Journal of Cancer Therapy
Vol. 4 No. 5 (2013) , Article ID: 34128 , 11 pages DOI:10.4236/jct.2013.45117
Toll-Like Receptors as Biomarkers of Gastric Carcinogenesis: Implications for Diagnosis, Prognosis and Treatment
![]()
1Department of Physiology, Cardiovascular Research & Development Unit, Faculty of Medicine, University of Porto, Porto, Portugal; 2Gastroenterology Department, Portuguese Oncology Institute, Porto, Portugal; 3Gastroenterology Department, Hospital de Braga, Braga, Portugal; 4CIDES/CINTESIS, Faculty of Medicine, University of Porto, Porto, Portugal.
Email: pedronunesml@gmail.com
Copyright © 2013 Pedro Pimentel-Nunes et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received March 3rd, 2013; revised April 5th, 2013; accepted April 12th, 2013
Keywords: Toll-Like Receptors; Gastric Cancer; TOLLIP; Carcinogenesis; Helicobacter pylori
ABSTRACT
Toll-like receptors (TLR) are essential for Helicobacter pylori (Hp) recognition and subsequent innate and adaptive immunity responses. TLR2 appears to be the receptor responsible for most of the immunologic reaction against Hp infection. However, TLR4, TLR9 and eventually TLR5 may also have a synergic effect with TLR2 against Hp. It has been shown that gastric Hp infection increases TLR expression in the gastric mucosa. Moreover, recent studies have shown that human gastric carcinogenesis is associated not only with increased expression of TLR but also with decreased expression of their inhibitors such as Toll-Interacting Protein (TOLLIP) and peroxisome proliferator-activated receptor (PPAR)-g. Indeed, gastric dysplasia and adenocarcinoma are associated with high expression levels of TLR and low levels of TOLLIP and PPAR-g, suggesting increased activation of these receptors throughout human gastric carcinogenesis. In this article we discuss how these novels findings could be used not only for the diagnosis and prognosis of gastric lesions associated with Hp infection but also for their treatment. Specifically, we discuss the potential use of TLR agonists in addition to antibiotics to improve eradication rates of Hp and of TLR antagonists to slow the progression of gastric preneoplastic lesions. We also discuss the potential value of TLR signalling blockers and quantification of tumoral TLR expression, respectively, in the treatment and prognosis of gastric cancer. In conclusion, TLRs can be an important link between Hp and the sequence of gastric carcinogenesis and they can be used as biomarkers of gastric carcinogenesis. In this article, future lines of investigation related with these novel scientific findings are proposed and discussed.
1. Introduction
Gastric pathology has some unique characteristics mainly because many gastric diseases have a strong association with a bacteria infection—Helicobacter pylori (Hp). Indeed, this bacteria discovered in the 1980s was rapidly associated to several gastric pathologies [1]. In 1994 it was clearly recognized that Hp was a major cause of gastroduodenal peptic ulcers and later that year the International Agency for Research on Cancer declared Hp to be a group I human carcinogen for gastric adenocarcinoma [2]. Hp is considered one of the oldest bacteria to infect humans with genetic studies identifying this bacterium in the first human populations, more than 58,000 years ago [1]. Even today with wide use of antibiotics, Hp is estimated to infect more than 50% of the World’s population, with prevalence’s as high as 90% in some developing countries [1].
Hp is a Gram-negative bacterium that adheres to the surface of gastric mucosa, without invasion of gastric epithelial cells, and that upon interaction with several innate immunity receptors such as Toll-like receptors (TLRs), causes inflammation of the mucosa that perpetuates as a chronic gastric inflammatory state [3,4]. In some patients this inflammation progresses leading to gastric atrophy and intestinal metaplasia, clearly established gastric premalignant gastric conditions [5-8]. Indeed, Correa was the first one to describe a multistep pathway for the intestinal-type gastric adenocarcinoma, where Hp is considered the initiator of the so-called Correa cascade of gastric carcinogenesis that involves chronic gastritis, atrophic gastritis, intestinal metaplasia, gastric dysplasia and finally, intestinal-type gastric adenocarcinoma [9-12]. The innate immune system is the first line of defense against several microbial agents, consisting of a diversity of components that initiate protective immunological responses [13,14]. Although many of these factors can prevent or destroy the invading pathogens non-specifically, we now know that the microbiological recognition by innate immunity is also a specific and highly coordinated process involving pattern recognition receptors (PRRs) that identify preserved structures of different pathogens, the so-called pathogen-associated molecular patterns (PAMPs) [13,14]. Moreover, this amazing specificity conferred by the recognition of PAMPs by PRRs is essential, not only for a more adequate initial control of a potential infection (innate immunity), but also for triggering the late antigen-specific acquired immunity (adaptive immunity), for controlling inflammation processes and for maintenance of a immunological homeostasis within the host [15,16].
The toll-like receptors (TLRs) are the most important class of pathogen-associated molecular patterns (PAMPs) receptors, with ten different TLRs being ubiquitously expressed in humans [14,17-19]. TLRs are surface molecules on eukaryotic cells, present in invertebrates and conserved in vertebrates, which were originally identified as homologs of Drosophila Toll molecule, an important component of antifungal defence mechanism [14,20,21]. The existence of several TLRs enables the innate immunity system to recognize different groups of pathogens while initiating appropriate and distinct immunological responses, according to the PAMP recognized [13,17, 22]. In normal physiological conditions TLRs do not recognize self-ligands. However, after tissue lesion they may recognize endogenous antigens, the so-called damage-associated molecular patterns (DAMPs), and contribute to promote sterile inflammation [23,24]. Although initially described in several immunological cells, various studies have shown that different human tissues express these receptors, with the degree of expression varying from tissue to tissue [14,17,18,20,21].
The structure of all TLRs is identical. TLRs are membrane-surface receptors consisting of a distinct leucine-rich repeat (LRR) extracellular domain that confers specificity to the receptor, a single transmembrane domain and a conserved toll/interleukin 1 (IL1) receptor (TIR) intracellular domain, homologous to the IL1 receptor [17,18]. In general, TLR2 recognizes PAMPs mainly from Gram positive bacteria, TLR4 is the recaptor for Gram negative bacteria lipopolysacharide (LPS), TLR5 recognizes bacteria flagellin, TLR3, TLR7 and TLR8 recognize viral components namely double (TLR3) and single-stranded RNA (TLR7/8), TLR9 recognizes unmethylated CpG DNA from bacteria and virus, and finally, TLR1 and TLR6 form heterodimers with TLR2 in order to sense tri-acyl (mycobacterium) and di-acyl lipopeptides (mycoplasma), respectively. In addition, TLR4 and TLR2 can detect a wide range of antigens not only from bacteria but also from fungus, parasites, virus (particularly TLR2) and DAMPs (particularly TLR4) [14,25].
The coupling of TLRs with its respective ligand initiates intracellular signalling pathways leading to the production of several inflammatory cytokines such as TNF-α, IL-1 and several others inflammatory molecules through the activation of nuclear factor-κB (NF-κB) [17,26-29]. Despite similar intracellular signalling pathways, the final result of stimulating different TLRs is not exactly the same depending not only of the activated receptor but also of the cell that is stimulated [30-32]. Moreover, because they are intrinsically related to inflammation but also to cell survival signalling, epithelial regeneration and cell proliferation, recent reports associate these receptors function to tumorigenesis [33,34]. Indeed, it appears that the perpetuation of TLRs signaling pathways, such as in Hp-induced chronic gastritis, confers oncogenic potential to the cells [17,26-29,33,34]. In that way, in the gastrointestinal tract, in order to prevent inadequate inflammatory responses to non-pathogenic antigens, a strict regulation of these receptors activity is fundamental for maintaining homeostasis [4,20,35-46]. For that reason, normal gastrointestinal mucosa expresses low levels of TLRs and high levels of several TLR-antagonists, like Toll-interacting protein (TOLLIP) and PPAR-γ, molecules that block TLR signalling pathways and NF-κB activation, respectively [4,20,35-46]. This further underscores a complex and not completely understood intracellular signalization for these receptors.
