Journal of Cancer Therapy
Vol.5 No.6(2014), Article ID:46251,14 pages DOI:10.4236/jct.2014.56068
Effects of Okadaic Acid, Retinoic Acid, and Phorbol Myristate Acetate Tumor Promoter on Oncogene Expression
John J. Wille1, Jong Y. Park2
1Bioderm Technologies, Inc., Trenton, USA
2Department of Cancer Epidemiology, Moffitt Cancer Center, Tampa, USA
Email: jjwille@aol.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 17 April 2014; revised 10 May 2014; accepted 18 May 2014
ABSTRACT
The effect of okadaic acid (OA) on proto-oncogene protein expression of c-neu, c-myc, v-rasH, EGFR, and phosphotyrosine-containing phosphoproteins (P-Tyr) was investigated in rapidly growing (RG) normal human keratinocytes (NHK) and in SV-40 virally-transformed keratinocytes (SVK) cultured in a growth factor supplemented serum-free medium as assessed by indirect immunofluorescence microscopy. P-Tyr positively stains cell surface antigens (cytoplasm) diffusely at monopolar sites in RG NHK cultures. OA-treatment intensifies cytoplasmic P-Tyr staining at localized monopolar intercellular focal adhesion (IFA) sites with reduced cytoplasmic staining. P-Tyr expression was predominate at IFA sites with little cytoplasmic staining in RG SVK cultures. OAtreatment increased monopolar P-Tyr staining and cytoplasmic staining. OA-treatment in RG NHK cultures intensified cytoplasmic staining of c-myc and EGFR (epidermal growth factor receptor) expression. OA-treatment in RG NHK and SVK cultures intensified c-neu staining at monopolar IFA sites and intensified c-neu staining at both cytoplasmic and bipolar IFA sites in RG SVK cells. OA was especially cytotoxic for SVK cells. RA treatment decreased c-neu expression in RG NHK cultures while TPA treatment has a lesser effect on both cytoplasmic and IFA sites. RA treatment also decreased P-Tyr staining in both NHK and SVK cells. Again, TPA had a lesser inhibitory effect on P-Tyr staining pattern. RA-treatment had a similar effect on P-Tyr staining of RG cultures of a mouse fibroblast cell line. These results confirm the generality of OA, RA and TPA on the regulation of oncogene expression in both normal and malignantly transformed keratinocytes.
Keywords:Epidermal Keratinocytes, Indirect Immunofluorescence Microscopy, Oncogene Protein Antibodies, Okadaic Acid, Phosphotyrosine Antibody, Retinoic Acid, SV-40 Transformed Keratinocytes, TPA, Tumor Promoter

1. Introduction
Oncogenes are genes that have the potential to cause cancer, and often are mutated or overexpressed in tumor cells. Activated oncogenes can suppress apoptosis, overcome environmentally-induced senescence, induce cellular immortalization, and uncontrolled proliferation. Normal cells often possess an analog DNA sequence or proto-oncogene that is not oncogenic unless mutated or overexpressed. They can be activated by mitogenic stimuli similar to their related oncogenes such as c-myc and c-neu, which also activate other plasma membraneassociated tyrosine protein kinases [1] . Ras protein belongs to family of regulatory GTPases involved in signaling pathways leading to cell proliferation. The polyoma DNA virus, Simian Virus 40 (SV-40) can also activate oncogene expression through its transcribed large T and middle T oncoproteins. These viral oncoproteins are complex with target proteins inhibiting tumor suppressor activities of p53 tumor-suppressor protein and the retinoblastoma protein [2] . The role of phosphotyrosine-binding domains in signal transduction and cancer has been well described [3] .
HER2/neu, known as receptor tyrosine protein kinase erbB-2, is a protein encoded by the ERBB2 gene or HER2, human epidermal growth factor receptor 2. Amplification or overexpression of HER2 plays a role in development and progression of breast cancer. Activation of HER2 proteins has been shown to form clusters in cell membranes that may play a role in tumorigenesis. Signaling through the erbB2 family of receptors promotes cell proliferation and suppresses apoptosis. In promoting carcinogenesis, erbB-2 is a potent oncogene when overexpressed in NIH/3T3 cells. The synergistic interaction of EGF receptor with c-neu leads to transformation of rodent fibroblasts. In normal keratinocytes, activation of heparin-binding EGF-like growth factor B receptors mediates hyperplasia [4] . In normal keratinocytes the HER family and the heparin-bonding EGF family member, heregulin are expressed differentially and modulate keratinocyte differentiation [5] . It has been reported that inhibition of erbB receptor family members protects keratinocytes from UV-B induced apoptosis [6] [7] .
myc expression regulates cell cycle clock function, promotes cell proliferation and can repress the expression of CDK inhibitors P15INK4B and p21CIP/ thereby, promoting continued cell cycle advance and molecular carcinogenesis. Constitutive expression of c-myc in murine keratinocytes (BALB/MK cells) reinitiated DNA synthesis by increasing sensitivity to EGF growth factor stimulation to cell proliferation [8] .
In this study, we employed normal human keratinocytes (NHK) and a tumor-derived cell line of SV-40 transfected and malignantly transformed human keratinocytes (SVK) to investigate the effect of two differently- acting tumor promoters, the phorbol ester (TPA), and okadaic acid (OA), and an anti-tumor promoter, all-transretinoic acid (t-RA) on oncogene expression. Previously, we reported that normal diploid human keratinocytes could be malignantly transformed by whole SV-40 virus [9] . We reported that SVK cells were immortal, anchorage-independent, expressed both keratin 10 protein and large T nuclear antigen, and were tumorigenic when injected in athymic mice. More importantly, we demonstrated that SVK cells were able to grow in serum-free medium without an EGF supplement. In order to verify the generality the effects of OA, RA and TPA on oncogene expression we extended the study to a well-studied mouse embryonic fibroblast line, C3H10T1/2. This cell type has been virally and chemically transformed and is thought to be an initiated cell line, making it an interesting target for co-carcinogenic agents employed here.
We have employed the technique of indirect immunofluorescence microscopy [10] to study the effects of OA, t-RA and TPA on oncogene expression. Earlier studies reported that OA, a known cytotoxin isolated from a marine sponge, was a tumor promoter [11] [12] with divergent pleiotropic activities. It paradoxically can revert the transformed phenotype of cells transformed by raf oncogene to the normal phenotype [13] . Unlike the phorbol ester-type tumor promoter, TPA, OA does not activate any known protein kinase-C (PK-C). Rather, OA suppresses the action of phosphatases 1 & 2A (PPT’s) [14] . OA interferes with interphase to mitotic-like microtubule dynamic instability, alters the shape and F-actin distribution and inhibits stimulus-dependent increases in cytoskeleton actin in human neutrophils [15] . It induces morphological changes, apoptosis and cell cycle alterations in many different normal and carcinoma human cell types [16] . It also inhibits protein synthesis in Vero cells through lipid peroxidation and production of oxygen reactive radicals [17] , and stimulates protein phosphorylation in intact cells [18] . Importantly, OA stimulates apoptosis through expression of Fas receptor and Fas ligand in human oral squamous carcinoma cells through activation of nuclear factor kappa B [19] . OA regulates EGR-1 transcription, blocks growth factor activation of extracellular signal-regulated kinase (ERK)1/2, elevates the activities of ERK1/2, c-jun N-terminal kinases (JNKs) and p38 mitogen activated protein kinases (MAPKs), and induces dephosphorylation of histone H1 in metaphase-arrested HeLa cells [20] . In papilloma-producing mouse keratinocytes, elevation of the latter kinase is correlated with AP-1 binding to TPA responsive element (TRE) [19] .
