned in S. pyogenes, but not in E. coli. However, we could not find an association with NADase-inactive spn allele and virulence in a mouse infection model. Conclusions: These results suggest that NADase-inactive spn allele has some toxic effect to E. coli, but not S. pyogenes. This effect may due to an as of yet unknown function attributable to NADase-inactive SPN.
1. Introduction
Streptococcus pyogenes is a gram-positive bacterium that infects the upper respiratory tract, including the tonsils and pharynx, and is responsible for post-infectious diseases such as rheumatic fever and glomerulonephritis. In addition, S. pyogenes causes severe invasive disease including necrotizing fasciitis [1-6]. The molecular mechanisms that the organism utilizes to cause these diseases are not yet elucidated fully. To analyze these mechanisms, it is important to characterize the virulence factors of S. pyogenes fully. S. pyogenes secretes several distinct proteins such as superantigens, DNases, streptokinase, cysteine proteinase SpeB, C5a peptidase, and streptococcal inhibitor of complement-mediated lysis (Sic) [7,8]. Several of these proteins have been identified as virulence factors and analyzed in detail, and others are still not yet fully characterized. NAD+ glycohydrolase (NADase, also known as SPN or Nga) is one of the secreted proteins which should be further characterized.
SPN is known as the host attacking enzymatic toxin produced by S. pyogenes that shows cytotoxic effects to keratinocytes in vitro experiment [9,10]. SPN is also demonstrated toxicity in bacterial cells. To counteract this toxicity, S. pyogenes encodes ifs gene whose product (IFS) is an endogenous inhibitor of NADase activity and is localized in the bacterial cytoplasmic compartment. Inside the S. pyogenes bacterial cell, SPN precursor exists as an inactive complex with IFS [11,12].
Although the SPN had been found long ago [13], initial studies on SPN were hindered by the fact that it was not possible to clone spn in Escherichia coli which is a commonly used bacterial host for genetic research (due to bacterial death) [11]. In the study conducted by Meehl et al. [11], in which ifs gene was discovered to exist as a spn-ifs operon, they were able to resolve this cloning toxicity issue when the spn gene was successfully cloned into a plasmid together with ifs (as a spn-ifs operon) and subsequently introduced into E. coli. The cytotoxicity of SPN is believed to depend on NADase activity; for example, the hypothesis has been put forth that depletion of cellular NAD+ through the enzymatic action of SPN induces host cell death [14]. Meanwhile, it has been demonstrated that some strains produce SPN that lack detectable NADase activity [11,15-17]. The presence of an aspartic acid instead of a glycine at amino acid residue 330 (G330D polymorphism) has been associated with loss of SPN NADase activity [18,19]. Additionally, ifs genes degrade into psedogenes in strains with the NADase-inactive SPN subtype, suggesting that the subtypes no longer lose the self-toxicity to require the functional IFS [11,19]. In contrast, the study using statistically sufficient number of NADase-inactive strains revealed that the spn alleles themselves never degrade into psedogenes [19]. Based on these findings suggesting that SPN has a hidden NADase-independent function [19], we were prompted to re-evaluate the role of NADase-inactive SPN in S. pyogenes pathogenesis.
2. Materials and Methods
2.1. Ethic Statement
All animal studies conducted comply with federal and institutional (the Committee on the Ethics of Animal Experiments of the Nagoya City University) guidelines. The protocol was approved by the Committee on the Ethics of Animal Experiments of the Nagoya City University (Permit Number: H23M-07). All efforts were made to minimize suffering.
2.2. Bacterial Strains
Streptococcus pyogenes strains 1529 and GT01 isolated as causative organisms from invasive diseases patients in Japan possess NADase active SPN [18,20]. S. pyogenes (GAS) strain SF370, which is prevalent as the database reference isolate (accession NC_002737), was provided by the courtesy of J. J. Ferretti [21,22]. Streptococcal strains were cultured in brain heart infusion (E-MC62, EIKEN Chemical Co., Tokyo, Japan) supplemented with 0.3% yeast extract (BD, Sparks, MD, USA) (BHI-Y) broth unless otherwise described.
2.3. Cloning Experiment of SpnSF370 Gene
The spnSF370 of S. pyogenes strain SF370 and the other control DNAs were amplified by PCR with Extaq DNA polymerase (Takara Bio, Ohtsu, Japan) using the corresponding primers listed in Table 1. pGEM®-T Easy vector system (Promega, Madison, WI, USA) was used for cloning of the PCR products purified by Gel extraction kit (Qiagen, Hilden, Germany). The high copy-number pGEM®-T Easy vector contains T7 and SP6 RNA poly-
Table 1. Sequences of primers used in this study.

Continued

aName of insert DNA fragment amplified by PCR (the expected size), used primer name and the sequence were described. The two PCR products of recASF370 and spy11931529 contain a part of the gene, respectively, whereas the others have the hole genes indicated. spnSF370 and recASF370 are PCR products from strain SF370. spn-ifsGT01, and covRSGT01 are PCR products derived from strain GT01. spy11931529, covRS1529, vicRK1529, and vicK1529 are PCR products derived from strain 1529. S. pyogenes strain SF370 encode NADase-inactive SPN, whereas strains 1529 and GT01 encode NA-Daseactive SPN.
merase promoters flanking a multiple cloning region within the α-peptide coding region of the enzyme β-galactosidase http://www.promega.com/~/media/files/resources/protocols/technical%20manuals/0/pgem-t%20and%20pgem-t%20easy%20vector%20systems%20protocol.pdf?la = en. In the pGEM®-T Easy vector system, recombinant clones are allowed to be directly identified by blue/white color screening on indicator plates.
