Paper Menu >>
Journal Menu >>
 J. Software Engineering & Applications, 2008, 1: 8-12 Published Online December 2008 in SciRes (www.SciRP.org/journal/jsea) Copyright © 2008 SciRes JSEA 1 A Bioinformatics-Inspired Adaptation to Ukkonen’s Edit Distance Calculating Algorithm and Its Applicability Towards Distributed Data Mining Bruce Johnson University of Tennessee 1508 Middle Drive Knoxville, TN 1-865-974-3461 Email: bjohnson@cs.utk.edu Received November 25 th , 2008; revised November 29 th , 2008; accepted December 1 st , 2008. ABSTRACT Edit distance measures the similarity between two strings (as the minimum number of change, insert or delete operations that transform one string to the other). An edit sequence s is a sequence of such operations and can be used to represent the string resulting from applying s to a reference string. We present a modification to Ukkonen’s edit distance calculating algorithm based upon representing strings by edit sequences. We conclude with a demonstration of how using this representation can improve mitochondrial DNA query throughput performance in a distributed computing environment. Keywords: Bioinformatics-Inspired Adaptation, Calculating Algorithm, Data Mining 1. Introduction Let ∑ be a finite alphabet and let ∑ * denote the collection of finite strings over ∑. Edit distance is a means of measuring similarity between a target and reference string in ∑ * by computing the minimum number of change, insert, or delete edit operations that transform one string into another. The edit distance is a metric [1] and is a means of measuring the similarity between two strings [2]. Wagner and Fischer presented one of the first algorithms for calculating edit distance [3]. Ukkonen improved upon Wagner and Fischer’s algorithm (using potentially less time and space) [4,5]. However, a significant performance bottleneck in Ukkonen’s algorithm is calculating the length of a longest common prefix (which we refer to as the degree of agreement) between two strings. Let alphabet ∑ d = {a, c, g, t}. ∑ d can be regarded as representing the molecules adenine, cytosine, guanine and thymine respectively. These molecules are collectively known as nucleotides. When covalently bonded together, these molecules become a polymer called a polynucleotide. Two polynucleotides can produce the well-known double helix shape of DNA. The determination of the order in which the nucleotides are covalently bonded together in a polynucleotide is called sequencing. The act of sequencing yields a string since each nucleotide in the given polynucleotide maps to one of the members of ∑ d . The mitochondria are organelles found throughout eukaryotic cells. They are responsible for the production of adenosine triphosphate (ATP), the primary currency by which a cell’s energy needs are trafficked [6]. Mitochondria possess DNA (mtDNA). This mtDNA is ultimately responsible for the production of the proteins which regulate the mitochondrion and produce ATP. We define an mtDNA string to be the string that results from sequencing one of the polynucleotides that comprise mtDNA. Anderson et al. [7] were the first scientists responsible for sequencing a human’s mtDNA. The mtDNA string they produced is a standard reference and is now known as the Cambridge Reference Sequence (CRS). Mitochondrial DNA is the subject of much research by forensic scientists because it has features that aid them in their identification of an individual [8]. 