Recent studies suggest that TLRs not only have an important role in Hp recognition but also in the progression of gastric lesions associated to this infection. In this article we will discuss the fundamental role of TLRs in the immunological response to HP infection and in the progression of gastric preneoplastic lesions and how this novel finding could potentially be used in clinical practice.
2. Helicobacter pylori Immune Recognition by Toll-Like Receptors
Several studies demonstrate that TLRs have an essential role in Hp recognition and subsequent innate and adaptive immunity against this bacterium [47-61]. After the first contact with the gastric mucosa Hp appears to interact with several TLRs inducing an immunological and inflammatory response [47]. Moreover, being Hp a bacterium that causes marked inflammation without invasion of gastric epithelial cells it looks clear that this first line of interaction is crucial for the subsequential cascade of inflammation induced by this bacteria [3]. However, which is the principal TLR responsible for this process is a question of some controversy.
Different TLRs may play a role in gastric immunologic response to Hp [4]. Although Hp is a Gram-negative bacteria that possesses LPS, the main ligand of TLR4, TLR4 does not appear to be the principal TLR responsible for the immunologic response to Hp [3]. Actually, TLR2, a TLR that recognizes several antigens from Gram-positive bacteria, appears to be the receptor responsible for most of the inflammatory changes occurring as the result of Hp infection [4,47,49].
Indeed, some studies showed that TLR2, but not TLR4, was required for Hp-induced NF-kappa B activation and cytokine production both by epithelial [56] and antigen presenting cells [57]. Moreover, cytotoxin-associated gene A (Cag A), an important virulence factor of Hp, promotes a higher production of inflammatory cytokines by TLR2 and not TLR4 signalling [58]. On the other hand, some studies also suggest that TLR4 recognizes several Hp antigens and, in that way, also plays an important role in Hp infection [59-61]. In fact, more conclusive studies demonstrate that either in epithelial or dendritic cells, TLR2 appears to be the principal receptor for recognition of Hp. However, this process depends also in some extent of TLR4 that acts in synergy with TLR2 [47-52]. The data regarding the role of TLR5 (an extracellular receptor for bacteria flagellin) on Hp recognition are highly controversial. Although Hp is a flagellated bacteria, it appears that the flagellin of ε-Proteobacteria, including Hp, is barely recognized by TLR5 [59-61]. Some initial studies suggested an interaction between Hp flagellin and TLR5 leading to production of several pro-inflammatory cytokines [49,62]. This was refuted by other studies demonstrating that TLR5 was unresponsive to Hp flagellin [59-61]. These studies suggested that non-recognition of Hp flagellin by TLR5 could be a mechanism for Hp immune evasion [59-61]. More recent studies suggested that TLR5 was important for Hp recognition and inflammatory response to these bacteria, adding more controversy about this subject [63]. Taking altogether, concerning TLR5, the data are contradictory and TLR5 may have none or little role in Hp recognition.
Concerning the intracellular TLRs, TLR9 is the only one that recognizes Hp DNA and appears to have a complementary and synergistic action with the other two receptors (TLR2 and TLR4) [49,64].
So, in conclusion, TLR2 by recognizing several Hp antigens appears to be the receptor responsible for most of the immunological process that occurs as the result of Hp infection. However, TLR4 and TLR9 recognize other Hp antigens and have a synergistic role with TLR2. The contradictory data regarding recognition of Hp flagellin by TLR5 suggest that this TLR has no or small role in Hp induced immune response.
3. Toll-Like Receptors and the Progression of Gastric Preneoplastic and Neoplastic Lesions
Although data associating TLRs with the progression of gastric lesions are scarcer than data concerning Hp-recognition by these receptors, recent studies suggest that TLRs may also have an important role in gastric carcinogenesis.
Initial studies showed that chronic Hp infection increased TLR4 expression and NF-κB activation [60,61]. Later on, using immunohistochemistry, our group and others showed that intestinal metaplasia, dysplasia and carcinoma cells expressed TLR2, TLR4, TLR5 and TLR9 in a more intense and diffuse way when comparing with normal mucosa [65,66]. Moreover, our group confirmed also at a genetic level this increased TLR expression through the progression of gastric carcinogenesis, particularly TLR2 and TLR4 [67]. More importantly, we demonstrated that not only TLR RNA and protein levels were increased but also TOLLIP and PPAR-y levels, important TLR antagonists, were significantly decreased through the progression of gastric preneoplastic and neoplastic lesions [67].
In fact, we demonstrated that epithelial cells from normal mucosa colonized by Hp (gastritis) express two times more TLR2 and 4 (but not TLR5) when compared with normal mucosa without Hp [67]. Furthermore, the expression of TLRs antagonists TOLLIP and PPAR-y in gastritis was decreased in 25% when compared with normal Hp negative mucosa [67]. This is very important since this decrease of TLRs antagonists not only suggests increase activation of TLRs pathways but it can directly interfere with TLRs protein levels. Actually, as we have shown in colon mucosa, a decrease in TOLLIP levels associates with higher TLR protein expression even with similar levels of RNA [68]. Indeed, besides antagonizing many intracellular kinases activated by TLRs, TOLLIP also blocks TLRs molecules and promotes traffic of newly synthesized proteins into endosomes leading to TLR early degradation, meaning that lower levels of TOLLIP are associated with more functional TLRs [69- 74].
Gastric intestinal metaplasia presents even lower PPAR-g and TOLLIP expression, with 30% to 40% less expression when comparing to normal mucosa, and 1.2 - 1.5 times more TLR2 and TLR4 expression now dispersed trough the entire cell [67]. In dysplasia and cancer the expression of TLR RNA is similar to intestinal metaplasia. However, PPAR-g and TOLLIP levels are even lower, almost half of the expression when compared to normal mucosa, and this translates in a more intense and diffuse TLR protein expression throughout all the cell [67]. In fact, this significantly decreased TOLLIP expression altered the distribution of TLR protein expression that was polarized at the cellular membrane (particularly basolateral membrane) in normal epithelial cells and diffuse trough the cytoplasm and membrane in intestinal metaplasia, dysplasia and cancer [65-67]. The effect of Hp on the genetic profile of the mucosa is higher in normal mucosa than in intestinal metaplasia since in normal mucosa Hp highly augments the expression of TLR2, TLR4, TNF-α and decreases TOLLIP and PPAR-y and in intestinal metaplasia it only augments in a low level TLR4 and TNF-α without a significantly change in the expression of TLRs antagonists. In dysplasia or cancer the presence of this bacterium does not appear to change the genetic profile of the mucosa [67].
In our opinion it is the progressive activation of the overexpressed TLRs induced by Hp that will eventually lead to aberrant transcription of CDX2 and phenotypic change to intestinal metaplasia (Figure 1), similarly to what was previously shown in biliary epithelium [75]. Intestinal metaplasia with higher TLRs and lower expression of its antagonists is probably more reactive to several PAMPs, including from other microorganisms. However, at this stage the presence of Hp can remain of some importance since the mucosa is still reactive to this bacterium that, as we have shown, doubles TNF-α levels in the mucosa and also increases TLR4 and CDX2 expression [67]. This increase in CDX2 induced by HP in intestinal metaplasia cells can be of some importance since CDX2 has been associated to progression of gastric lesions and cancer [76,77].
Progressive activation of TLRs in intestinal metaplasia will eventually lead to dysplasia and cancer, characterized by even lower TOLLIP expression and consequently diffuse TLR expression throughout the entire cell. At this stage, we believe that this intense expression of TLRs make the cell more reactive to several PAMPs from different microorganisms, even commensal ones, and even DAMPs and, so, the effect of Hp becomes negligible (Figure 1).