For spnSF370-cloning, we obtained three recombinants that have the correct size insert in the corresponding plasmids (named pGEM-spnSF37026, pGEM-spnSF37032, and pGEM-spnSF37013; see Result section for additional detail on the creation of these plasmids).
2.4. Nucleotide Substitution by Inverse PCR
Primestar Taq DNA polymerase (Takara) was used for the inverse PCR described previously [18]. Primers used are listed in Table 1. PCR product was self-ligated and used to transform E. coli strain DH5α.
2.5. Construction of pLZ12-Km2 Derivatives
The inserts of pGEM-spnSF37026, pGEM-spnSF37032, and pGEM-spnSF37013 were digested with EcoRI, and subcloned into pLZ12-Km2 to yield pLZ-spnSF37026, pLZspnSF37032, and pLZ-spnSF37013, respectively.
2.6. Creation of Spn Mutant of Strain SF370
E. coli JM109 was used to propagate plasmid constructions. Non-polar inactivated mutant of spn was constructed via double-crossover allelic replacement in the chromosome of S. pyogenes SF370. To construct the plasmid for the spn knockout mutant, the 5’ end of spn (fragment 1) was amplified with oligonucleotide primers ngaGT-n1 (5’-GGCTAGCGAACAGATGTGAAGGTTCTG-3’) with an NheI restriction site and ngaGT-c1 (5’-TCCCCCGGGTTTCTCATGTAAACCACCT-3’) with an SmaI restriction site, and the 3’ end of spn (fragment 2) was amplified with ngaGT-n2 (5’-TCCCCCGGGATAGGAAGTAACAATATGT-3’) with an SmaI restriction site and ngaGT-c2 (5’-GGACTAGTATGTTAGCTTTCAATTGGGT-3’) with an SpeI restriction site. Oligonucleotides ngaGT-n1, ngaGT-c1, ngaGT-n2 and ngaGT-c2 contained a restriction site for NheI, SmaI, SmaI and SpeI, respectively, (shown in underline in the primer sequence). Fragment 2 was digested with SmaI and SpeI for insertion into multi-cloning site 2 of the pFW12 plasmid [23]. The resulting plasmid was then digested with NheI and SmaI, and both the spc2 DNA fragment containing aad9 (promoter less spectinomycin resistant gene), which was obtained from a SmaI digested fragment of pSL60-2 [24], and the NheI-SmaI-digested fragment 1 were inserted. This plasmid, pFW12::(spn::aad9), was a suicide vector for S. pyogenes. For the preparation of competent cells, strain SF370 was harvested at earlyto mid-log phase (OD660 = 0.4 to 0.5) and washed twice with 0.5 M sucrose buffer. The constructed suicide vector pFW12::(spn::aad9) was used to transform strain SF370 by electroporation. The conditions of electroporation were 1.25 kV/mm, 25 μF capacitance and 200 Ω resistance, using Gene Pulser II (Bio-Rad, Hercules, CA, USA). After incubation at 37˚C for 3 h, competent cells were spread onto BHI agar plates containing 0.3% yeast extract and spectinomycin (final concentration 100 μg/ml). Selected colonies on the plates were cultured. Cultured bacteria were washed once with saline, resuspended in 10 mM Tris, 1 mM EDTA and boiled for 10 min. Genomic DNA was obtained from the supernatant of boiled bacteria. The double-crossover replacement was analyzed using genomic DNA by PCR and successful double-crossover replacement was further confirmed by DNA sequencing.
2.7. Mouse Model of Invasive Skin Tissue Infection
The ability of S. pyogenes to cause local skin lesions and necrosis in mice after skin inoculation was assessed using a procedure similar to that described elsewhere [25]. In brief, 3-week-old female ICR mice (10 - 12 g) were anesthetized with sevoflurane, and the skin of the left flank was bared by separating hair with alcohol swab, unless otherwise indicated. Bacteria (0.2 ml; 2 × 107 cfu per mouse) grown in BHI-Y were injected with a 27- gauge needle just under the surface of the skin so that a superficial bleb was raised immediately below the skin surface. The number of colony-forming units injected was verified for each experiment by plating bacteria on BHI-Y or sheep blood agar plates (with or without kanamycin) and counting colony-forming units. Lesion sizes (length × width) were measured, with the length determined as the longest dimension of the lesion at day 3 or at the death time point.
Bacteria were recovered from the mice which survived until day 8. For mouse blood samples: 100 µl of blood from the heart was spread on a sheep blood agar plate. For the spleen samples: Spleen was homogenized and suspended with 100 µl of PBS and spread on a sheep blood agar plate. β-hemolysis positive colonies were calculated. Two colonies per plate were randomly selected and inoculated into BHI-Y broth supplemented with or without 50 µg/ml spectinomycin for microscopic analysis to confirm coccus morphology and chain arrangements characteristic of Streptoccoal species.
All animal procedures were approved by the Institutional Animal Care and Use Committee at Nagoya City University.
2.8. Statistical Analysis
Data collected for virulence to mouse (survival days) were assessed using a log-rank comparison described previously [20]. R software was used for statistical analysis http://bioinf.wehi.edu.au/software/russell/logrank/. P-value ≤ 0.05 was considered significant.