1) It is widely distributed throughout a given cell 2) It is always inherited from a child’s mother 3) It is conservative, i.e., the edit distance between the CRS and a target mtDNA string is very small in comparison to their lengths. The first feature means that intact mtDNA can likely be extracted from some piece of human detritus such as hair or fingernails. The second feature means that it is likely that the mtDNA possessed by maternally related individuals is the same. This feature is particularly advantageous for individuals who seek to determine whether the remains of a body belong to their sibling. With regard to the third feature, we will show that since mtDNA is conservative, the performance of the longest common prefix calculation for Ukkonen’s edit distance calculating algorithm can be improved by representing strings as edit sequences. We will show how this feature can improve mtDNA query throughput performance in a distributed computing environment. 2. Preliminaries 2.1 Definitions We begin by defining edit operations (to streamline exposition, they may be referred to simply as operations).  A Bioinformatics-Inspired Adaptation to Ukkonen’s Edit Distance Calculating Algorithm 9 and Its Applicability Towards Distributed Data Mining Copyright © 2008 SciRes JSEA A nontrivial change operation has the form of ac σ and acts on string α = α 0 … α l (provided 0≤a≤l ) to produce β = β 0 … β l where α α σ β β = ≠ =i i i i if fi , , In other words, symbol α a at (address) a is changed to symbol σ . An insert operation has the form of ai σ and acts on string α = α 0 … α l (provided 0≤a≤l ) to produce β = β 0 … β l+1 where α α α σ α β >if if <if , ,1 , i i i a i i i = = − In other words, symbol σ has been inserted into string α at address a. A delete operation has the form of ad and acts on string α = α 0 … α l (provided 0≤a≤l) to produce β = β 0 … β l-1 where α α α α β >if <if , 1 , i i i i i = + In other words, symbol α a has been deleted from string α . A sequence of edit operations is referred to as an edit sequence. The concatenation of edit sequence s with t is denoted s|t. Given edit operation e, the function &() returns e’s address, (i.e. &(ad ) = a), the function τ() returns e’s type (i.e. τ (ai σ ) =i) and the function δ() returns the symbol to be inserted or changed, i.e. δ(ac σ ) = σ . A change operation e is called trivial (with respect to α ) if it acts as the identity function on α (i.e. e ( α ) = α ). To indicate that is trivial (when is understood) it may be written as at σ . The notation [expression] is defined as falseisexpif trueisexpif ,0 ,1 ][exp ression ression ression = Given strings α , β ∈ ∑ * , an edit sequence s taking α to β (i.e. s( α ) = β ) is produced by Wagner and Fischer’s algorithm [1]. Their algorithm-which we refer to as WF - first proceeds by calculating a (n+1) × (m+1) distance matrix D as follows (where | α | = n and | β | = m). otherwise 0 0 ),],[min(1 , , 1,,,1111,1 , = = ≠−+ = −−−−−− iif jif DDD j i D jijijiji ji βα Next, an edit sequence s (transforming α into β ) is obtained by the recursive function S S(∅) = ε S(D) = e | S(D′) where ∅ denotes the empty matrix (0 rows, 0 columns), ε denotes the empty edit sequence, and D′ is either the result of removing the last D (if case 1 applied), removing the last column from D if case 2 applied or removing both the last row and last column from D (if case 3 or 4 is applied). 