Retinoids are essential for normal epidermal differentiation and a deficiency may cause hyperkeratonic metaplasia [21] . They modulate many different cellular interactions often with divergent pleiotropic effects on growth and differentiation. t-RA inhibits the proliferation of many different untransformed, transformed and tumor cell lines [22] including C3H10T1/2 and normal keratinocytes examined here. RA reversibly arrests normal keratinocytes in the G1 phase of the cell cycle [23] and increases the number of EGF receptors in 3T6 fibroblast cell and many untransformed, and spontaneous and chemically-transformed fibroblast cell lines. Retinoids modulate terminal differentiation and response of tumor promoters in mouse epidermal cell cultures. Retinoids reverse benzo[α]pyrene-induced keratinization in normal hamster tracheal explants [24] . RA suppresses TPAinduced ornithine decarboxylase enzyme induction (ODC) [25] and mitigates tumor promotion in the two-stage model of mouse skin tumorigenesis [26] . A DNA antisense construct of the open-reading frame of the α-nuclear RA receptor induced apoptosis in an immortalized keratinocyte cell line, indicating that RA-linked transcription cascade is vital to maintain keratinocyte stem cell function [27] .
Phorbol ester type tumor promoters such as 12-tetradecanoyl phorbol 13-acetate (TPA), the active principle of croton oil tumor promoter is a potent activator of protein kinase-C [28] . TPA inhibits the clonal growth of normal human keratinocytes, inhibits DNA synthesis and arrests cells in both the G1 and G2 phases of the cell cycle, and induces terminal keratinizing squamous differentiation of NHK cells [29] . We previously reported that OA elicits a distinct set of oncoproteins associated with the onset tumor promotion. In this report, we investigated the effect of TPA on oncogene expression in both NHK and a tumorigenic cell line of SV-40 transfected human keratinocytes, and contrasted it relative to the effect of OA on EGFR, c-neu, v-rasH, c-myc and on the expression of phosphotyrosine-containing phosphoproteins [30] . The technique of indirect immunofluorescence microscopy was employed to study the effects of OA, t-RA and TPA on oncogene and proto-oncogene expression.
2. Materials and Methods
2.1. Materials
All primary antibodies (c-neu, c-myc, v-rasH, EGFR and P-Tyr) were obtained from Oncogene Sciences (Manhasset, NY). All other organic compounds and tissue culture supplies were purchased from Sigma-Aldrich Co (St. Louis, MO). Okadaic acid was obtained as a gift from Dr. F. O. Cope (National Cancer Institute, Tokyo, JN).
2.2. Cell Culture
Normal human keratinocytes (NHK) were prepared as previously reported and were cultured in a modification of MCDB 151 (HECK 109), supplemented with 5 µg/ml insulin 5 ng/ml EGF, and 0.2% bovine pituitary extract [31] . The origin and derivation of an SV40 virus-transfected cell line and its malignantly transformed cell line of normal human diploid keratinocytes (SVK) were as we previously described [9] . They were cultured in the same medium supplemented with insulin only. OA was added to cell culture medium at a final concentration of 1nM for 7 hours; RA or tumor promoter, TPA was added at a final concentration of 2 × 10−6 M, 100 ng/ml for 24 hours, respectively.
2.3. Indirect Immunofluorescence Assay
As we previously described, [10] cells attached by 3 days of culture growth to sterile 12-mm glass coverslips and fixed with 3.7% formalin in 1X PBS. To assess the effect of dephosphorylation on the phosphorylation status of cells treated with various test agents cells grown on glass chips were incubated under appropriate conditions suggested by the supplier to a solution of alkaline phosphatase enzyme (AP+) prior to fixation. The fixed cells were then washed in 1X PBS and prepared for indirect immunofluorescence microscopy by first staining for 30 minutes with primary antibody, and then with a fluorescence-conjugated second antibody to either mouse or rat IgG, and recorded with a fluorescence microscope (Zeiss). Control cells not exposed to primary antibody but stained with the appropriate second IgG-FITC-conjugated antibody were used as a negative control for fluorescent stain. Titers of the working solutions in 1X PBS of both first and second antibody were as suggested by the supplier. Minimum 10 random fields were examined and recorded before selection of typical representative figures.
3. Results
3.1. Effect of Okadaic Acid Treatment
3.1.1. Expression of Phosphotyrosine (P-Tyr) in Normal and SVK Cells
The typical patterns of P-Tyr expression in untreated (a), OA-treated (b) RG cultures of normal keratinocytes, and in untreated (c), OA treated (d) cultures of SVK keratinoctyes were presented (Figure 1). Most of the larger (average diameter was 7.2 ± 0.1 µM) untreated cycling basal keratinocytes (a) display intense cytoplasmic staining with some evidence of preferential monopolar localization (arrowhead), while smaller cells have only unipolar cytoplasmic staining. Treatment with OA (1 nM for 7 hours) greatly enhances P-Tyr staining with a definite unipolar cone-shaped localization. Moreover, OA-treated cells are smaller in size (average diameter was 5.2 ± 0.1 µM) and are aggregated in to clumps with the cone-shaped unipolar cytoplasmic staining oriented in a parallel manner (b). By contrast, untreated cultures of rapidly cycling SVK keratinocytes (c) display highly intense P-Tyr staining restricted to spherically-shaped uniand bipolar staining pattern. A similar P-Tyr staining pattern was observed for cultures of OA-treated SVK keratinocytes (d) where staining occurred as lens-shaped uniand bipolar staining and a fairly intense and uniform particulate-type cytoplasmic staining. OA-treated SVK cells appear to be larger and more flattened than corresponding untreated SVK cells.