3. Results
3.1. Cloning of SpnSF370 Gene into a pGEM®-T Easy Vector
S. pyogenes strain SF370 is a representative among NADase negative strains. In order to evaluate the role of NADase-inactive SPN, we firstly attempted to clone the spnSF370 gene of the strain SF370 into a pGEM®-T Easy vector in E. coli strain DH5α without support of the ifsSF370 gene. The vector and the strain DH5α are compatible with the blue/white color screening for recombinants. Typically, we see about half of the transformants showing white color under our experimental condition when a DNA insert without toxicity to E. coli is used. We show data from two representative experiments in Table 2 (40.9% and 44.4% white colonies for recASF370 and spy11931529, respectively; these genes have been cloned for our other studies around the same time as this study). In contrast, the four cloning experiments using the insert encoding spnSF370 showed only 8.7%, 27.6%, 31.8% and 0.8% white colonies (Table 2). Eighty-five of the white colonies derived from the spnSF370-insert were randomly selected for further plasmid analysis (named as pGEM-spnSF3701 to 85), and only three colonies (4%) possessed the correct size insert (1.5 kbp) in the corresponding plasmids (pGEM-spnSF37026, pGEM-spnSF37032, and pGEM-spnSF37013; see Table 3 and Figure S1). In contrast, in the experiments using control inserts, more than 50% of white colonies had the correct size inserts: 94%, 67%, 100%, 100%, 86%, 57% and 88% for spn-ifsGT01, recASF370, spy11931529, covRSGT01, covRS1529, vicRK1529 and vicK1529, respectively (Table 3).
For the spnSF370, the insert of the three plasmids (pGEM-spnSF37026, pGEM-spnSF37032, and pGEMspnSF37013) were sequenced, and the following mutations were found: substitution of the start codon ATG to ACG in pGEM-spnSF37026, the second codon AGA (Arg) to TGA (stop) in pGEM-spnSF37032, and an adenine nucleotide was substituted to a guanine (G) at nucleotide 34 upstream from the adenine (A) of the start codon in pGEM-spnSF37013, respectively (see most right column in Table 3). We propose these mutations may have been introduced for the following reasons: 1) A spontaneous mutation is often inserted in DNA fragment amplified by PCR or 2) spnSF370 gene is toxic to E. coli cells, so mutations to make the gene inactive were given for a natural survival advantage. In order to examine those possibilitiesTable 2. The numbers of blue/white colonies.
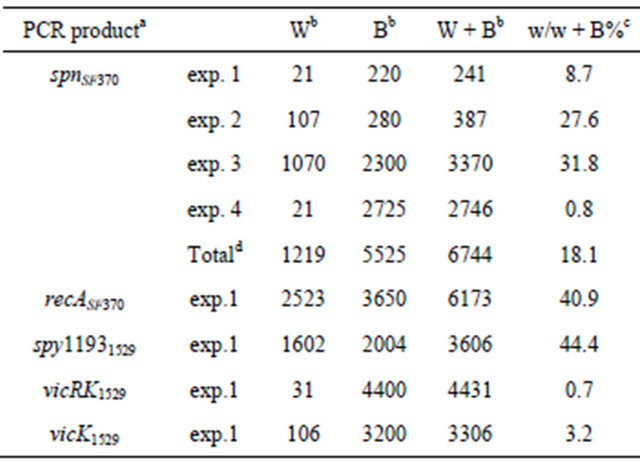
aPCR products of recASF370 and spy11931529 contain a part of the gene, respectively, whereas the others have the hole genes indicated. bThe numbers of white (W), blue (B), and the total (W + B) colonies. c% white colonies. dsum of the colony numbers from 4 experiments.
Table 3. Cloning of the insert containing spnSF370 gene into pGEM-T easy vector.

aAs a control for spnSF370, seven examples spn-ifsGT01, recASF370, spy11931529, covRSGT01, covRS1529, vicRK1529, and vicK1529 were shown. See Table 1 about information for the insert DNAs. bZ-test was used in order to compare with spnSF370. cNumber of insert without any mutation/number of sequenced insert DNA.
we attempted to change the mutated nucleotides back to the original nucleotides using the inverse PCR method described previously [18]. We performed inverse PCR with primers nga(SF370)-F and nga(SF370)-R (Tables 1 and 4) constructed to substitute the mutated second codon of the spnSF370 on pGEM-spnSF37032 to the original
Table 4. Physical maps of primers used for inverse PCR were shown.

The mutated nucleotides were attempted to change back to the original nucleotides. Primers used for the inverse PCR were shown by arrows. The primer’ nucleotide sequences were also shown in Table 1. Unsuccessful substitutions were shown in italic type. aOriginal nucleotide sequence of the junction site. The adenine and thymine nucleotides which were substituted in No. 32 and No. 26, respectively, were shown as bold “A” and “T”. The start (ATG) and second (AGA) codons were underlined. bThe mutated thymine nucleotide of the spnSF370 on pGEM-spnSF37032 was shown as bold “T”. cThe mutated cytosine nucleotide of the spnSF370 on pGEM-spnSF37026 was shown as bold “C”. dOriginal nucleotide sequence of the junction site. The adenine (A) nucleotide, which was substituted in No. 13, was shown in bold type. eThe mutated guanine nucleotide on the pGEM-spnSF37013 was shown as bold “G”. fOriginal sequence of the junction site. The adenine nucleotide, which was substituted in No. 11 and No. 28, was shown as bold “A”. The 267th codon (ACC) was underlined. gThe mutated guanine nucleotide of the vicK1529 on pGEM-vicRK152911 was shown as bold “G”. hThe mutated guanine nucleotide of the vicK1529 on pGEM-vicK152928 was shown as bold “G”.