4case 3case 2case 1case otherwise 1if 1if 1if )1( )1( )1( , 1,1, 1,, ,1, 1 1 1 ' ' ' −− − − − − − += += += − − − = mnmn mnmn mnmn n m m DD DD DD tm cm im md e α β β Given edit sequence s = S transforming α into β , the function r (s, α , β ) returns the reduced edit sequence s r (with respect to α ). Example: let s = e 0 e 1 e 2 e 3 e 4 e 5 = 0tg 1tt 2d 2ig 3tt 4ct. Then, s r = r (s, α , β ) = e 0′ e 1′ e 2′ = 2d 2ig 4ct. Note that 1) 0′=2, 1′=3 and 2′=5 2) both s and s r map α to β 3) s r uses the minimum number of edit operations to transform α to β Edit sequence s = S(D) has the following properties. 1) No edit sequence mapping α to β is shorter than r(s, α , β ). 2) Addresses of edit operations found in s are nondec- reasing. 3) If e j is a delete edit operation in s, then &(e j )= &(e j+1 ). 4) If e j is an insert or change edit operation in s, then e j + 1 has an address that differs from e j by one. 2.2 Characteristics of Reduced and Non-reduced Edit Sequences Given edit sequence s, define 〈s〉 by Given s r ( α ) = β the length of β can be recovered by | β | = α + 〈s〉 Let ρ t be a subsequence of s consisting of trivial change operations, maximal with respect to containment, such that the addresses of successive members differ by one. Such a subsequence ρ t is called a trivial change queue. Example: s =0ta 1tc 2ia 3ct 4tt; ρ t = 0ta 1tc. Let ρ c be a subsequence of s consisting of nontrivial change operations, maximal with respect to containment, such that the addresses of successive members differ by one. Such a subsequence ρ c is called a nontrivial change queue. Example: s = 0ca 1cc 2ia 3ct 4tt; ρ c = 0ca 1cc. Let ρ i be a subsequence of s consisting of insert operations, maximal with respect to containment, such that the addresses of successive members differ by one. Such a subsequence ρ i is called an insert queue. Example: s = 0ia 1ic 2ca 3ct 4tt; ρ i = 0ia 1ic. Let ρ d be a subsequence of s consisting of delete operations, maximal with respect to containment, such that the addresses of successive members do not differ. Such a subsequence ρ d is called a delete queue. Example: s = 0d 0d 0t t 1ca; ρ d = 0d 0d. The length of a change or insert queue ρ = e y … e z is given by | ρ | = &(e z )-&(e y ) + 1. 2.3 Recovering Elements of s Using s r = r(s, α αα α , β ββ β ) Given s r = r(s, α , β ), we can recover the trivial change queues removed from s while producing s r . We will first  10 A Bioinformatics-Inspired Adaptation to Ukkonen’s Edit Distance Calculating Algorithm and Its Applicability Towards Distributed Data Mining Copyright © 2008 SciRes JSEA consider how to find the locations and then the symbols associated with trivial change queues. A trivial change queue ρ t may be a prefix, a suffix or neither a prefix nor a suffix of s r . In order to find the addresses of members of ρ t , there are three cases to consider. Case 1: Queue ρ t = e k … e l is a prefix of s r : Queue ρ t is a prefix of s