3.1.2. Expression of Epidermal Growth Factor Receptor (EGFR) in Normal and SVK Cells
The typical patterns of EGFR expression in untreated (a), OA-treated (b) RG cultures of normal keratinocytes, and in untreated (c), OA treated (d) cultures of SVK keratinoctyes were presented (Figure 2). Cytoplasmic staining was moderately intense in untreated cycling NHK cells with a polarized localization of the EGFR stain (a). Average cell size (diameter) of untreated NHK was 8.0 µM). OA-treated NHK cells (b) displayed elevated intense cytoplasmic and nuclear EGFR staining. OA treatment cultures of SVK cells resulted in cellular aggre-
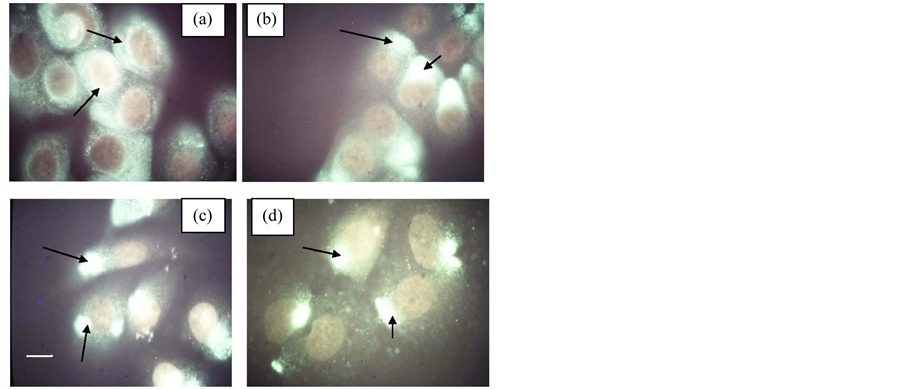
Figure 1. A panel of four photomicrographs of NHK ((a) & (b)) and SVK ((c) & (d)) cultures immunofluorescence (I)-stained with P-Tyr antibodies. (a) untreated NHK (two arrows point to two cells with intense perinuclear and cytoplasmic I-stain; (b) OA-treated NHK culture (two arrows point to cone-shaped monopolar caps with intense I-stain; (c) untreated SVK culture (two arrows point to monopolar and bipolar cytoplasmic areas showing intense I-stain); (d) OA-treated SVK culture (two arrows point to monopolar cytoplasmic areas showing intense I-stain near points of cell-cell contact). Total magnification, 1650×. Scale bar (white) 1 cm = 1.6 µM.

Figure 2. A panel of four photomicrographs of NHK ((a) & (b)) and SVK ((c) & (d)) cultures I-stained with EGFR antibodies. (a) untreated NHK culture (two arrows point to two cells with intensely I-stained but diffuse monopolar areas); (b) OA-treated NHK culture (two arrows points to I-stained nuclei in cells with increased cytoplasmic staining); (c) several untreated SVK cells showing very low levels of I-staining; (d) OA-treated SVK culture showing a group of three cells (note: left set of two arrows pointing to intense cytoplasmic I-staining, and top set of two arrows pointing to I-stained nuclei). Total magnification, 2700×. Scale bar (white) 1 cm = 2.7 µM.
gation and smaller cell size (diameter) of 6.8 µM as noted above for SVK cells stained with P-Tyr antibody. By contrast, EGFR antibody staining of the cytoplasm of SVK cells was greatly diminished (c), but was elevated in both the cytoplasm and nuclei of OA-treated SVK cells. Cellular aggregation also occurred in OA-treated SVK cells.
3.1.3. Expression c-myc Proto-Oncogene in Normal and SVK Cells
The typical patterns of c-myc proto-oncogene expression in untreated (a), OA-treated (b) RG cultures of normal keratinocytes, and in untreated (c), OA treated (d) cultures of SVK keratinoctyes were presented (Figure 3). c-myc staining of the cytoplasm was relatively low in untreated cycling NHK cells (a) with discrete particulate staining in the nucleus (arrowhead). Average cell size (diameter) was 7.8 µM. By contrast, OA-treatment of NHK cells (b) is elevated in both cytoplasmic and nuclear c-myc expression and induced cellular aggregation with and average cell size (diameter) was 5.7 µM. Cytoplasmic and nuclear staining was highly intense in untreated cycling SVK cells (c). OA-treatment of SVK cells (d) resulted in severe rounding up, vacuolization and evidence of cytolysis; c-myc staining was intense in both nuclei and cytoplasm of OA-treated SVK cells.
3.1.4. Expression of c-neu Proto-Oncogene in Normal and SVK Cells
The typical patterns of c-neu proto-oncogene expression in untreated (a), OA-treated (b) RG cultures of normal keratinocytes, and in untreated (c), OA-treated (d) cultures of SVK keratinoctyes were presented (Figure 4). The cytoplasm of untreated normal cycling keratinocytes (a) was positively stained with a uniform particulate-type staining pattern (a) with evidence of intercellular focal adhesions (IFA) bordering the cell periphery. The average cell size (diameter) was 8.7 µM. OA-treated cycling NHK cell (b) displayed an intense coneshaped unipolar staining pattern. It also increased cell rounding with diminished overall cytoplasmic staining with an average cell size (diameter) of 5.8 µM. Cycling SVK cells (c) displayed both overall cytoplasmic staining and intense unipolar and multipolar globular staining predominantly localized at cell-to cell contact points at the cell periphery reminiscent of delocalized IFAs (arrowhead). OA-treated SVK cells displayed elevated and intense cytoplasmic c-neu staining (d) with evidence of both unipolar and bipolar localized staining. OA-treated SVK cells were larger and flattened relative to untreated SVK cells.

Figure 3. A panel of four photomicrographs of NHK ((a) & (b)) and SVK ((c) & (d)) cultures stained with c-myc antibodies. (a) untreated NHK culture (arrows point to two cells with evidence of particulate nuclear I-stain and low levels of cytoplasmic I-staining); (b) OA-treated NHK culture (note: two left arrows point to elevate cytopladsmic I-stain; right set of two arrows point to elevated levels of nuclear I-stain); (c) untreated SVK culture (top arrows point to elevated nuclear I-stain and bottom set of arrows point to intense monopolar I-stain); (d) OA-treated SVK culture ( note: arrows point to intense I-stained nuclei surrounded by vacuolated and lysed cytoplasmic areas). Total magnification, 2600×. Scale bar (white) 1 cm = 2.6 µM.
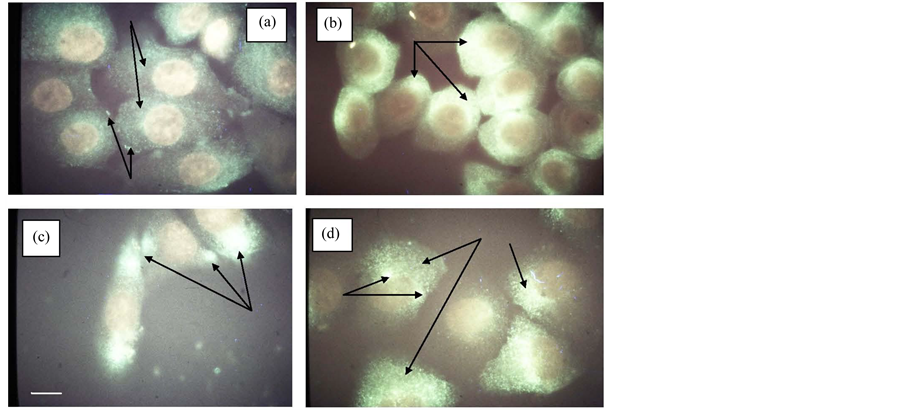
Figure 4. A panel of four photomicrographs of NHK ((a) & (b)) and SVK ((c) & (d)) cultures stained with c-neu antibodies. (a) untreated NHK culture (note: top two arrows point to intense particulate-type I-stained cytoplasm, and bottom two arrows point to I-stained intercellular focal (IFA) areas between adjacent cells); (b) OAtreated NHK culture (note: three arrows point to alignment of intensely I-stained monopolar caps and reduced cytoplasmic I-staining); (c) untreated SVK culture (note: three arrows point to intensely I-stained monoand bipolar caps that are oriented toward adjacent cells); (d) OA-treated SVK culture (note: left two arrows point to bipolar IFA areas not oriented to any adjacent cell; two middle arrows point to elevated levels of I-stained cytoplasm, and right single arrow points to a diffuse monopolar I-stained cap. Total magnication, 2900×. Scale bar (white) 1 cm = 2.9 µM.