codon AGA (R). The amplification product was selfligated and used to transform E. coli strain DH5α. Plasmids were prepared from randomly selected 14 transformants (named as 32 - 1 to 32 - 14). Five plasmids (32 - 2, 32 - 4, 32 - 8, 32 - 9 and 32 - 12) appeared to possess the correct size insert, whereas the other nine (32 - 1, 32 - 3, 32 - 5, 32 - 6, 32 - 7, 32 - 10, 32 - 11, 32 - 13, and 32 - 14) had the smaller size inserts based on the result seen during agarose gel electrophoresis (data not shown). In addition to the inserts of the five passed plasmids (32 - 2, 32 - 4, 32 - 8, 32 - 9 and 32 - 12), one of the dropped-out plasmids (32 - 1) which was added as a representative (internal) negative-control were sequenced. As shown in Table 4, the 32 - 1 had a large deletion and the other five contained a nucleotide mutation or deletion at the junction site. Additionally for pGEM-spnSF37026, and pGEMspnSF37013, we attempted to change the mutated nucleotides back to the original nucleotides by using same method with primers nga (SF370)-F and nga (SF370)-R, or primers nga(SF370)-F2 and nga(SF370)-R2 (Table 4). Two of the seven pGEM-spnSF37026 derivative plasmids (26 - 1and 26 - 2) prepared from randomly selected transformants (named as 26 - 1 to 26 - 7) appeared to possess the correct size insert. However, both plasmids had a nucleotide deletion at the junction site (Table 4). For pGEM-spnSF37013, four (13 - 1, 13 - 2, 13 - 4, and 13 - 5) of seven plasmids prepared from randomly selected transformants appeared to possess the correct size insert. However, one plasmid (13 - 4) was same as the original pGEM-spnSF37013 and the other three possessed a nucleotides deletion or a nucleotide mutation at the junction site (Table 4). We have observed successful substitution of a corresponding nucleotide for more than 50% of the transformants checked in other recent experiments. Two representative examples are shown below. As described above (see Table 3), the four and the seven types of pGEM-T easy derivatives having vicRK1529 and vicK1529 have been previously constructed, respectively (named as pGEM-vicRK152911, pGEM-vicRK152912pGEM-vicRK152913, pGEM-vicRK152915 and pGEMvicK152922, pGEM-vicK152923, pGEM-vicK152924, pGEMvicK152925, pGEM-vicK152926, pGEM-vicK152927, pGEMvicK152928: each plasmid has a mutation(s) in somewhere of vicRK1529 or vicK1529). Among the plasmids, pGEMvicRK152911 and pGEM-vicK152928 which have both a mutation changing the codon 276 of vicK1529 gene from a ACC (encoding “T”) to a GCC (encoding “A”) were used for the control experiments performed with primers vicK-F and vicK-R (Table 4). We observed a successful substitution of the corresponding nucleotide in one of two transformants (11 - 1 and 11 - 2) analyzed for pGEM-vicRK152911, two of two transformants (28 - 1 and 28 - 2) in pGEM-vicK152928 (Tables 4 and 5). In the case of spnSF370 gene, we never observed the expected nucleotide change when using any of three plasmids as template described above (Tables 4 and 5). These results suggest that spnSF370 gene is toxic to E. coli cells.
In strain SF370, the ifsSF370 allele has a nonsense mutation in the codon for leucine 24 to produce a truncated open reading frame [11]. In order to determine whether the truncated ifsSF370 open reading frame can successfully inhibit the toxicity of spnSF370 gene in E. coli cells, we attempted to clone a spn-ifsSF370 operon into the pGEM®- T Easy vector. The original forward primer ngaGTn1Nhe previously used for cloning of spnSF370 gene and three altered reverse primers (nga-c8xho, IFS-R(EcoRI), and slo-c2) to include ifs were tested (Table 1). However, we did not obtain any recombinants having the expected insert in the size (data not shown).
3.2. Cloning SpnSF370 Gene into pLZ12-Km2 Vector
We attempted to clone spnSF370 by using plasmid pLZ12- Km2 instead of pGEM®-T Easy. pLZ12-Km2 which has a rolling circle type of replication can be successfully maintained in both E. coli and S. pyogenes [26]. Firstly, each insert DNA of pGEM-spnSF37026, pGEM-spnSF37032, and pGEM-spnSF37013 was subcloned into pLZ12-Km2 (named as pLZ-spnSF37026, pLZ-spnSF37032, and pLZspnSF37013 respectively). By using these plasmids as template for inverse PCR, we attempted to change the mutated nucleotides back to original nucleotides. Ligated DNA was introduced into E. coli strain DH5α and S. pyogenes strain 1529. We obtained a handful of and many transformants in E. coli and S. pyogenes, respecttively. Therefore, all E. coli transformants obtained were investigated, while only a subset of transformants in S. pyogenes were further investigated. We did not observe successful substitution in any of the E. coli transformants
Table 5. Substitution of the mutated nucleotides back to the original one.