Although it is now clear that the progression of gastric lesions is associated with increase expression of TLRs and lower expression of its antagonists, which suggests overactivation of TLRs signalling pathways, we do not know for sure if these receptors are functional and leading to the production pro-inflammatory and pro-oncogenic mediators at these later stages of gastric carcinogenesis. However, as gastric metaplastic, dysplastic and carcinoma cells express several TLRs, an interaction with Hp and with other microorganisms is probable [66]. Indeed, in cell lines studies it was shown that several PAMPs via interaction with epithelial TLRs can induce gastric carcinoma-promoting factors, such as IL-1, IL-8 and angiogenic factors [56,66,78]. Moreover, Hp appears to augment the growth of gastric cancer cells through the LPS-TLR4 pathway, promoting proliferation and progression of gastric cancers [79]. Other studies showed that Hp activation of NF-κB through TLR2 and TLR9 was able to promote cyclooxygenase-2 (COX-2) overexpression, invasiveness and angiogenesis of gastric cells [27,80]. Other suggested that PAMPs from other microorganisms may also promote tumor growth via TLR2
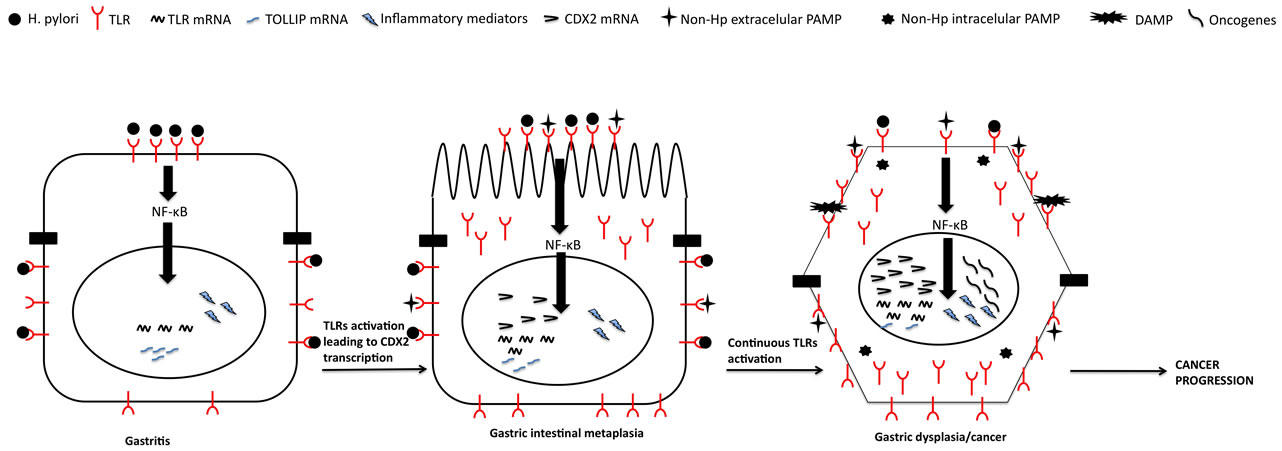
Figure 1. TLRs progressive activation in gastric carcinogenesis. Sequence from normal mucosa to gastric cancer involving progressive TLRs activation, initially only to Helicobacter pylori but later on eventually to several different PAMPs and even DAMPs. Helicobacter pylori-dependent TLRs chronic activation throughout the years leads to chronic inflammation that eventually leads to aberrant transcription of CDX2 with phenotipical change to intestinal metaplasia. At this point other agents may promote chronic inflammation with production of oncogenic mediators that eventually will lead to dysplasia/ cancer.
signaling, independently of Hp [81]. So, these in vitro studies gave indirect evidence that the overexpressed TLRs that we saw in human gastric biopsies are in fact functional and may play a crucial role in gastric carcinogenesis.
In some way confirming this very important role of TLRs in gastric carcinogenesis, some studies described an association of TLR4, TLR2 and even TLR5 polymorphisms with the risk of developing gastric preneoplastic lesions and gastric cancer [82-86].
In conclusion, although more data is needed in this area, several in vitro, translational and polymorphisms human studies suggest that TLRs may have a crucial role in gastric carcinogenesis.
4. Potential Diagnostic and Prognostic Value of Toll-Like Receptors in Gastric Lesions
Scarce scientific evidence exists concerning the clinical application of the previous described data on the role of TLRs for Hp recognition and progression of gastric lesions. However, we believe that in the future this knowledge can acquire an important role not only for the diagnosis of gastric preneoplastic lesions but particularly for the prognostic of gastric neoplasias.
Concerning the diagnosis of gastric lesions, we have shown that TLRs immunohistochemistry quantification could be used for the differentiation of the different gastric lesions [65]. When adding the relative expression of TLR2, 4 and 5, we obtained several expression scores (from 0 to 9), with different scores associating to different lesions. For instance, the presence of an expression score of 1 seems to leads to a very low false negative rate (<1%) for lesions as severe as dysplasia. On the other hand, to adequately identify dysplasia a score of 8 seems to be very useful as it leads to a very low false positive rate (4%) in patients with precancerous conditions and also to a low false positive rate (missing invasiveness of 9%) when distinguishing dysplasia from invasive cancer [65]. In practical terms we may say that we could use the fact that gastric dysplastic cells have a strong expression of TLRs for an accurate diagnosis of gastric dysplasia. Indeed, sometimes when there are marked inflammatory and/or regenerative epithelia changes it is difficult for the pathologist to differentiate between these benign alterations from dysplasia. In this particular case we believe that TLRs immunohistochemistry quantification may help in the differentiation of inflammation (low to moderate expression) with dysplasia (very high TLRs expression).
Moreover, we may speculate that this different TLR expression in gastric lesions may also help the endoscopic diagnostic of lesions. Current guidelines still recommend histology as the gold standard, however, it was recognized that new endoscopy technologies might help the diagnosis of gastric lesions [87]. In fact, our group demonstrated that narrow-band imaging (NBI) was highly accurate for the diagnosis of gastric preneoplastic and neoplastic lesions [88]. However, the problem of these new techniques is that they can be cumbersome and, until now, they rely on previous mucosal changes identified on standard white-light endoscopy, and only then they can be applied. We may speculate that antibodiesmarked TLRs that release, for example, fluorescence waves upon interaction with white light would help the diagnosis of lesions, by creating fluorescence hot-spots for dysplasia that could easily be recognized in a standard endoscopy. We believe that future studies should have this in mind.
As for predicting gastric cancer risk or prognosis, all that can be said is only speculative, since to our knowledge no single study analyzed if TLRs epithelial or tumoural expression has prognostic/predicting value or not. Nevertheless, as we have discussed it appears that TLRs are active in tumoural cells producing several inflammatory, survival, angiogenic and oncogenic mediators that may facilitate tumorogenesis and cancer progression. So, in theory, TLR expression in tumours may in fact have prognostic value and may act as biomarkers, with worse prognosis seen in tumours with higher TLR expression. Indeed, in colorectal cancer it has been shown that higher TLR4 expression confers worse prognosis [89]. Moreover, several TLRs polymorphisms, including from TLR2 and 4, were associated with colorectal cancer development and prognosis [90]. In oesophageal adenocarcinoma increased TLR9 expression was also associated with adverse prognosis [91]. If TLR expression can influence prognosis of other gastrointestinal tumours, we believe that it can alter also the prognosis of gastric cancer. Future studies are needed to evaluate this aspect.