r if &(e 0′ ) > 0. Furthermore, k = &(e k ) = 0 and l = &(e l ) = &(e 0′ ) - 1. Case 2: Queue ρ t = e k … e l is a suffix of s r : Queue ρ t is a suffix of s r if the last edit operation, e m ′ , in s r has address &(e m′ ) < n = |β| - 1. Furthermore, k = m ′ + 1, &(e k ) = &(e m ′ ) + [τ (e m′ ) ≠ d ], &(e l ) = n and l = m′ + (&(e l ) – &(e m ′ + 1 ) + 1). Case 3: Queue ρ t = e k … e l is neither a prefix nor a suffix of s r : Queue ρ t is neither a prefix nor a suffix of s r if the consecutive edit operations e j ′ and e (j+1)′ in s r have addresses &(e j′ ) < &(e (j+1)′ ) – [τ(e j′ ) = d ]. Furthermore, k = j′ + 1 and l = ( j+1)′ – 1 where &(e k ) = &(e j ′ ) + [τ(e j ′ ) = d] and &(e l ) = &(e (j+1)′ ) – 1. Now that we know how to find the addresses of members of trivial change queues, we need to find their symbols. Given s r = r(s, α , β ). Let cell D i,j have a column whose address is that of a trivial change operation. Let function ni( j) return the number of insert edit operations in s r whose addresses are less than j. Let function nd( j) return the number of delete edit operations in s r whose addresses are less than or equal to j. In order to find the symbols in trivial change queues, we discovered that nd( j) – ni( j) = i – j. Since nd( j) – ni( j) = i – j it follows that α i = α j + nd(j) – ni(j) . If e = at α i then we can say that e = at α j + nd(j) – ni(j) . Since the address of e is equal to the column j labeled by D i,j , we can say that e = jt α j + nd(j) – ni(j) . Hence, given α and s r , we can acquire the address and symbol associated with each trivial change operation in s. Given element β x , let t r = Partition (s r , x) return edit sequence t r whose elements are comprised of those elements of s r whose addresses are greater than or equal to x. Let e = GetOp(s r , y) return the first edit operation found in s r whose address is greater than or equal to y. Let ρ t be a trivial change queue, the following pseudocode ρ t = Recover(s r , x) shows the procedure for finding trivial change queues in s r . The code is initialized by a call to Partition (s r , x). ρ t = Recover (t r , x) 1. e = GetOp(t r , x) 2. if (e == e 0′ && &(e 0′ ) > 0) //Case 1 2.1. k = 0 2.2. l = &(e l ) 2.3. return ( ρ t = e k … e l ) 3. if (e == e m′ && &(e m′ ) < n = |β| - 1) //Case 2 3.1. k = m′ + 1 3.2. l = m′ + (&(e l ) – &(e m ′ + 1 ) + 1) 3.3. return ( ρ t = e k … e l ) 4. if (e==e j ′ && &(e j ′ )<&(e (j+1)′ )–[τ(e j ′ ) == d ]) //Case 3 4.1. k = j′ + 1 4.2. l = ( j+1)′ – 1 4.3. return ( ρ t = e k … e l ) 5. return ∅ 3. Calculating the Degree of Agreement Using Edit Sequences 3.1 Motivation for Using Reduced Edit Sequences At this point, it is productive to ask why we care about reduced edit sequences. Let reference string α be the CRS, target strings β and γ be mtDNA strings and let s r1 =r (s 1 , α , β ) and s r2 = s (s 2 , α , γ ). Edit sequences s r1 and s r2 (and reference string α ) can be used as a means of representing β and γ , respectively. This is significant because large, conservative target strings are represented by edit sequences that are substantially smaller. Hence, calculating the edit distance between β and γ by using α , s r1 and s r2 , may lead to a more efficient utilization of distributed computing resources for calculating edit distance by increasing network throughput. Furthermore, using α , s r1 and s r2 can afford forensic experts seeking to find a match for an mtDNA string the ability to store and carry large numbers of mtDNA sequences. 