3.1.5. Expression of Proto-Oncogene Analogue of v-rasH in Normal and SVK Keratinocytes
The expression of proto-oncogene version of v-rasH in in RG NHK cells (a), RG SVK cells (b) and the effect of OA on RG SVK cells (Figure 5). RG NHK cells display a high constitutive level of cytoplasmic v-rasH protooncogene expression with evidence of peri-nuclear localization. OA-treatment of NHK cells was not performed. Untreated RG SVK cells display an even higher constitutive level of cytoplasmic and nuclear v-rasH proto- oncogene expression. OA-treated SVK cells express an elevated nuclear v-rasH proto-oncogene expression with evident signs of cytolytic damage (remnant pieces of cytoplasm surrounding the cell).
3.2. Effect of t-RA and TPA
3.2.1. Expression of P-Tyr in Normal and SVK Keratinocytes
The typical patterns of P-Tyr expression for RG NHK cells without ((a) and (b)) and with t-RA treatment ((c) and (d)), and for NHK cells treated with TPA ((e) and (f)) (Figure 6). Panels (a), (c) and (e) show the P-Tyr expression before treatment with alkaline phosphatase (AP) enzyme while panels (b), (d) and (f) show P-Tyr staining after AP enzyme treatment. Normal cycling NHK cells (a) display both uniform cytoplasmic stain and a fan-shaped fringe of intensely stain at the advancing cell front with localization at cell-to cell points of contact associated with IFAs. This pattern is similar in AP-treated cells (b) accompanied with increased cytoplasmic staining but much diminished and disturbed peripheral IFA staining. The average cell size (diameter) was 8.7 µM. The effect of t-RA on cycling NHK cells (c) appears to be localized and diminished at the IFA sites which no longer associate with cell-cell points of contact. AP treatment of t-RA-treated NHK cells (d) further reveals decreased overall cytoplasmic staining as well as reduced IFA staining. TPA-treated cycling NHK (e) cells exhibit increased cytoplasmic P-Tyr staining and fragmented yet elevated and diffuse IFA-type staining, which is restricted to peripheral sites away from cell-cell contacts. TPA-treated NHK were larger than untreated cells (average cell size was 12 µM). AP post-treatment of TPA-treated NHK cells exhibit increased cytoplasmic P-Tyr staining in larger flattened cells along with multipolar intensely staining IFAs.
The effect of t-RA (b) and TPA (c) on P-Tyr expression on cycling SVK cells was shown as compared with untreated cells (Figure 7). Cycling SVK cells are smaller on average (3.0 µM) than RG NHK cells. Cycling SVK cells display intense cytoplasmic staining with no evidence of polarized localization of stain (a). t-RAtreated SVK cells (b) reveals drastic overall decrease in cytoplasmic P-Tyr staining and were larger and flatter cells (average diameter was 6.0 µM). TPA-treated SVK cells (c) were also larger (average diameter was 6.2 µM) and flatter than cycling RG SVK cells. They display less intense cytoplasmic P-Tyr staining relative to untreated cells and evidence of disruption in cell-cell contacts.
3.2.2. t-RA and TPA Effects on Expression of c-neu in Normal Keratinocytes
The typical patterns of c-neu expression for cycling NHK cells without ((a) and (b)) and with t-RA treatment ((c) and (d)), and for NHK cells treated with TPA ((e) and (f)) (Figure 8). Untreated cycling NHK cells displayed moderate cytoplasmic c-neu staining with some evidence of unipolar orientation in cells not pretreated with AP. The average cell diameter was 8.1 µM. AP-treatment of normal cycling NHK cells exhibited markedly increased cytoplasmic staining and the appearance of intensely stained unipolar IFA sites (b). All-trans-RA treatment of cycling NHK cells (c) displayed increased cytoplasmic c-neu staining with the appearance of low level c-neu



Figure 5. A panel of three photomicrographs of NHK culture (a) and SVK cultures ((b) & (c)) stained with v-rasH antibodies. (a) untreated NHK culture (note single arrow points to intense perinuclear and diffuse cytoplasmic I-staining); B) untreated SVK culture (two arrows point to intense I-staining of nuclei surrounded by low level of cytoplasmic I-stain); (c) OA-treated SVK culture (bottom arrow points to intense nuclear I-stain; the middle two arrow point to lytic remnants that are intensely iIstained cytoplasmic fragments). Total magnification, 3050×. Scale bar (white) 1 cm = 2.1 µM.
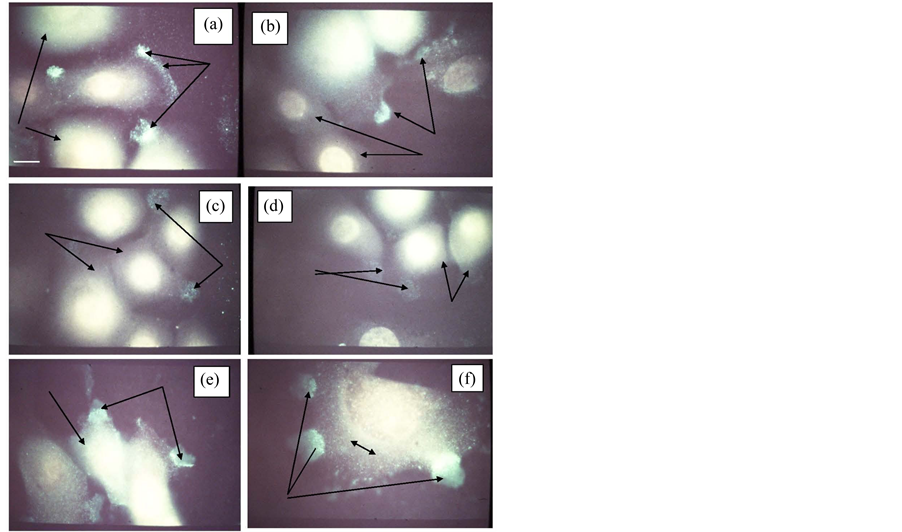
Figure 6. A panel of six photomicrographs untreated NHK cultures ((a) & (b)), t-RA-treated NHK cultures ((c) & (d)), and TPA-treated NHK cultures stained with P-Tyr antibodies. (a) untreated NHK culture (note: left two arrows point to diffuse I-stained cytoplasm; the right three arrows point to monopolar IFA plaques present before alkaline phosphatase (AP) enzyme treatment); (b) untreated NHK culture after AP enzyme treatment (note: bottom two arrows point to low level of I-stained cytoplasm; the middle two arrows point to remnants of I-stained IFA plaques); (c) t-RA-treated NHK cultures before AP treatment (note: left two arrows point to diffuse low level of I-stai; the right two arrows point to low level of IFA plaques I-stain not aligned toward adjacent cells); (d) t-RA treated NHK culture after AP enzyme treatment (note: left arrows point to diffuse low level of I-stained IFA plaques; the right two arrows point to diminished cytoplasmic I-stain; (e) TPA-treated NHK culture before AP enzyme treatment (note: single left arrow points to intense cytoplasmic Istaining; the right two arrow point to diffuse but intense I-stained IFA plaques); (f) TPA-treated NHK culture after AP enzyme treatment (note: left three arrows point to diffuse but intense I-stained multipolar IFA plaques; the middle double arrow points to intense I-stained cytoplasm). Toal magnification, 2900×. Scale bar 1 cm = 2.9 µM.