5. Potential Therapeutic Value of the Modulation of Toll-Like Receptors
Taking together the important role of TLRs for Hp recognition and immune response as well as the potential role in the progression of gastric lesion we may preview several lines for therapy (Table 1). The first one is vaccination—can TLR modulation help in the creation of an efficacious vaccine against Hp infection? The true is that until now no vaccine, prophylactic or therapeutic, using whole inactivated Hp, Hp proteins or recombinant vaccines, has shown to be efficacious against this infection in humans [92,93]. In theory, TLRs agonists as adjuvant to the vaccine can improve efficacy rates. However, until now, to our knowledge no study has evaluated this aspect.
Other line of potential research is using TLRs agonists
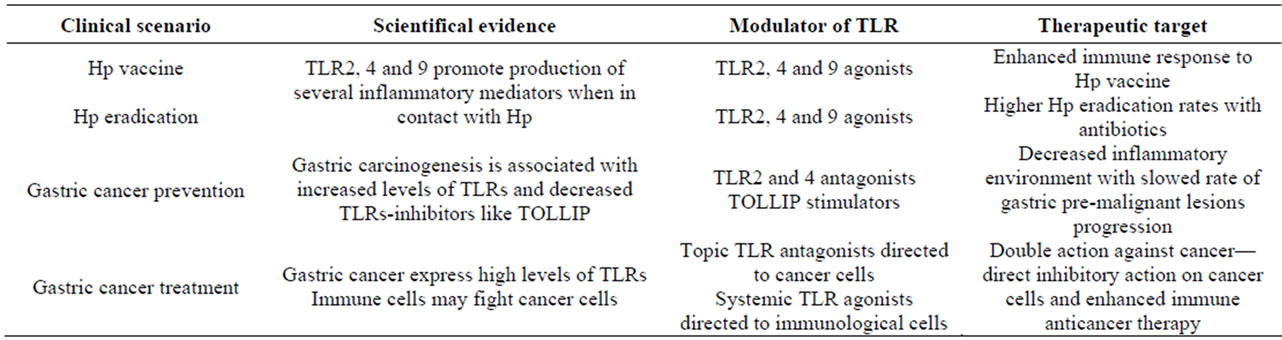
Table 1. Potential therapeutic targets for Hp-TLRs interaction in clinical practice.
as adjuvant not to a vaccine but to antibiotherapy for treatment of Hp established infection. Indeed, current eradication rates with standard multidrug regimens are not ideal. We may speculate that using TLRs agonists with antibiotics may increase eradication rates and may help to overcome antibiotic resistance of these bacteria. However, once more, until know no study has evaluated this aspect. We believe that both for vaccination and to therapy future studies should evaluate the role of TLRs agonists. However, we should keep in mind that the effect of these substances is not linear since some studies show that stimulation of these receptors may in some circumstances have an anti-inflammatory effect. Indeed, although TLR2 is considered the most important receptor for HP recognition and subsequent immune response it appears that under some circumstances TLR2 can induce production of anti-inflammatory cytokines and, in this way, inducing immunotolerance [49]. Furthermore, TLR9, another important recognition receptor for HP may have an anti-inflammatory effect on the early phase of gastritis [94]. So, although TLR stimulation may have a role in Hp vaccine and eradication, these aspects should be kept in mind in future studies.
Another potential line of treatment, and eventually even more important one, is the role of TLR modulation for prevention of gastric cancer in patients with premalignant lesions. As we have seen progression of gastric lesions is associated with increasing levels of TLRs and diminished TLRs inhibitors. The question is: If we block TLRs or if we are able to increase its inhibitors (TOLLIP) can we prevent cancer development? No study has investigated this question, however, in theory, if we block TLRs we will decrease its stimulation and consequently diminish production of pro-inflammatory and pro-oncogenic mediators. Although a direct antagonism of TLRs can be feasible, particularly if these substances act only topically in the gastrointestinal tract, a substance that can modulate TLRs activation by augmenting TOLLIP levels, for example with probiotics, would be even more attractive since the risk of infection would be theoretically lower with these substances. This could be a very important line of research since at this moment, besides surveillance, we have nothing more to offer to these patients.
Finally, since TLR stimulation on malignant cells leads to production of cancer promoting factors, we must consider that TLR antagonists may a role in gastric cancer treatment. Interestingly, TLR agonists and not antagonists have been used for cancer treatment as a form of immunotherapy. However, if TLR agonists may promote an immune reaction against cancer, the true is that a direct effect on tumours may have an opposite effect. So, in our opinion a strategy of a topical action of TLR antagonism directly on cancer cells with a systemic TLR activation of immune cells could promote a double attack against cancer. This is a strategy that deserves further study since gastric cancer is one of the most lethal cancers in the world.
6. Conclusions
As we have seen TLRs have a crucial role in Hp recognition and subsequent immune response against this bacteria. Furthermore, they may have an essential role in the progression of gastric lesions associated with Hp leading to cancer since the progression of gastric lesions is associated with increasing levels of TLRs. Although the role of TLRs in the physiopathology of gastric lesions is established, scarce humans studies exist concerning the potential use of Hp-TLRs interaction in clinical practice. In this article we suggested that TLRs may act as biomarkers of gastric carcinogenesis and several lines for future research considering the important role of TLRs in gastric disease were presented. Namely, we suggested that these receptors should be evaluated in the prognosis of gastric cancer. Moreover, we provide several lines for translational and human research using TLRs modulation not only for vaccination and HP eradication but also for the prevention of the progression of gastric preneoplastic lesions and also to the treatment of gastric cancer.
In conclusion, TLRs can be the link between Hp and the sequence of gastric carcinogenesis. Future lines of investigation should study these novel and enthusiastic scientific findings in order to obtain more weapons in the fight against gastric cancer.
7. Acknowledgements
The authors thank Portuguese Oncology Institute of Porto, Portugal for allowing the creation of this line of research.
REFERENCES
- D. A. Pleura and S. E. Crowe. “Helicobacter pylori,” In: L. Feldman, L. Friedman and J. Brandt, Eds., Sleisenger and Fordtran’s Gastrointestinal and Liver Disease, Saunders Elsevier, Philadelphia, 2010, pp. 833-843.
- World Health Organization and International Agency for Research on Cancer, “Schistosomes, Liver Flukes and Helicobacter pylori,” IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, Lyon, 7-14 June 1994, pp. 1-241.
- M. Clyne and B. Drumm, “Adherence of Helicobacter pylori to Primary Human Gastrointestinal Cells,” Infection and Immunity, Vol. 61, No. 10, 1993, pp. 4051-4057.
- P. Pimentel-Nunes, J. B. Soares, R. Roncon-Albuquerque Jr., M. Dinis-Ribeiro and A. F. Leite-Moreira, “Toll-Like Receptors as Therapeutic Targets in Gastrointestinal Diseases,” Expert Opinion on Therapeutic Targets, Vol. 14, No. 4, 2010, pp. 347-368. doi:10.1517/14728221003642027
- P. Laurèn, “The Two Histological Main Types of Gastric Carcinoma: Diffuse and So-Called Intestinal-Type Carcinoma,” Acta Pathologica Microbiologica Scandinavica, Vol. 64, No. 1, 1965, pp. 31-49.
- A. Fukao, S. Hisamichi, N. Ohsato, N. Fujino, N. Endo and M. Iha, “Correlation between the Prevalence of Gastritis and Gastric Cancer in Japan,” Cancer Causes Control, Vol. 4, No. 17, 1993, p. 20.
- C. R. Kapadia, “Gastric Atrophy, Metaplasia and Dysplasia. A Clinical Perspective,” Journal of Clinical Gastroenterology, Vol. 36, No. 5, 2003, pp. S29-S36. doi:10.1097/00004836-200305001-00006
- R. M. Genta, “Review Article: Gastric Atrophy and Atrophic Gastritis—Nebulous Concepts in Search of a Definition,” Alimentary Pharmacology & Therapeutics, Vol. 12, Supplement 1, 1998, pp. 17-23. doi:10.1111/j.1365-2036.1998.00003.x
- P. Correa, “Human Gastric Carcinogenesis: A Multistep and Multifactorial Process—First American Cancer Society Award Lecture on Cancer Epidemiology and Prevention,” Cancer Research, Vol. 52, No. 24, 1992, pp. 6735- 6740.