3.2 Our Algorithm Let β and γ be target strings of lengths m and n, respectively. Let s r1 = r (s 1 , α , β ) and s r2 = s (s 2 , α , γ ) and let (0≤x 1 ≤m-1) and (0≤x 2 ≤n-1). We want to know the length of the longest common prefix of the substrings β x1 … β m - 1 and γ x2 … γ n-1 (i.e. the degree of agreement between β and γ ). We will now consider how the degree of agreement between β and γ can be calculated using reduced edit sequences that represent β and γ by examining how our algorithm deals with the different types of edit operations that comprise our edit sequences used to represent our strings. Case 1: x 1 or x 2 is the address of a member of a delete queue. In this case, we do not have any symbols to compare; hence, we will simply traverse to the end of the respective queues. Case 2: x 1 and x 2 are the addresses of members of trivial change queues ρ 1 and ρ 2 , respectively. Let l 1 be the last member of ρ 1 and let l 2 be the last member of ρ 2 . Let e 1 be a member of ρ 1 and let e 2 be a member of ρ 2 where e 1 =x 1 cα w , e 2 =x 2 cα y , w=x 1 +nd 1 (x 1 ) –ni 1 (x 1 ) and y=x 2 +nd 2 (x 2 ) – ni 2 (x 2 ). If w = y, then δ((x 1 + n) tα w + n ) = δ((x 2 + n)tα y + n ) for 0 ≤ n ≤ min(g, h), where g = |{e 1 … l 1 }| and h = |{e 2 … l 2 }|. Hence, the degree of agreement will be min(g, h). Case 3: x 1 and x 2 are the addresses of members of ρ 1 and ρ 2 , respectively and neither ρ 1 nor ρ 2 are trivial change queues nor delete queues. Let e j and e k be members of ρ 1 and ρ 2 respectively, and let &(e j ) = x 1 and &(e k ) = x 2 . Let e y and e z be the last members of queues ρ 1 and ρ 2 , respectively. Let r be the degree of agreement between β and γ . We compare the symbols associated with these queues sequentially using the following loop. 1. r = 0 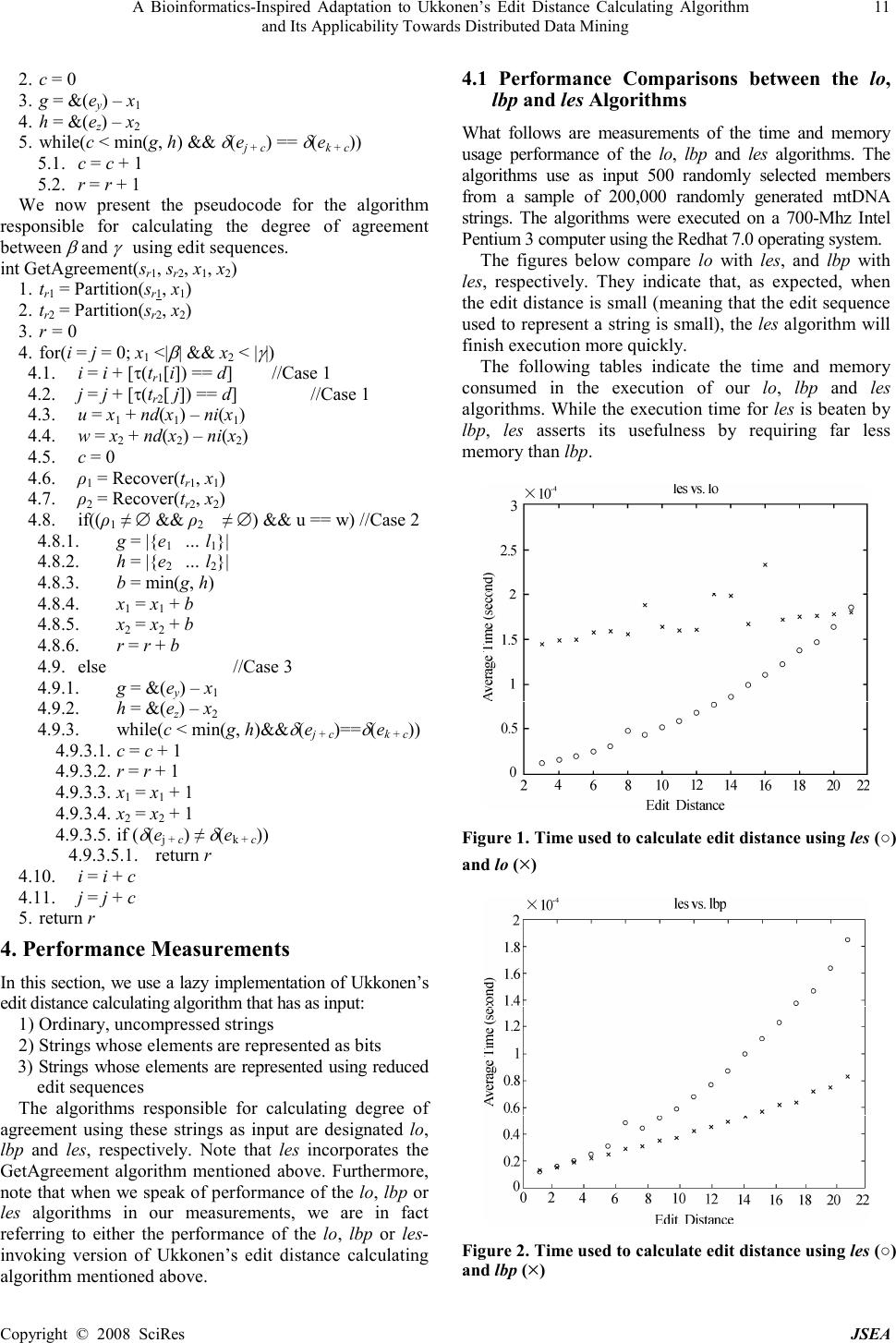 A Bioinformatics-Inspired Adaptation to Ukkonen’s Edit Distance Calculating Algorithm 11 and Its Applicability Towards Distributed Data Mining Copyright © 2008 SciRes JSEA 2. c = 0 3. g = &(e y ) – x 1 4. h = &(e z ) – x 2 5. while(c < min(g, h) && δ (e j + c ) == δ (e k + c )) 5.1. c = c + 1 5.2. r = r + 1 We now present the pseudocode for the algorithm responsible for calculating the degree of agreement between β and γ using edit sequences. int GetAgreement(s r1 , s r2 , x 1 , x 2 ) 1. t r1 = Partition(s r1 , x 1 ) 2. t r2 = Partition(s r2 , x 2 ) 3. r = 0 4. for(i = j = 0; x 1 <| β | && x 2 < | γ |) 4.1. i = i + [τ(t r1 [i]) == d] //Case 1 4.2. j = j + [τ(t r2 [ j]) == d] //Case 1 4.3. u = x 1 + nd(x 1 ) – ni(x 1 ) 4.4. w = x 2 + nd(x 2 ) – ni(x 2 ) 4.5. c = 0 4.6. ρ 1 = Recover(t r1 , x 1 ) 4.7. ρ 2 = Recover(t r2 , x 2 ) 4.8. if((ρ 1 ≠ ∅ && ρ 2 ≠ ∅) && u == w) //Case 2 4.8.1. g = |{e 1 … l 1 }| 4.8.2. h = |{e 2 … l 2 }| 4.8.3. b = min(g, h) 4.8.4. x 1 = x 1 + b 4.8.5. x 2 = x 2 + b 4.8.6. r = r + b 4.9. else //Case 3 4.9.1. g = &(e y ) – x 1 4.9.2. h = &(e z ) – x 2 4.9.3. while(c < min(g, h)&& δ (e j + c )== δ (e k + c )) 4.9.3.1. c = c + 1 4.9.3.2. r = r + 1 4.9.3.3. x 1 = x 1 + 