Figure 7. A panel of three photomicrographs of untreated SVK (a), t-RA-treated SVK culture (b) and TPA-treated SVK cultures stained with P-Tyr antibodies. (a). untreated SVK culture (note: three arrows pointing to intensely I-stained cytoplasm among tightly nestled cells); (b) t-RA-treated SVK culture (note: two arrows point to very reduced cytoplasmic I-stain); (c) TPA-treated SVK culture (note: two arrows point todiaspersed cytoplasmic I-staining). Total magnification, 2100×. Scale bar (white) 1 cm = 2.1 µM.
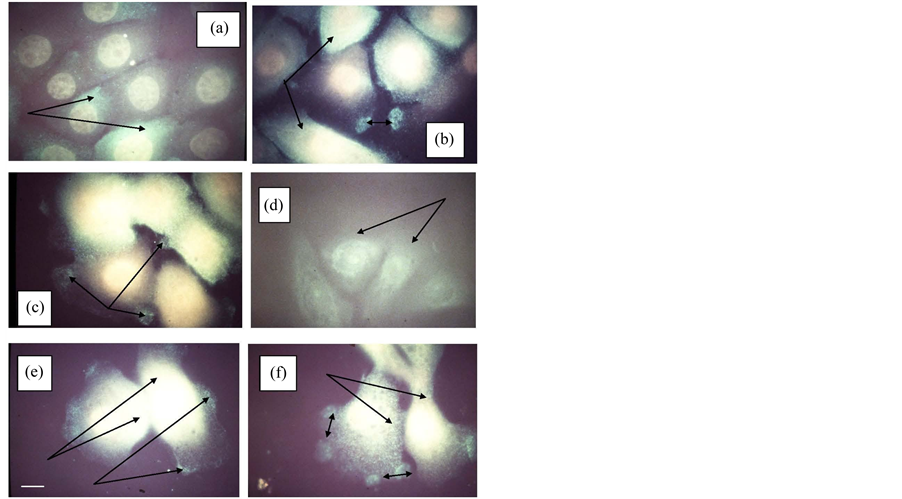
Figure 8. A panel of six photomicrographs of untreated NHK ((a) & (b)), t-RA-treated ((c) & (d)), and TPA treated ((e) & (f)) NHK cultures stained with c-neu antibodies. (a) untreated NHK culture before AP enzyme treatment (note: two arrows point two larger cells that show moderate to intense I-stained monopolar cytoplasm): (b) untreated NHK culture after AP enzyme treatment (note: left two arrows point to intensely I-stained cytoplasm and a biheaded arrow pointing to intense I-stained IFA plaques between adjacent cells); (c) t-RA-treated NHK culture before AP enzyme treatment (note: three arrow arrows point to low I-stained levels of diffuse IFA cytoplasmic areas surrounded by high levels of I-stained cytoplasm); (d) t-Ra-treated NHK culture after AP treatment (note: two arrows pointing to two cells showing very low levels of I-stained cytoplasm). Total magnification, 2700×. Scale bar (white) 1 cm = 2.7 µM.
staining at IFA sites not engaged in cell-to-cell contacts. Cytoplasmic c-neu staining of t-RA-treated NHK cells was significantly reduced relative to no AP treatment of cycling NHK cells (d). TPA-treated cells were larger (12 µM) relative to both RA treated and normal cycling cells. Cytoplasmic c-neu staining of TPA treated cycling NHK cells was elevated relative to untreated cells and there were multipolar IFA sites not engaged in cell-to-cell contacts (e). Finally, TPA-treated cycling NHK cells treated with AP revealed elevated cytoplasmic c-neu staining as well as semi-diffuse multipolar IFA sites (f).
3.2.3. t-RA and TPA Effect on Expression of P-Tyr in Mouse C3H10T1/2 Mouse Fibroblast Cells
The typical pattern of P-Tyr expression for cycling 10T1/2 mouse cells without ((a) and (b)) and with t-RA ((c) and (d)), or TPA treatment ((e) and (f)) are shown in Figure 9. Cycling mouse embryonic fibroblasts were larger (14 µM) than normal human keratinocytes. There was extensive cytoplasmic P-Tyr staining for untreated cells not AP-treated (a). AP post-treatment of untreated cycling cells (b) revealed reduction in overall cytoplasmic staining but greatly enhanced staining of multipolar IFA sites (b). RA-treatment did not alter the cell size relative to normal cycling cells. All-t-RA treatment of cycling cells greatly reduced cytoplasmic staining except for elongated filamentous structures (c). AP-treatment of t-RA treated cycling cells reduced overall cytoplasmic staining except for peri-nuclear filamentous structures (d). TPA treatment of cycling cells exhibited cytoplasmic P-Tyr staining with some evidence of stained multipolar polarized IFA sites and general disoriented cell enlargement average cell diameter was 16 µM (e). AP post-treated TPA-treated cycling cells exhibited a lower level of cytoplasmic staining relative to untreated AP treated cells but similar levels of disorganized multipolar IFA sites.