- P. B. Ernst and B. D. Gold, “The Disease Spectrum of Helicobacter pylori: The Immunopathogenesis of Gastroduodenal Ulcer and Gastric Cancer,” Annual Review of Microbiology, Vol. 54, No. 1, 2000, pp. 615-640. doi:10.1146/annurev.micro.54.1.615
- T. Ihamaki, M. Sankkonen and M. Siurala, “Long Term Observation of Subjects with Normal Mucosa and with Superficial Gastritis: Results of 23 - 27 Years Follow-Up Examination,” Scandinavian Journal of Gastroenterology, Vol. 13, No. 7, 1978, pp. 771-775. doi:10.3109/00365527809182189
- M. C. Ormiston, M. W. Gear and B. W. Codling, “Five Year Follow-Up Study of Gastritis,” Journal of Clinical Pathology, Vol. 35, No. 7, 1982, pp. 757-760. doi:10.1136/jcp.35.7.757
- S. Akira and H. Hemmi, “Recognition of Pathogen-Associated Molecular Patterns by TLR Family,” Immunology Letters, Vol. 85, No. 2, 2003, pp. 85-95. doi:10.1016/S0165-2478(02)00228-6
- K. Takeda, T. Kaisho and S. Akira, “Toll-Like Receptors,” Annual Review of Immunology, Vol. 21, No. 1, 2003, pp. 335-376. doi:10.1146/annurev.immunol.21.120601.141126
- G. M. Barton and R. Medzhitov, “Control of Adaptive Immune Responses by Toll-Like Receptors,” Current Opinion in Immunology, Vol. 14, No. 3, 2002, pp. 380-383. doi:10.1016/S0952-7915(02)00343-6
- G. Napolitani, A. Rinaldi, F. Bertoni, F. Sallusto and A. Lanzavecchia, “Selected Toll-Like Receptor Agonist Combinations Synergistically Trigger A T Helper Type 1- Polarizing Program in Dendritic Cells,” Nature Immunology, Vol. 6, No. 8, 2005, pp. 769-776. doi:10.1038/ni1223
- S. Akira and K. Takeda, “Toll-Like Receptor Signaling,” Nature Reviews Immunology, Vol. 4, No. 7, 2004, pp. 499-511. doi:10.1038/nri1391
- T. Kawai and S. Akira, “TLR Signaling,” Cell Death & Differentiation, Vol. 13, No. 5, 2006, pp. 816-825. doi:10.1038/sj.cdd.4401850
- F. L. Rock, G. Hardiman, J. C. Timans, R. A. Kastelein and J. F. Bazan, “A Family of Human Receptors Structurally Related to Drosophila Toll,” Proceedings of the National Academy of Sciences of the United States of America Vol. 95, No. 2, 1998, pp. 588-593. doi:10.1073/pnas.95.2.588
- P. A. Hopkins and S. Sriskandan, “Mammalian Toll-Like Receptors: To Immunity and Beyond,” Clinical & Experimental Immunology, Vol. 140, No. 3, 2005, pp. 395-407. doi:10.1111/j.1365-2249.2005.02801.x
- H. Kumar, T. Kawai and S. Akira, “Toll-Like Receptors and Innate Immunity,” Biochemical and Biophysical Research Communications Vol. 388, No. 4, 2009, pp. 621-625. doi:10.1016/j.bbrc.2009.08.062
- S. Akira, S. Uematsu and O. Takeuchi, “Pathogen Recognition and Innate Immunity,” Cell, Vol. 124, No. 4, 2006, pp. 783-801. doi:10.1016/j.cell.2006.02.015
- B. Beutler, “Neo-Ligands for Innate Immune Receptors and the Etiology of Sterile Inflammatory Disease,” Immunological Reviews, Vol. 220, No. 1, 2007, pp. 113-128. doi:10.1111/j.1600-065X.2007.00577.x
- M. T. Lotze, H. J. Zeh, A. Rubartelli, L. J. Sparvero, A. A. Amoscato, N. R. Washburn, M. E. Devera, X. Liang, M. Tor and T. Billiar, “The Grateful Dead: Damage-Associated Molecular Pattern Molecules and Reduction/Oxidation Regulate Immunity,” Immunological Reviews, Vol. 220, No. 1, 2007, pp. 60-81. doi:10.1111/j.1600-065X.2007.00579.x
- T. Kaisho and S. Akira, “Pleiotropic Function of TollLike Receptors,” Microbes and Infection, Vol. 6, No. 15, 2004, pp. 1388-1394. doi:10.1016/j.micinf.2004.08.019
- M. Fukata, A. Chen, A. Klepper, S. Krishnareddy, A. S. Vamadevan, L. S. Thomas, R. Xu, H. Inoue, M. Arditi, A. J. Dannenberg, et al., “Cox-2 is Regulated by Toll-Like Receptor-4 (TLR4) Signaling: Role in Proliferation and Apoptosis in the Intestine,” Gastroenterology, Vol. 131, No. 3, 2006, pp. 862-877. doi:10.1016/j.micinf.2004.08.019
- Y. J. Chang, M. S. Wu, J. T. Lin and C. C. Chen, “Helicobacter pylori-Induced Invasion and Angiogenesis of Gastric Cells Is Mediated by Cyclooxygenase-2 Induction through TLR2/TLR9 and Promoter Regulation,” The Journal of Immunology, Vol. 175, No. 12, 2005, pp. 8242-8252.
- J. A. Spitzer, M. Zheng, J. K. Kolls, C. Vande Stouwe and J. J. Spitzer, “Ethanol and LPS Modulate NF-KappaB Activation, Inducible NO Synthase and COX-2 Gene Expression in Rat Liver Cells in Vivo,” Frontiers in Bioscience, Vol. 7, 2002, pp. a99-a108. doi:10.2741/spitzer
- I. T. Lee, C. W. Lee, W. H. Tung, S. W. Wang, C. C. Lin, J. C. Shu and C. M. Yang, “Cooperation of TLR2 with MyD88, PI3K, and Rac1 in Lipoteichoic Acid-Induced cPLA2/CO X-2-Dependent Airway Inflammatory Responses,” American Journal of Pathology, Vol. 176, No. 4, 2010, pp. 1671-1684. doi:10.2353/ajpath.2010.090714
- M. Palazzo, S. Gariboldi, L. Zanobbio, G. F. Dusio, S. Selleri, M. Bedoni, A. Balsari and C. Rumio, “Cross-Talk among Toll-Like Receptors and Their Ligands,” International Immunology, Vol. 20, No. 5, 2008, pp. 709-718. doi:10.1093/intimm/dxn027
- F. Re and J. L. Strominger, “IL-10 Released by Concomitant TLR2 Stimulation Blocks the Induction of a Subset of Th1 Cytokines That Are Specifically Induced by TLR4 or TLR3 in Human Dendritic Cells,” The Journal of Immunology, Vol. 173, No. 12, 2004, pp. 7548-7555.