1 4.9.3.4. x 2 = x 2 + 1 4.9.3.5. if ( δ (e j + c ) ≠ δ (e k + c )) 4.9.3.5.1. return r 4.10. i = i + c 4.11. j = j + c 5. return r 4. Performance Measurements In this section, we use a lazy implementation of Ukkonen’s edit distance calculating algorithm that has as input: 1) Ordinary, uncompressed strings 2) Strings whose elements are represented as bits 3) Strings whose elements are represented using reduced edit sequences The algorithms responsible for calculating degree of agreement using these strings as input are designated lo, lbp and les, respectively. Note that les incorporates the GetAgreement algorithm mentioned above. Furthermore, note that when we speak of performance of the lo, lbp or les algorithms in our measurements, we are in fact referring to either the performance of the lo, lbp or les- invoking version of Ukkonen’s edit distance calculating algorithm mentioned above. 4.1 Performance Comparisons between the lo, lbp and les Algorithms What follows are measurements of the time and memory usage performance of the lo, lbp and les algorithms. The algorithms use as input 500 randomly selected members from a sample of 200,000 randomly generated mtDNA strings. The algorithms were executed on a 700-Mhz Intel Pentium 3 computer using the Redhat 7.0 operating system. The figures below compare lo with les, and lbp with les, respectively. They indicate that, as expected, when the edit distance is small (meaning that the edit sequence used to represent a string is small), the les algorithm will finish execution more quickly. The following tables indicate the time and memory consumed in the execution of our lo, lbp and les algorithms. While the execution time for les is beaten by lbp, les asserts its usefulness by requiring far less memory than lbp. Figure 1. Time used to calculate edit distance using les (○) and lo ( × ) Figure 2. Time used to calculate edit distance using les (○) and lbp ( × )  12 A Bioinformatics-Inspired Adaptation to Ukkonen’s Edit Distance Calculating Algorithm and Its Applicability Towards Distributed Data Mining Copyright © 2008 SciRes JSEA 4.2 Query throughput Performance Comparisons in a Distributed Computing Environment Using the lo, lbp and les Algorithms A query is defined as an mtDNA string submitted by a client to a server. Query satisfaction is defined as the determination of which mtDNA strings residing on a server fall within an edit distance threshold of the query. Query throughput is defined as the number of edit distance calculations performed in a second while satisfying a query. The following tables provide performance measurements in terms of query strings submitted per second and queries satisfied per second for the lo, lbp and les algorithms in a LAN and WAN distributed computing environment. The algorithms used as input 200,000 randomly generated mtDNA strings. The queries were transmitted on a 1GB LAN where each network node was a 3.2-Ghz Intel Pentium 4 computer using the Debian GNU/Linux 3.1 operating system. The queries were also transmitted on a 54MB wireless WAN where the client and server were 2.2-Ghz and 2.4-Ghz Table 1. Time consumption (microseconds) les lbp lo Average 79 43 172 Minimum 12 13 145 Maximum 185 83 234 Table 2. Memory consumption (bytes) les lbp lo Average 