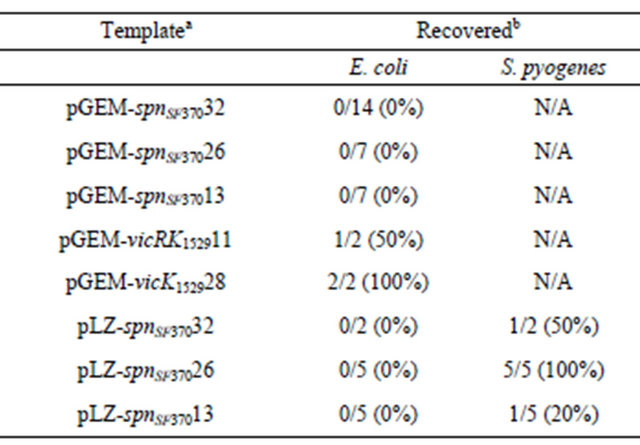
aPlasmids used for inverse PCR. bNumber of plasmid having the successful substitution/number of the analyzed plasmid (%). N/A: not applicable.
regardless of the plasmid templates used (Table 5). We did observe successful substitution in the S. pyognenes transformants for all three of the plasmid templates used (Table 5). Finally, the spnSF370 gene was successfully cloned only when S. pyogenes strain 1529 was used as host. These results suggest that the insert DNA encoding spnSF370 has some toxic effect to E. coli, but not S. pyogenes.
3.3. Construction and Analysis of SF370∆spn
The toxicity of spnSF370 could be related with Riddle et al.’s claim that SPN has a NADase-independent function. In order to further explore this hypothesis, spnSF370 gene was replaced with an antibiotics marker and the resulting strain SF370∆spn was used to infect mice.
When the SF370∆spn was inoculated in mouse skin, the mortality (36%) of the infected mice was not significantly different from the infection with the parental strain SF370 (14%) (p = 0.214, see Table 6). In addition, there were not significant differences in the lesion sizes of mouse skin infected with either SF370∆spn or strain SF370 (data not shown). Furthermore, bacteria were recovered from blood and spleen of the surviving mice on day 8 (Table 7). The bacterial number recovered from the mice infected with SF370∆spn was not reduced compared with the case infected with the parental strain SF370.
4. Discussion
Riddle et al. suggested that SPN has both NADase-dependent and NADase-independent function [19]. Cloning of a target gene is the first step in studying the function of genes in many biological researches. Therefore, in order to explore what is the NADase-independent function, we attempted to clone the spnSF370 gene encoding the NADase-inactive SPN in Escherichia coli. We initially expected that cloning this gene into E. coli would be simple, because it was believed that the toxicity of SPN for bacterial cells is associated with the NADase activity [11,19]. But in actuality, we were not able to clone the gene in E. coli, suggesting that spnSF370 gene has some NADase-independent toxicities to E. coli cells compared with several controls used in this study. Therefore, we were forced to explore another strategy to achieve successful cloning of the gene. We tested ifsSF370 gene as the first strategy, because spn alleles encoding NADaseactive SPN subtype have ever been cloned by aid of ifs gene in E. coli [11,12,20]. Consequently, spn-ifsSF370 also was not able to be cloned in E. coli. This result could be explained by the fact that ifsSF370 has been previously shown to degrade into a peudogene [19]. Additionally, we took into account that IFS does not necessarily provide a perfect suppression of the self-toxicity of the
Table 6. Virulence (Mortality) to mouse of SF370∆spn.

Mortality was determined on Day 8 (P = 0.214 for comparison of survival days).
Table 7. Bacterial number (CFU) recovered from the survived mice.
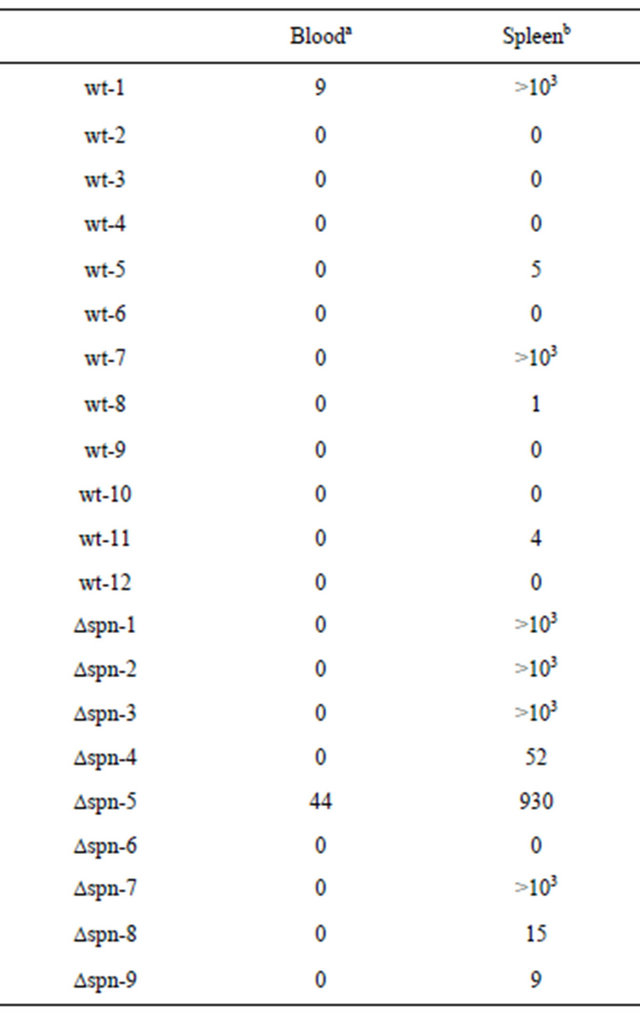
The mice, which survived until day 8 in Table 6, were used. a100 µl of blood from heart was spread on a sheep blood agar plate. bSpleen was homogenized and suspended with 100 µl of PBS. All the PBS (100 µl) was spread on a sheep blood agar plate. Two colonies per plate were randomly selected, and we observed coccus morphology and chain arrangements characteristic of Streptoccoal species by using a microscope. In addition, the colonies derived from the mice challenged with SF370Δspn were spectinomycinresistant, whereas the colonies from the mice challenged with the parental strain SF370 were sensitive to spectinomycin.