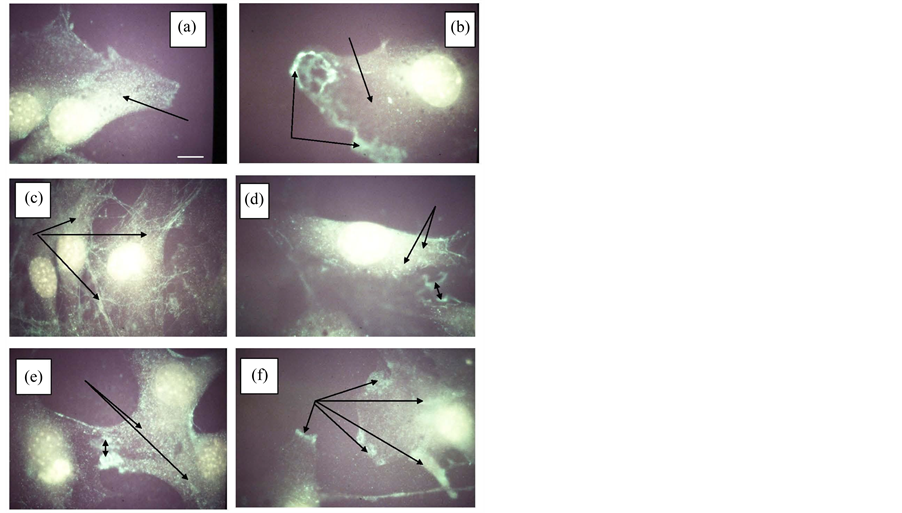
Figure 9. A panel of six photomicrographs of C3H10T1/2 mouse fibroblast untreated ((a) & (b)), t-RA-treated ((c) & (d)), and TPA-treated ((e) & (f)) cultures stained with P-Tyr antibodies. (a) untreated C3H10T1/2 cells before AP enzyme treatment (note: arrow points to intese I-stain of the cytoplasm); (b) untreated fibroblast cells after AP enzyme treatment (note: top arrow points to low level of I-stain of the cytoplasm; left two arrows point to two prominently I-stained IFA areas); (c) t-RA-treated fibroblast cells before AP enzyme treatment (note: three arrows pointing to cytoplasmic I-stained fibrous networks present within and between two cells); (d) t-RA-treated fibroblast cells after AP enzyme treatment (note: top two arrows point to an apparent collapse of I-stained fibrous network as seen in C; the double-headed arrow points to remnant IFA structures between two cells); (e) TPA-treated fibroblast cells before AP enzyme treatment (note: two single-headed arrows pointing to intense particulate type I-stained cytoplasm; the double-headed arrow points to two IFA areas between adjacent cells); (f) TPA-treated fibroblast cells after AP enzyme treatment (note: the five diffw3rent single-headed arrows that point to multiple I-stained IFA 10. plaques extending between and from other cells-cell contacts). Total magnification, 2700×. Scale bar (white) 1 cm = 2.7 µM.
4. Discussion
The staining pattern and level of expression of proto-oncogenes, oncoproteins and phosphotyrosine-containing phosphoproteins in normal untreated RG NHK and RG SVK cells form the basis for interpreting the results of treatment with tumor-promoters OA and TPA, and t-RA. We introduced the enzymatic dephosphorylation of oncogene expression by AP treatments to uncover possible alteration in sensitivity of the phosphorylation status of oncogene expression. AP-treatment altered oncogene phosphorylation patterns in untreated and both RA and TPA-treated NHK as well as in C3H10T1/2 fibroblast cells. AP treatments altered c-neu expression with an elevated cytoplasmic staining and the appearance of fragmented IFA sites. AP treatment of P-Tyr stained and untreated cycling NHK increased cytoplasmic staining again with fragmented peripheral IFA staining. By contrast, AP treatment decreased cytoplasmic c-neu oncoprotein staining and reduced IFA staining in t-RA treated NHK cells. AP treatment also increased cytoplasmic P-Tyr staining in larger flattened cells along with fragmented multipolar staining of IFAs in untreated NHK and for C3H10T1/2 fibroblast cells. We interpret these AP-induced changes as demonstrating the resistance of cytoplasmically-located phosphorylated c-neu oncoprotein and P-Tyr stained protein sites and conversely the susceptibility of the phosphorylated state of c-neu at IFA loci and at cell-cell contact sites. AP-treatment had opposing effect on the dephosphorylation of c-neu expression consistent with its overall down -regulation of oncogene expression.
In general, normal cycling keratinocytes express lower levels of the select proto-oncogene and related oncoproteins. For example, c-neu was expressed primarily in larger cycling cells, in telophase and mitotic cells, but appears to be is absent in smaller, perhaps, early G1 cells. This agrees with an earlier report that c-neu is located in the suprabasal layer cells of intact skin [32] . The constitutive level of oncoproteins evaluated by immunocytochemical staining procedures in cycling SVK cells is elevated relative to normal cycling keratinocyte. Expression of large and middle T proteins is associated with elevated levels of RAS oncogene and the acquisition of anchorage-independent growth exhibited by SVK cells reported here and elsewhere [2] [9] .
This report provides evidence that OA-treatment elevates select oncogene-related proteins and modifies the phosphorylated status of cell surface phosphotyrosine-containing proteins. Figure 10 is a schematic representation of the proposed mechanism by which OA exerts its effect on oncogene expression via inhibition of PP2A (red lines) through the RAS/MEKK1 pathway and blockade of the JNK/ERK pathway. The first blockade would elevate RAS protein which we document here. Elevation of growth factor-related oncogenes in both NHK and SVK also indicates a role for OA-sensitive PP2A in the regulation of keratinocyte proliferation. The second blockage ultimately leads to apoptosis and accompanying morphological changes in both normal and SVK keratinocytes reported here and by others [14] of its damaging effects on cytoskeleton proteins. We show that it induces polarized cytoplasmic localization of EGFR, and c-neu, c-myc oncogene expression consistent with a shift of staining pattern to IFA proteins sites. The elevated expression of c-neu gene product and increased localization of P-Tyr to IFA sites in both NHK and SVK suggest that OA-sensitive phosphatases play important roles in phosphorylation events occurring at IFA sites. An alternative account of the effects of OA on induction of polarized tyrosine kinase staining pattern is that it induces patching and capping familiar to the effects of antibody cross-linking with cell surface antigen accompanying cytoplasmic flow in lymphocytes [33] . However, this hypothesis fails to explain the polarized location of IFA sites observed in untreated NHK cells, which truly represent the role of phosphotyrosine kinases in intercellular interactions.
The responses to OA and TPA were quite different. TPA treatments causes a slight down regulation of C-NEU and C-MYC oncogene and P-Tyr—containing proteins, whereas OA-treatment greatly elevated the number, integrity and localization of membrane associated IFA sites. Figure 10 presents a schematic representation of the effect of tumor promoter (TPA). TPA exerts its action through the calcium/PKC pathway activation of RAS signaling pathways (blue arrows), and ultimately in activation of DNA transcription factors (TF) and down regulation of c-neu, c-myc and EGFR oncoproteins, and ultimately on the induction of keratinocyte differentiation through activation of the AP-1 transcription pathway.