- F. Re and J. L. Strominger, “Heterogeneity of TLR-Induced Responses in Dendritic Cells: From Innate to Adaptive Immunity,” Immunobiology, Vol. 209, No. 1-2, 2004, pp. 191-198. doi:10.1016/j.imbio.2004.03.005
- M. Fukata and M. T. Abreu, “Role of Toll-Like Receptors in Gastrointestinal Malignancies,” Oncogene, Vol. 27, No. 2, 2008, pp. 234-243. doi:10.1038/sj.onc.1210908
- M. Fukata and M. T. Abreu, “Pathogen Recognition Receptors, Cancer and Inflammation in the Gut,” Current Opinion in Pharmacology, Vol. 9, No. 6, 2009, pp. 680- 687. doi:10.1016/j.coph.2009.09.006
- P. Desreumaux, L. Dubuquoy, S. Nutten, M. Peuchmaur, W. Englaro, K. Schoonjans, B. Derijard, B. Desvergne, W. Wahli, P. Chambon, et al., “Attenuation of Colon Inflammation through Activators of the Retinoid X Receptor (RXR)/Peroxisome Proliferator-Activated Receptor Gamma (PPARgamma) Heterodimer. A Basis for New Therapeutic Strategies,” The Journal of Experimental Medicine, Vol. 193, No. 7, 2001, pp. 827-838. doi:10.1084/jem.193.7.827
- R. A. Gupta, D. B. Polk, U. Krishna, D. A. Israel, F. Yan, R. N. DuBois and R. M. Peek Jr., “Activation of Peroxisome Proliferator-Activated Receptor Gamma Suppresses Nuclear Factor Kappa B-Mediated Apoptosis Induced by Helicobacter pylori in Gastric Epithelial Cells,” The Journal of Biological Chemistry, Vol. 276, No. 33, 2001, pp. 31059-31066. doi:10.1074/jbc.M104141200
- D. Kelly, J. I. Campbell, T. P. King, G. Grant, E. A. Jansson, A. G. Coutts, S. Pettersson and S. Conway, “Commensal Anaerobic Gut Bacteria Attenuate Inflammation by Regulating Nuclear-Cytoplasmic Shuttling of PPARGamma and RelA,” Nature Immunology, Vol. 5, No. 1, 2004, pp. 104-112.
- G. Melmed, L. S. Thomas, N. Lee, S. Y. Tesfay, K. Lukasek, K. S. Michelsen, Y. Zhou, B. Hu, M. Arditi and M. T. Abreu, “Human Intestinal Epithelial Cells Are Broadly Unresponsive to Toll-Like Receptor 2-Dependent Bacterial Ligands: Implications for Host-Microbial Interactions in the Gut,” Journal of Immunology, Vol. 170, No. 3, 2003, pp. 1406-1415.
- J. M. Otte, E. Cario and D. K. Podolsky, “Mechanisms of Cross Hyporesponsiveness to Toll-Like Receptor Bacterial Ligands in Intestinal Epithelial Cells,” Gastroenterology, Vol. 126, No. 4, 2004, pp. 1054-1070. doi:10.1053/j.gastro.2004.01.007
- F. Y. Liew, D. Xu, E. K. Brint and L. A. O’Neill, “Negative Regulation of Toll-Like Receptor-Mediated Immune Responses,” Nature Reviews Immunology, Vol. 5, No. 6, 2005, pp. 446-458.
- C. F. Ortega-Cava, S. Ishihara, M. A. Rumi, K. Kawashima, N. Ishimura, H. Kazumori, J. Udagawa, Y. Kadowaki and Y. Kinoshita, “Strategic Compartmentalization of Toll-Like Receptor 4 in the Mouse Gut,” Journal of Immunology, Vol. 170, No. 8, 2003, pp. 3977-3985.
- M. T. Abreu, L. S. Thomas, E. T. Arnold, K. Lukasek, K. S. Michelsen and M. Arditi, “TLR Signaling at the Intestinal Epithelial Interface,” Journal of Endotoxin Research, Vol. 9, No. 5, 2003, pp. 322-330.
- M. T. Abreu, P. Vora, E. Faure, L. S. Thomas, E. T. Arnold and M. Arditi, “Decreased Expression of Toll-Like Receptor-4 and MD-2 Correlates with Intestinal Epithelial Cell Protection against Dysregulated Proinflammatory Gene Expression in Response to Bacterial Lipopolysaccharide,” Journal of Immunology, Vol. 167, No. 3, 2001, pp. 1609-1616.
- E. Furrie, S. Macfarlane, G. Thomson and G. T. Macfarlane, “Toll-Like Receptors-2, -3 and -4 Expression Patterns on Human Colon and Their Regulation by Mucosal-Associated Bacteria,” Immunology, Vol. 115, No. 4, 2005, pp. 565-574. doi:10.1111/j.1365-2567.2005.02200.x
- T. H. Chuang and R. J. Ulevitch, “Triad3A, an E3 Ubiquitin-Protein Ligase Regulating Toll-Like Receptors,” Nature Immunology, Vol. 5, No. 5, 2004, pp. 495-502. doi:10.1038/ni1066
- A. Mansell, R. Smith, S. L. Doyle, P. Gray, J. E. Fenner, P. J. Crack, S. E. Nicholson, D. J. Hilton, L. A. O’Neill and P. J. Hertzog, “Suppressor of Cytokine Signaling 1 Negatively Regulates Toll-Like Receptor Signaling by Mediating Mal Degradation,” Nature Immunology, Vol. 7, No. 2, 2006, pp. 148-155.
- R. Rad, L. Brenner, A. Krug, P. Voland, J. Mages, R. Lang, S. Schwendy, W. Reindl, A. Dossumbekova, W. Ballhorn, et al., “Toll-Like Receptor-Dependent Activation of Antigen-Presenting Cells Affects Adaptive Immunity to Helicobacter pylori,” Gastroenterology, Vol. 133, No. 1, 2007, pp. 150-163.
- R. Takenaka, K. Yokota, K. Ayada, M. Mizuno, Y. Zhao, Y. Fujinami, S. N. Lin, T. Toyokawa, H. Okada, Y. Shiratori, et al., “Helicobacter pylori Heat-Shock Protein 60 Induces Inflammatory Responses through the Toll-Like Receptor-Triggered Pathway in Cultured Human Gastric Epithelial Cells,” Microbiology, Vol. 150, No. 12, 2004, pp. 3913-3922. doi:10.1099/mic.0.27527-0
- R. Rad, W. Ballhorn, P. Voland, K. Eisenacher, J. Mages, L. Rad, R. Ferstl, R. Lang, H. Wagner, R. M. Schmid, et al., “Extracellular and Intracellular Pattern Recognition Receptors Cooperate in the Recognition of Helicobacter pylori,” Gastroenterology, Vol. 136, No. 7, 2009, pp. 2247-2257. doi:10.1053/j.gastro.2009.02.066
- K. Uno, K. Kato, T. Atsumi, T. Suzuki, J. Yoshitake, H. Morita, S. Ohara, Y. Kotake, T. Shimosegawa and T. Yoshimura, “Toll-Like Receptor (TLR) 2 Induced through TLR4 Signaling Initiated by Helicobacter pylori Cooperatively Amplifies iNOS Induction in Gastric Epithelial Cells,” American Journal of Physiology Gastrointestinal and Liver Physiology, Vol. 293, No. 5, 2007, pp. G1004-G1012. doi:10.1152/ajpgi.00096.2007
- M. Obonyo, M. Sabet, S. P. Cole, J. Ebmeyer, S. Uematsu, S. Akira and D. G. Guiney, “Deficiencies of Myeloid Differentiation Factor 88, Toll-Like Receptor 2 (TLR2), or TLR4 Produce Specific Defects in Macrophage Cytokine Secretion Induced by Helicobacter pylori,” Infection and Immunity, Vol. 75, No. 5, 2007, pp. 2408-2414. doi:10.1128/IAI.01794-06