337.6 8494 33777 Minimum 300 8494 33777 Maximum 372 8494 33777 Table 3. LAN throughput performance (strings submitted/second) les lbp lo Average 3.3e4 1.2e3 310 Minimum 2.9e4 1.1e3 295 Maximum 3.5e4 1.3e3 326 Table 4. LAN query throughput performance les lbp lo Average 1.7e4 1.2e3 310 Minimum 5.8e3 1.1e3 295 Maximum 3.4e4 1.3e3 326 Table 5. WAN throughput performance (strings submitted/second) les lbp lo Average 9.1e3 353 88 Minimum 7.8e3 340 84 Maximum 9.6e3 362 92 Table 6. WAN query throughput performance les lbp lo Average 9.1e3 353 88 Minimum 7.8e3 340 84 Maximum 9.6e3 362 92 Intel Pentium 4 computers, respectively, and were each using the Windows XP operating system. Network performance was measured using Jperf 2.0 [9]. We see that when queries are submitted in a distributed computing environment, the les algorithm can accept more query strings transmitted and therefore allows our les algorithm to achieve greater query throughput than either the lbp or les algorithms. 5. Conclusions This decade has witnessed three major disasters-the 9/11 attacks, the Indian Tsunami and hurricane Katrina. In the wake of such disasters, identifying people who have perished is of paramount importance. The usefulness of the les algorithm is asserted by the fact that it consumes far less memory than competing algorithms lo and lbp. This means that greater information throughput may be achieved on a network and thus greater use of distributed computational resources is facilitated. Moreover, this means that forensic experts can store far more mtDNA sequences using the les algorithm than they could if they were using the mtDNA strings required by lo or lbp algorithms. Having the ability to store a huge number of mtDNA sequences by forensic experts could prove to be a boon by those forensic experts charged with the duty of identifying the remains of people after a major disaster. Having the ability to draw from a vast database of mtDNA strings increases the likelihood that a match can be made between the mtDNA collected and the mtDNA stored in a database. 6. Acknowledgements We would like to thank Dr. Michael Vose for his kind mentorship and guidance. REFERENCES [1] M. D. Vose, “A formal analysis of edit distance,” UT CS Technical Report ut-cs-04-517, February 2004. [2] R. O. Duda and P. E. Hart, Pattern Classification (2nd ed.). Wiley Interscience, 2000. [3] A. Wagner and M. I. Fischer, “The string-to-string correction problem,” Journal of the ACM, 21(1) (Jan. 1974), pp. 168-173, 1974. [4] E. Ukkonen, “Algorithms for approximate string matching,” International Control 64, pp. 100-118, 1985. [5] E. Ukkonen, “On approximate string matching,” International Conference Fundamentals of Computation Theory, Lecture Notes in Computer Science, pp. 158:487-495, 1983. [6] N. Campbell and J. Reese, Biology (6th ed.), Addison Wesley, 1997. [7] S. Anderson, et al., “Sequence and organization of the human mitochondrial genome,” Nature, 290(5806) (April 9, 1981), pp. 457-265, 1981. [8] K. L. Monson, et al., “The mtDNA population database: An integrated software and database resource for forensic comparison,” Forensic Science Communications, 4(2), April 2002. DOI=http://www.fbi.gov/hq/lab/fsc/ backissu/april2002/miller1.htm. [9] http://iperf.sourceforge.net. |