We found that RA treatment completely abolishes cytoplasmic and polarized expression of both c-neu and
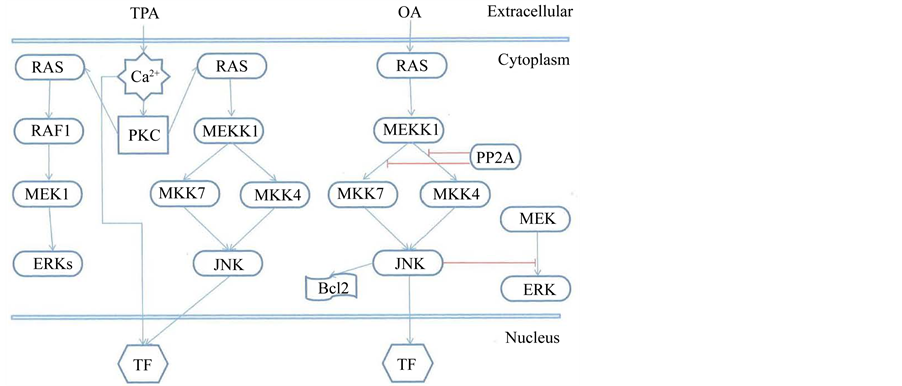
Figure 10. A schematic figure depicting the route TPA (left side) and OA (right side) take on activating (blue arrows) and repressing (red arrows) oncogene signal transduction pathways affecting the Ca2+/PKC and RAS oncogene pathways., respectively (see text for further explanation).
c-myc oncoproteins, and P-Tyr phosphoproteins in both normal and SV-40 keratinocytes. RA-treatment also alters cell morphology which is characteristic of agents which cause keratinocyte growth arrest, cell enlargement and cell flattening. We propose that these effects are mediated through RAS action on proto-oncogenes and tyrosine protein kinases, and that they account for our previous report that RA reverses the effect of carcinogenic agents that induces aryl hydrocarbon arylase [24] and eliminate alcohol-stimulated elevation of cytochrome P450 in human keratinocytes [34] . This proposal is supported by a recent report that t-RA inhibits proliferation of SCL-1 cells, induces G1 cell-cycle arrest with apoptotic effects through inhibition of cyclin D1/cyclin- dependent kinase (CDK4) and cyclin E/CDK2, increases expression of the cyclin-dependent kinase inhibitors p21 and p27, decreases the phosphorylation of ERK1/2 and JNK1/2, and inhibits AP-1 transcriptional activity [20] .
5. Conclusion
In summary, the effect of OA, RA and TPA is not specific for human epidermal keratinocyte and their signal transduction pathways. We observed similar effects of RA and TPA on P-Tyr staining pattern in C3H10T1/2 cells, which mimic those observed for NHK and SVK cells. In particular, the results show the generality of phosphotyrosine-containing phosphosprotein expression at cytoplasmic and IFA membrane sites. We interpret these results as demonstrating a role of anti-tumor (RA) and tumor promoters (OA and TPA) on activation and suppression of proto-oncogenes and in mediating proliferation and differentiation of mammalian tissue culture cells.
References
- Croce, C.M. (2008) Oncogenes and Cancer. The New England Journal of Medicine, 358, 502-511. http://dx.doi.org/10.1056/NEJMra072367
- Butel, J.S. and Lednicky, J.A. (1999) Cell and Molecular Biology of Simian Virus 40: Implications for Human Infections and Disease. Journal of the National Cancer Institute, 91, 119-134. http://dx.doi.org/10.1093/jnci/91.2.119
- Yaffe, M.B. (2002) Phosphotyrosine-Binding Domains in Signal Transduction. Nature Reviews Molecular Cell Biology, 3, 177-186. http://dx.doi.org/10.1038/nrm759
- Varani, J., Zeigler, M., Dame, M.K., Kang, S., Fisher, G.J., Voorhees, J.J., Stoll, S.W. and Elder, J.T. (2001) HeparinBinding Epidermal-Growth-Factor-Like Growth Factor Activation of Keratinocyte ErbB Receptors Mediates Epidermal Hyperplasia, a Prominent Side-Effect of Retinoid Therapy. Journal of Investigative Dermatology, 117, 1335-1341. http://dx.doi.org/10.1046/j.0022-202x.2001.01564.x
- De Potter, I.Y., Poumay, Y., Squillace, K.A. and Pittelkow, M.R. (2001) Human EGF Receptor (HER) Family and Heregulin Members Are Differentially Expressed in Epidermal Keratinocytes and Modulate Differentiation. Experimental Cell Research, 271, 315-328. http://dx.doi.org/10.1006/excr.2001.5390
- Lewis, D.A., Zweig, B., Hurwitz, S.A. and Spandau, D.F. (2003) Inhibition of erbB Receptor Family Members Protects HaCaT Keratinocytes from Ultraviolet-B-Induced Apoptosis. Journal of Investigative Dermatology, 120, 483-488. http://dx.doi.org/10.1046/j.1523-1747.2003.12060.x
- Montemurro, F. and Scaltriti, M. (2014) Biomarkers of Drugs Targeting HER-Family Signalling in Cancer. The Journal of Pathology, 232, 219-229. http://dx.doi.org/10.1002/path.4269
- Reiss, M., Dibble, C.L. and Narayanan, R. (1989) Transcriptional Activation of the c-myc Proto-Oncogene in Murine Keratinocytes Enhances the Response to Epidermal Growth Factor. Journal of Investigative Dermatology, 93, 136-141. http://dx.doi.org/10.1111/1523-1747.ep12277384
- Wille, J.J. (1989) Malignant Transformation of Normal Human Keratinocytes by SV-40 Virus. The Annals of the New York Academy of Sciences, 567, 307-310.
- Wilke, M.S., Hsu, B.M., Wille Jr., J.J., Pittelkow, M.R. and Scott, R.E. (1988) Biologic Mechanisms for the Regulation of Normal Human Keratinocyte Proliferation and Differentiation. American Journal of Pathology, 131, 171-181.