NADase-active SPN as described bellow. We had attempted to clone spnGT01 gene encoding NADase-active SPN by aid of ifsGT01 gene in E. coli in the previous study [20]. For this experiment, four different forward primers were used to amplify the spn-ifsGT01 genes with the reverse primer slo2 (Figure S2). While the first forward primer (Nga-n4Eco) does not contain any upstream DNA sequences encoding a potential ribosome-binding site, the other three would contain longer upstream DNA sequences (118, 185 and 287 bp respectively as shown in the Figure S2). For the latter three primers, we did not obtain any transformants containing the prospective plasmids. Using the Nga-n4Eco we obtained the resulting 13 transformants having the plasmids (pNGIe1 to 13, respectively) in which only the coding regions of spnGT01 were cloned. In addition, all the spnGT01 genes (of pNGIe1 to 13) were oriented in the opposite direction as the lacUV5 promoter on the pGEM®-T Easy vector. These selections for the upstream DNA sequences length and the orientation of the cloned spnGT01 may decrease the amount of NADase produced, because it have been already shown that at least addition of 16 bp and 26 bp upstream DNA sequences to spnGT01 resulted in the increased production of NADase activity in our previous study [20]. In that study, therefore, we hypothesized that plasmids producing NADase at lower level were selected for due to the potential toxicity of over produced NADase to bacterial cell. However we were not able to explain the reason why ifsGT01 gene did not sufficiently suppress the potential toxicity. Now, we propose a hypothesis that IFS GT01 could not inhibit the potential NADase-independent (self-) toxicity of SPNGT01 because IFS GT01 was inhibitor of NADase activity.
It is possible that S. pyogenes has some mechanism to manage the NADase-independent toxic properties of SPN as well as IFS for the NADase-dependent toxic property. Therefore, we attempted to use S. pyogenes as a host for cloning of spnSF370 gene. For this experiment, we used the E. coli-Streptococcus shuttle vector pLZ12-Km2 which copy number would be intermediate (personal communication with Dr. June R. Scott). The spnSF370 gene was successfully cloned in S. pyogenes, but not in E. coli. These results suggested that S. pyogenes has a mechanism for management of NADase-independent toxic properties of SPN that is lacking in E. coli.
The toxicity of spnSF370 could be related with Riddle et al.’s claim that SPN has a NADase-independent function. In order to further investigate the hypothetical function, we used the experimental mouse infection model. Based on our findings, we could not find any direct evidence for the hypothetical function. This may be related with the limitation of this experimental model, since humans are the only natural host for S. pyogenes. However, there was an unexpected result. It seemed that spnSF370 mutants survived better in the spleen (Table 7). About this, we have only one positive idea. Strain SF370 is not hyper virulent, compared with clinical isolates from severe invasive disease. NADase inactive SPNSF370 might contribute to the low virulence of the SF370. Hyper virulence is not only strategy in order to survive in host, because non-pathogenic, but not enterohemorrhagic, E. coli is living persistently in all human gut.
5. Conclusion
We have presented further supportive evidence that SPN has a NADase-independent function.
6. Acknowledgements
We thank Laura Leverton for critical reading of the manuscript and Hideyuki Matsui for technical assistance. This study was supported by JSPS KAKENHI Grant number 21790425 and KK24590531, a grant from Ohyama Health Foundation, and a grant from the 24th General Assembly of the Japanese Association of Medical Sciences (Medical Science Promotion Fund).