- A. P. Gobert, J. C. Bambou, C. Werts, V. Balloy, M. Chignard, A. P. Moran and R. L. Ferrero, “Helicobacter pylori Heat Shock Protein 60 Mediates Interleukin-6 Production by Macrophages via a Toll-Like Receptor (TLR)- 2-, TLR-4-, and Myeloid Differentiation Factor 88-Independent Mechanism. The Journal of Biological Chemistry, Vol. 279, No. 1, 2004, pp. 245-250. doi:10.1074/jbc.M307858200
- S. K. Lee, A. Stack, E. Katzowitsch, S. I. Aizawa, S. Suerbaum and C. Josenhans, “Helicobacter pylori Flagellins Have very Low Intrinsic Activity to Stimulate Human Gastric Epithelial Cells via TLR5,” Microbes and Infection, Vol. 5, No. 15, 2003, pp. 1345-1356. doi:10.1016/j.micinf.2003.09.018
- E. Andersen-Nissen, K. D. Smith, K. L. Strobe, S. L. Barrett, B. T. Cookson, S. M. Logan and A. Aderem, “Evasion of Toll-Like Receptor 5 by Flagellated Bacteria,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 102, No. 26, 2005, pp. 9247-9252. doi:10.1073/pnas.0502040102
- A. T. Gewirtz, Y. Yu, U. S. Krishna, D. A. Israel, S. L. Lyons and R. M. Peek Jr., “Helicobacter pylori Flagellin Evades Toll-Like Receptor 5-Mediated Innate Immunity,” The Journal of Infectious Diseases, Vol. 189, No. 10, 2004, pp. 1914-1920. doi:10.1086/386289
- M. F. Smith Jr., A. Mitchell, G. Li, S. Ding, A. M. Fitzmaurice, K. Ryan, S. Crowe and J. B. Goldberg, “Toll-Like Receptor (TLR) 2 and TLR5, but Not TLR4, Are Required for Helicobacter pylori-Induced NF-kappa B Activation and Chemokine Expression by Epithelial Cells,” The Journal of Biological Chemistry, Vol. 278, No. 35, 2003, pp. 32552-32560. doi:10.1074/jbc.M305536200
- A. Amedei, A. Cappon, G. Codolo, A. Cabrelle, A. Polenghi, M. Benagiano, E. Tasca, A. Azzurri, M. M. D’Elios, G. Del Prete, et al., “The Neutrophil-Activating Protein of Helicobacter pylori Promotes Th1 Immune Responses,” The Journal of Clinical Investigation, Vol. 116, No. 4, 2006, pp. 1092-1101. doi:10.1172/JCI27177
- L. Mandell, A. P. Moran, A. Cocchiarella, J. Houghton, N. Taylor, J. G. Fox, T. C. Wang and E. A. Kurt-Jones, “Intact Gram-Negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus Bacteria Activate Innate Immunity via Toll-Like Receptor 2 but Not Toll-Like Receptor 4,” Infection and Immunity, Vol. 72, No. 11, 2004, pp. 6446-6454. doi:10.1128/IAI.72.11.6446-6454.2004
- C. Basak, S. K. Pathak, A. Bhattacharyya, S. Pathak, J. Basu and M. Kundu, “The Secreted Peptidyl Prolyl Cis, Trans-Isomerase HP0175 of Helicobacter pylori Induces Apoptosis of Gastric Epithelial Cells in a TLR4- and Apoptosis Signal-Regulating Kinase 1-Dependent Manner,” Journal of Immunology, Vol. 174, No. 9, 2005, pp. 5672-5680.
- S. Ishihara, M. A. Rumi, Y. Kadowaki, C. F. Ortega-Cava, T. Yuki, N. Yoshino, Y. Miyaoka, H. Kazumori, N. Ishimura, Y. Amano, et al., “Essential Role of MD-2 in TLR4-Dependent Signaling during Helicobacter pyloriAssociated Gastritis,” Journal of Immunology, Vol. 173, No. 2, 2004, pp. 1406-1416.
- T. Kawahara, Y. Kuwano, S. Teshima-Kondo, T. Kawai, T. Nikawa, K. Kishi and K. Rokutan, “Toll-Like Receptor 4 Regulates Gastric Pit Cell Responses to Helicobacter pylori Infection,” The Journal of Medical Investigation, Vol. 48, No. 3-4, 2001, pp. 190-197.
- A. M. Torok, A. H. Bouton and J. B. Goldberg, “Helicobacter pylori Induces Interleukin-8 Secretion by TollLike Receptor 2- and Toll-Like Receptor 5-Dependent and -Independent Pathways,” Infection and Immunity, Vol. 73, No. 3, 2005, pp. 1523-1531. doi:10.1128/IAI.73.3.1523-1531.2005
- S. K. Pachathundikandi, S. Brandt, J. Madassery and S. Backert, “Induction of TLR-2 and TLR-5 Expression by Helicobacter pylori Switches cagPAI-Dependent Signalling Leading to the Secretion of IL-8 and TNF-Alpha,” PloS One, Vol. 6, No. 5, 2011, p. e19614. doi:10.1371/journal.pone.0019614
- A. E. Anderson, M. L. Worku, W. Khamri, K. B. Bamford, M. M. Walker and M. R. Thursz, “TLR9 Polymorphisms Determine Murine Lymphocyte Responses to Helicobacter: Results from a Genome-Wide Scan,” European Journal of Immunology, Vol. 37, No. 6, 2007, pp. 1548-1561. doi:10.1002/eji.200636562
- P. Pimentel-Nunes, L. Afonso, P. Lopes, R. RonconAlbuquerque Jr., N. Goncalves, R. Henrique, L. MoreiraDias, A. F. Leite-Moreira and M. Dinis-Ribeiro, “Increased Expression of Toll-like Receptors (TLR) 2, 4 and 5 in Gastric Dysplasia,” Pathology & Oncology Research, Vol. 17, No. 3, 2011, pp. 677-683. doi:10.1007/s12253-011-9368-9
- B. Schmausser, M. Andrulis, S. Endrich, H. K. MullerHermelink and M. Eck, “Toll-Like Receptors TLR4, TLR5 and TLR9 on Gastric Carcinoma Cells: An Implication for Interaction with Helicobacter pylori,” International Journal of Medical Microbiology, Vol. 295, No. 3, 2005, pp. 179-185. doi:10.1016/j.ijmm.2005.02.009
- P. Pimentel-Nunes, N. Goncalves, I. Boal-Carvalho, L. Afonso, P. Lopes, R. Roncon-Albuquerque Jr., R. Henrique, L. Moreira-Dias, A. F. Leite-Moreira and M. DinisRibeiro, “Helicobacter pylori Induces Increased Expression of Toll-Like Receptors and Decreased Toll-Interacting Protein in Gastric Mucosa that Persists throughout Gastric Carcinogenesis,” Helicobacter, Vol. 18, No. 1, 2013, pp. 22-32.
- P. Pimentel-Nunes, N. Goncalves, I. Boal-Carvalho, L. Afonso, P. Lopes, R. Roncon-Albuquerque Jr., J. B. Soares, E. Cardoso, R. Henrique, L. Moreira-Dias, et al., “Decreased Toll-Interacting Protein and Peroxisome Proliferator-Activated Receptor Gamma Are Associated with Increased Expression of Toll-Like Receptors in Colon Carcinogenesis,” Journal of Clinical Pathology, Vol. 65, No. 4, 2012, pp. 302-308. doi:10.1136/jclinpath-2011-200567
- Y. Bulut, E. Faure, L. Thomas, O. Equils and M. Arditi, “Cooperation of Toll-Like Receptor 2 and 6 for Cellular Activation by Soluble Tuberculosis Factor and Borrelia burgdorferi Outer Surface Protein A Lipoprotein: Role of Toll-Interacting Protein and IL-1 Receptor Signaling Molecules in Toll-Like Receptor 2 Signaling,” Journal of Immunology, Vol. 167, No. 2, 2001, pp. 987-994.