- Suganuma, M., Fujiki, H., Suguri, H., Yoshizawa, S., Hirota, M., Nakayasu, M., Ojika, M., Wakamatsu, K., Yamada, K. and Sugimura, T. (1988) Okadaic Acid: An Additional Non-Phorbol-12-Tetradecanoate-13-Acetate-Type Tumor Promoter. Proceedings of the National Academy of Sciences, 85, 1768-1771. http://dx.doi.org/10.1073/pnas.85.6.1768
- Fujiki, H., Suganuma, M., Yoshizawa, S., Nishiwaki, S., Winyar, B. and Sugimura, T. (1991) Mechanisms of Action of Okadaic Acid Class Tumor Promoters on Mouse Skin. Environmental Health Perspectives, 93, 211-214. http://dx.doi.org/10.1289/ehp.9193211
- Sakai, R., Ikeda, I., Kitani, H., Fujiki, H., Takaku, F., Rapp, U., Sugimura, T. and Nagao, M. (1989) Flat Reversion by Okadaic Acid of raf and ret-II Transformants. Proceedings of the National Academy of Sciences, 86, 9946-9950. http://dx.doi.org/10.1073/pnas.86.24.9946
- Garcia, A., Cayla, X., Guergnon, J., Dessauge, F., Hospital, V., Rebollo, M.P., Fleischer, A. and Rebollo, A. (2003) Serine/Threonine Protein Phosphatases PP1 and PP2A Are Key Players in Apoptosis. Biochimie, 85, 721-726. http://dx.doi.org/10.1016/j.biochi.2003.09.004
- Boudreau, R.T. and Hoskin, D.W. (2005) The Use of Okadaic Acid to Elucidate the Intracellular Role(s) of Protein Phosphatase 2A: Lessons from the Mast Cell Model System. International Immunopharmacology, 5, 1507-1518. http://dx.doi.org/10.1016/j.intimp.2005.05.007
- Valdiglesias, V., Laffon, B., Pásaro, E. and Méndez, J. (2011) Okadaic Acid Induces Morphological Changes, Apoptosis and Cell Cycle Alterations in Different Human Cell Types. Journal of Environmental Monitoring, 13, 1831-1840. http://dx.doi.org/10.1039/c0em00771d
- Matias, W.G., Traore, A., Bonini, M., Sanni, A. and Creppy, E.E. (1999) Oxygen Reactive Radicals Production in Cell Culture by Okadaic Acid and Their Implication in Protein Synthesis Inhibition. Human & Experimental Toxicology, 18, 634-639. http://dx.doi.org/10.1191/096032799678839473
- Haystead, T.A., Sim, A.T.R., Carling, D., Honnor, R.C., Tsukitani, Y., et al. (1989) Effects of the Tumour Promoter Okadaic Acid on Intracellular Protein Phosphorylation and Metabolism. Nature, 337, 78-81. http://dx.doi.org/10.1038/337078a0
- Haneji, T., Hirashima, K., Teramachi, J. and Morimoto, H. (2013) Okadaic Acid Activates the PKR Pathway and Induces Apoptosis through PKR Stimulation in MG63 Osteoblast-Like Cells. International Journal of Oncology, 42, 1904-1910.
- Zhang, M.L., Tao, Y., Zhou, W.Q., Ma, P.C., Cao, Y.P., et al. (2014) All-Trans Retinoic Acid Induces Cell-Cycle Arrest in Human Cutaneous Squamous Carcinoma Cells by Inhibiting the Mitogen-Activated Protein Kinase-Activated Protein 1 Pathway. Clinical and Experimental Dermatology, 39, 354-360. http://dx.doi.org/10.1111/ced.12227
- Vahlquist, A., Lee, J.B., Michaëlsson, G. and Rollman, O. (1982) Vitamin A in Human Skin: II Concentrations of Carotene, Retinol and Dehydroretinol in Various Components of Normal Skin. Journal of Investigative Dermatology, 79, 94-97. http://dx.doi.org/10.1111/1523-1747.ep12500033
- Lotan, R. and Nicolson, G.L. (1977) Inhibitory Effects of Retinoic Acid or Retinyl Acetate on the Growth of Untransformed, Transformed, and Tumor Cells in Vitro. JNCI Journal of the National Cancer Institute, 59, 1717-1722.
- Wille, J.J.B.J., Park, J.Y. and Triggs, G. (2002) Retinoid Stimulation of Autocrine Growth Factor Controlled Keratinocyte Proliferation. Wound Healing Society, Baltimore.
- Wille, J.J. and Chopra, D.P. (1988) Reversal by Retinoids of Keratinization Induced by Benzo[alpha]pyrene in Normal Hamster Tracheal Explants: Comparison with the Assay Involving Organ Culture of Tracheas from Vitamin A-Deficient Hamsters. Cancer Letters, 40, 235-246. http://dx.doi.org/10.1016/0304-3835(88)90082-1
- Verma, A.K., Rice, H.M., Shapas, B.G., and Boutwell, R.K. (1978) Inhibition of 12-O-tetradecanoylphorbol-13-acetateInduced Ornithine Decarboxylase Activity in Mouse Epidermis by Vitamin A Analogs (Retinoids). Cancer Research, 38, 793-801.
- Verma, A.K., Shapas, B.G., Rice, H.M. and Boutwell, R.K. (1979) Correlation of the Inhibition by Retinoids of Tumor Promoter-Induced Mouse Epidermal Ornithine Decarboxylase Activity and of Skin Tumor Promotion. Cancer Research, 39, 419-425.
- Cope, F.O. and Wille, J.J. (1989) Retinoid Receptor Antisense DNAs Inhibit Alkaline Phosphatase Induction and Clonogenicity in Malignant Keratinocytes. Proceedings of the National Academy of Sciences of the United States of America, 86, 5590-5594. http://dx.doi.org/10.1073/pnas.86.14.5590
- Iizuka, H., Sakai, H. and Tamura, T. (1989) Effects of the Tumor Promoter, Phorbol 12-Myristate, 13-Acetate, on the Epidermal Adenylate Cyclase System: Evidence for Adenylate Cyclase-Regulation by Protein Kinase C. Journal of Investigative Dermatology, 93, 387-391. http://dx.doi.org/10.1111/1523-1747.ep12280284
- Wille Jr., J.J., Pittelkow, M.R. and Scott, R.E. (1985) Normal and Transformed Human Prokeratinocytes Express Divergent Effects of a Tumor Promoter on Cell Cycle-Mediated Control of Proliferation and Differentiation. Carcinogenesis, 6, 1181-1187. http://dx.doi.org/10.1093/carcin/6.8.1181
- Wille, J. (2004) Effect of Okadiac Acid on Oncogene Expression in Normal and Transformed Human Keratinocytes. Proceedings of the American Association for Cancer Research, 27-31 March 2004, Orlando, Abstract #: 2778.
- Wille Jr., J.J., Pittelkow, M.R., Shipley, G.D. and Scott, R.E. (1984) Integrated Control of Growth and Differentiation of Normal Human Prokeratinocytes Cultured in Serum-Free Medium: Clonal Analyses, Growth Kinetics, and Cell Cycle Studies. Journal of Cellular Physiology, 121, 31-44. http://dx.doi.org/10.1002/jcp.1041210106
- Maguire Jr., H.C., Jaworsky, C., Cohen, J.A., Hellman, M., Weiner, D.B. and Greene, M.I. (1989) Distribution of Neu (C-erbB-2) Protein in Human Skin. Journal of Investigative Dermatology, 92, 786-790. http://dx.doi.org/10.1111/1523-1747.ep12696796
- Taylor, R.B., Duffus, W.P.H., Raff, M.C. and de Petris, S. (1971) Redistribution and Pinocytosis of Lymphocyte Surface Immunoglobulin Molecules Induced by Anti-Immunoglobulin Antibody. Nature New Biology, 233, 225-229. http://dx.doi.org/10.1038/newbio233225a0
- Wille, J. and Park, J.Y. (2012) Retinoid and Ethanol-Sensitive Benzo[Alpha]pyrene Induction of Cytochrome P-450 in Human Keratinocytes. Journal of Cancer Therapy, 3, 1080-1085. http://dx.doi.org/10.4236/jct.2012.36141