REFERENCES
- L. A. Cone, D. R. Woodard, P. M. Schlievert and G. S. Tomory, “Clinical and Bacteriologic Observations of a Toxic Shock-Like Syndrome Due to Streptococcus pyogenes,” New England Journal of Medicine, Vol. 317, No. 3, 1987, pp. 146-149. doi:10.1056/NEJM198707163170305
- C. W. Hoge, B. Schwartz, D. F. Talkington, R. F. Breiman, E. M. MacNeill and S. J. Englender, “The Changing Epidemiology of Invasive Group A Streptococcal Infections and the Emergence of Streptococcal Toxic ShockLike Syndrome. A Retrospective Population-Based Study,” JAMA, Vol. 269, No. 3, 1993, pp. 384-389. doi:10.1001/jama.1993.03500030082037
- B. Schwartz, R. R. Facklam and R. F. Breiman, “Changing Epidemiology of Group A Streptococcal Infection in the USA,” Lancet, Vol. 336, No. 8724, 1990, pp. 1167- 1171. doi:10.1016/0140-6736(90)92777-F
- D. L. Stevens, “Invasive Group A Streptococcal Infections: The Past, Present and Future,” Pediatric Infectious Disease Journal, Vol. 13, No. 6, 1994, pp. 561-566. doi:10.1097/00006454-199406000-00033
- M. Minami, Y. Wakimoto, M. Matsumoto, H. Matsui, Y. Kubota, A. Okada, M. Isaka, I. Tatsuno, Y. Tanaka and T. Hasegawa, “Characterization of Streptococcus pyogenes Isolated from Balanoposthitis Patients Presumably Transmitted by Penile-Oral Sexual Intercourse,” Current Microbiology, Vol. 61, No. 2, 2010, pp. 101-105. doi:10.1007/s00284-010-9581-x
- T. Hasegawa, S. N. Hashikawa, T. Nakamura, K. Torii and M. Ohta, “Factors Determining Prognosis in Streptococcal Toxic Shock-Like Syndrome: Results of a Nationwide Investigation in Japan,” Microbes and Infection, Vol. 6, No. 12, 2004, pp. 1073-1077. doi:10.1016/j.micinf.2004.06.001
- M. W. Cunningham, “Pathogenesis of Group A Streptococcal Infections,” Clinical Microbiology Revews, Vol. 13, No. 4, 2000, pp. 470-511. doi:10.1128/CMR.13.3.470-511.2000
- R. J. Olsen, S. A. Shelburne and J. M. Musser, “Molecular Mechanisms Underlying Group A Streptococcal Pathogenesis,” Cellular Microbiology, Vol. 11, No. 1, 2009, pp. 1-12. doi:10.1111/j.1462-5822.2008.01225.x
- A. L. Bricker, C. Cywes, C. D. Ashbaugh and M. R. Wessels, “NAD+-Glycohydrolase Acts as an Intracellular Toxin to Enhance the Extracellular Survival of Group A Streptococci,” Molecular Microbiology, Vol. 44, No. 1, 2002, pp. 257-269. doi:10.1046/j.1365-2958.2002.02876.x
- J. C. Madden, N. Ruiz and M. Caparon, “Cytolysin-Mediated Translocation (CMT): A Functional Equivalent of Type III Secretion in Gram-Positive Bacteria,” Cell, Vol. 104, No. 1, 2001, pp. 143-152. doi:10.1016/S0092-8674(01)00198-2
- M. A. Meehl, J. S. Pinkner, P. J. Anderson, S. J. Hultgren and M. G. Caparon, “A Novel Endogenous Inhibitor of the Secreted Streptococcal NAD-Glycohydrolase,” PLoS Pathogens, Vol. 1, No. 4, 2005, p. e35. doi:10.1371/journal.ppat.0010035
- H. Kimoto, Y. Fujii, S. Hirano, Y. Yokota and A. Taketo, “Genetic and Biochemical Properties of Streptococcal NAD-Glycohydrolase Inhibitor,” Journal of Biological Chemistry, Vol. 281, No. 14, 2006, pp. 9181-9189. doi:10.1074/jbc.M506879200
- R. Lutticken, D. Lutticken, D. R. Johnson and L. W. Wannamaker, “Application of a New Method for Detecting Streptococcal Nicotinamide Adenine Dinucleotide Glycohydrolase to Various M Types of Streptoccus pyogenes,” Journal of Clinical Microbiology, Vol. 3, No. 5, 1976, pp. 533-536.
- A. Michos, I. Gryllos, A. Hakansson, A. Srivastava, E. Kokkotou and M. R. Wessels, “Enhancement of Streptolysin O Activity and Intrinsic Cytotoxic Effects of the Group A Streptococcal Toxin, NAD-Glycohydrolase,” Journal of Biological Chemistry, Vol. 281, No. 12, 2006, pp. 8216- 8223. doi:10.1074/jbc.M511674200
- D. Ajdic, W. M. McShan, D. J. Savic, D. Gerlach and J. J. Ferretti, “The NAD-Glycohydrolase (nga) Gene of Streptococcus pyogenes,” FEMS Microbiology Letters, Vol. 191, No. 2, 2000, pp. 235-241. doi:10.1111/j.1574-6968.2000.tb09345.x
- T. Karasawa, K. Yamakawa, D. Tanaka, Y. Gyobu and S. Nakamura, “NAD(+)-Glycohydrolase Productivity of Haemolytic Streptococci Assayed by a Simple Fluorescent Method and Its Relation to T Serotype,” FEMS Microbiolgy Letters, Vol. 128, No. 3, 1995, pp. 289-292. doi:10.1111/j.1574-6968.1995.tb07538.x
- P. D. Lazarides and A. W. Bernheimer, “Association of Production of Diphosphopyridine Nucleotidase with Serological Type of Group A Streptococcus,” Journal of Bacteriology, Vol. 74, No. 3, 1957, pp. 412-413.
- I. Tatsuno, J. Sawai, A. Okamoto, M. Matsumoto, M. Minami, M. Isaka, M. Ohta and T. Hasegawa, “Characterization of the NAD-Glycohydrolase in Streptococcal Strains,” Microbiology, Vol. 153, No. 12, 2007, pp. 4253-4260. doi:10.1099/mic.0.2007/009555-0
- D. J. Riddle, D. E. Bessen and M. G. Caparon, “Variation in Streptococcus pyogenes NAD+ Glycohydrolase Is Associated with Tissue Tropism,” Journal of Bacteriology, Vol. 192, No. 14, 2010, pp. 3735-3746. doi:10.1128/JB.00234-10
- I. Tatsuno, M. Isaka, M. Minami and T. Hasegawa, “NADase as a Target Molecule of in Vivo Suppression of the Toxicity in the Invasive M-1 Group A Streptococcal Isolates,” BMC Microbiology, Vol. 10, 2010, p. 144. doi:10.1186/1471-2180-10-144
- J. J. Ferretti, W. M. McShan, D. Ajdic, D. J. Savic, G. Savic, K. Lyon, C. Primeaux, S. Sezate, A. N. Suvorov, S. Kenton, H. S. Lai, S. P. Lin, Y. Qian, H. G. Jia, F. Z. Najar, Q. Ren, H. Zhu, L. Song, J. White, X. Yuan, S. W. Clifton, B. A. Roe and R. McLaughlin, “Complete Genome Sequence of an M1 Strain of Streptococcus pyogenes,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 98, No. 8, 2001, pp. 4658-4663. doi:10.1073/pnas.071559398
- A. N. Suvorov and J. J. Ferretti, “Physical and Genetic Chromosomal Map of an M Type 1 Strain of Streptococcus pyogenes,” Journal of Bacteriology, Vol. 178, No. 18, 1996, pp. 5546-5549.
- A. Podbielski, B. Spellerberg, M. Woischnik, B. Pohl and R. Lutticken, “Novel Series of Plasmid Vectors for Gene Inactivation and Expression Analysis in Group A Streptococci (GAS),” Gene, Vol. 177, No. 1, 1996, pp. 137- 147. doi:10.1016/0378-1119(96)84178-3
- S. Lukomski, N. P. Hoe, I. Abdi, J. Rurangirwa, P. Kordari, M. Liu, S. J. Dou, G. G. Adams and J. M. Musser, “Nonpolar Inactivation of the Hypervariable Streptococcal Inhibitor of Complement Gene(sic) in Serotype M1 Streptococcus pyogenes Significantly Decreases Mouse Mucosal Colonization,” Infection and Immunity, Vol. 68, No. 2, 2000, pp. 535-542. doi:10.1128/IAI.68.2.535-542.2000
- C. D. Ashbaugh, H. B. Warren, V. J. Carey and M. R. Wessels MR, “Molecular Analysis of the Role of the Group A Streptococcal Cysteine Protease, Hyaluronic Acid Capsule, and M Protein in a Murine Model of Human Invasive Soft-Tissue Infection,” Journal of Clinical Investigation, Vol. 102, No. 3, 1998, pp. 550-560. doi:10.1172/JCI3065
- N. Okada, I. Tatsuno, E. Hanski, M. Caparon and Sasakawa C, “Streptococcus pyogenes Protein F Promotes Invasion of HeLa Cells,” Microbiology, Vol. 144, No. 11, 1998, pp. 3079-3086. doi:10.1099/00221287-144-11-3079
Supplement
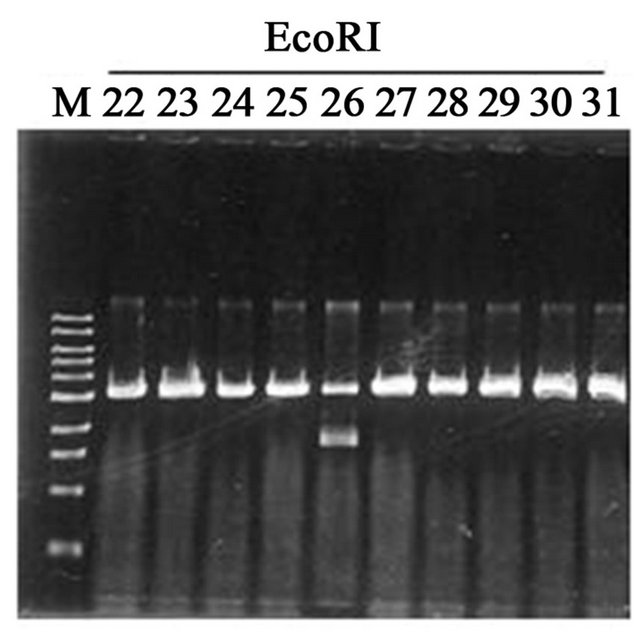
Figure S1. Cloning of SpnSF370 gene into a pGEM®-T Easy Vector. Representative plasmids (pGEM-spnSF37022 to 31) were shown. The pGEM-spnSF37026 (lane number 26) possessed the correct size insert (1.5 kbp).
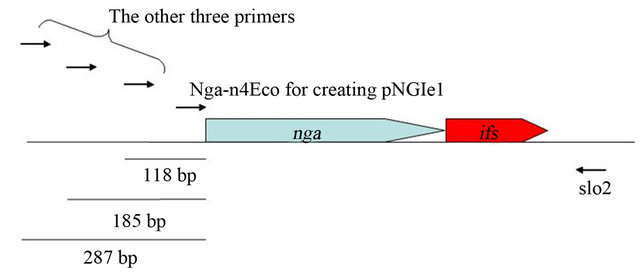
Figure S2. Physical map of spn-ifsGT01 genes. Used primers were shown as arrows.