- G. Zhang and S. Ghosh, “Negative Regulation of TollLike Receptor-Mediated Signaling by Tollip,” The Journal of Biological Chemistry, Vol. 277, No. 9, 2002, pp. 7059- 7065. doi:10.1074/jbc.M109537200
- B. Brissoni, L. Agostini, M. Kropf, F. Martinon, V. Swoboda, S. Lippens, H. Everett, N. Aebi, S. Janssens, E. Meylan, et al., “Intracellular Trafficking of Interleukin-1 Receptor I Requires Tollip,” Current Biology, Vol. 16, No. 22, 2006, pp. 2265-2270. doi:10.1016/j.cub.2006.09.062
- A. Didierlaurent, B. Brissoni, D. Velin, N. Aebi, A. Tardivel, E. Kaslin, J. C. Sirard, G. Angelov, J. Tschopp and K. Burns, “Tollip Regulates Proinflammatory Responses to Interleukin-1 and Lipopolysaccharide,” Molecular and Cellular Biology, Vol. 26, No. 3, 2006, pp. 735-742. doi:10.1128/MCB.26.3.735-742.2006
- Y. Katoh, H. Imakagura, M. Futatsumori and K. Nakayama, “Recruitment of Clathrin onto Endosomes by the Tom1-Tollip Complex,” Biochemical and Biophysical Research, Vol. 341, No. 1, 2006, pp. 143-149. doi:10.1016/j.bbrc.2005.12.156
- Y. Katoh, Y. Shiba, H. Mitsuhashi, Y. Yanagida, H. Takatsu and K. Nakayama, “Tollip and Tom1 Form a Complex and Recruit Ubiquitin-Conjugated Proteins onto Early Endosomes,” The Journal of Biological Chemistry, Vol. 279, No. 23, 2004, pp. 24435-24443. doi:10.1074/jbc.M400059200
- H. Ikeda, M. Sasaki, A. Ishikawa, Y. Sato, K. Harada, Y. Zen, H. Kazumori and Y. Nakanuma, “Interaction of Toll-Like Receptors with Bacterial Components Induces Expression of CDX2 and MUC2 in Rat Biliary Epithelium in Vivo and in Culture,” Laboratory Investigation, Vol. 87, No. 6, 2007, pp. 559-571.
- X. S. Feng, Y. F. Wang, S. G. Hao, Y. Ru, S. G. Gao and L. D. Wang, “Expression of CDX2 and Villin in Gastric Cardiac Intestinal Metaplasia and the Relation with Gastric Cardiac Carcinogenesis,” Asian Pacific Journal of Cancer Prevention, Vol. 13, No. 1, 2012, pp. 247-250. doi:10.7314/APJCP.2012.13.1.247
- J. M. Kang, B. H. Lee, N. Kim, H. S. Lee, H. E. Lee, J. H. Park, J. S. Kim, H. C. Jung and I. S. Song, “CDX1 and CDX2 Expression in Intestinal Metaplasia, Dysplasia and Gastric Cancer,” Journal of Korean Medical Science, Vol. 26, No. 5, 2011, pp. 647-653. doi:10.3346/jkms.2011.26.5.647
- R. M. Strieter, “Chemokines: Not Just Leukocyte Chemoattractants in the Promotion of Cancer,” Nature Immunology, Vol. 2, No. 4, 2001, pp. 285-286. doi:10.1038/86286
- K. Chochi, T. Ichikura, M. Kinoshita, T. Majima, N. Shinomiya, H. Tsujimoto, T. Kawabata, H. Sugasawa, S. Ono, S. Seki, et al., “Helicobacter pylori Augments Growth of Gastric Cancers via the Lipopolysaccharide-TollLike Receptor 4 Pathway whereas Its Lipopolysaccharide Attenuates Antitumor Activities of Human Mononuclear Cells,” Clinical Cancer Research, Vol. 14, No. 10, 2008, pp. 2909-2917. doi:10.1158/1078-0432.CCR-07-4467
- Y. J. Chang, M. S. Wu, J. T. Lin, B. S. Sheu, T. Muta, H. Inoue and C. C. Chen, “Induction of Cyclooxygenase-2 Overexpression in Human Gastric Epithelial Cells by Helicobacter pylori Involves TLR2/TLR9 and c-SrcDependent Nuclear Factor-KappaB Activation,” Molecular Pharmacology, Vol. 66, No. 6, 2004, pp. 1465-1477. doi:10.1124/mol.104.005199
- B. Huang, J. Zhao, S. Shen, H. Li, K. L. He, G. X. Shen, L. Mayer, J. Unkeless, D. Li, Y. Yuan, et al., “Listeria monocytogenes Promotes Tumor Growth via Tumor Cell Toll-Like Receptor 2 Signaling,” Cancer Research, Vol. 67, No. 9, 2007, pp. 4346-4352. doi:10.1158/0008-5472.CAN-06-4067
- J. G. de Oliveira, A. F. Rossi, D. M. Nizato, K. Miyasaki and A. E. Silva, “Profiles of Gene Polymorphisms in Cytokines and Toll-Like Receptors with Higher Risk for Gastric Cancer,” Digestive Diseases and Sciences, Vol. 58, No. 4, 2012, pp. 978-988.
- D. Santini, S. Angeletti, A. Ruzzo, G. Dicuonzo, S. Galluzzo, B. Vincenzi, A. Calvieri, F. Pizzagalli, N. Graziano, E. Ferraro, et al., “Toll-Like Receptor 4 Asp299Gly and Thr399Ile Polymorphisms in Gastric Cancer of Intestinal and Diffuse Histotypes,” Clinical & Experimental Immunology, Vol. 154, No. 3, 2008, pp. 360-364. doi:10.1111/j.1365-2249.2008.03776.x
- G. L. Hold, C. S. Rabkin, W. H. Chow, M. G. Smith, M. D. Gammon, H. A. Risch, T. L. Vaughan, K. E. McColl, J. Lissowska, W. Zatonski, et al., “A Functional Polymorphism of Toll-Like Receptor 4 Gene Increases Risk of Gastric Carcinoma and Its Precursors,” Gastroenterology, Vol. 132, No. 3, 2007, pp. 905-912. doi:10.1053/j.gastro.2006.12.026
- T. Tahara, T. Arisawa, F. Wang, T. Shibata, M. Nakamura, M. Sakata, I. Hirata and H. Nakano, “Toll-Like Receptor 2 -196 to 174del Polymorphism Influences the Susceptibility of Japanese People to Gastric Cancer,” Cancer Science, Vol. 98, No. 11, 2007, pp. 1790-1794. doi:10.1111/j.1349-7006.2007.00590.x
- H. M. Zeng, K. F. Pan, Y. Zhang, L. Zhang, J. L. Ma, T. Zhou, H. J. Su, W. Q. Li, J. Y. Li, M. Gerhard, et al., “Genetic Variants of Toll-Like Receptor 2 and 5, Helicobacter pylori Infection, and Risk of Gastric Cancer and Its Precursors in a Chinese Population,” Cancer Epidemiology, Biomarkers & Prevention, Vol. 20, No. 12, 2011, pp. 2594-2602.
- M. Dinis-Ribeiro, M. Areia, A. C. de Vries, R. Marcos-Pinto, M. Monteiro-Soares, A. O’Connor, C. Pereira, P. Pimentel-Nunes, R. Correia, A. Ensari, et al., “Management of Precancerous Conditions and Lesions in the Stomach (MAPS): Guideline from the European Society of Gastrointestinal Endoscopy (ESGE), European Helicobacter Study Group (EHSG), European Society of Pathology (ESP), and the Sociedade Portuguesa de Endoscopia Digestiva (SPED),” Endoscopy, Vol. 44, No. 1, 2012, pp. 74-94. doi:10.1055/s-0031-1291491
- P. Pimentel-Nunes, M. Dinis-Ribeiro, J. B. Soares, R. Marcos-Pinto, C. Santos, C. Rolanda, R. P. Bastos, M. Areia, L. Afonso, J. Bergman, et al., “A Multicenter Validation of an Endoscopic Classification with Narrow Band Imaging for Gastric Precancerous and Cancerous Lesions,” Endoscopy, Vol. 44, No. 3, 2012, pp. 236-246. doi:10.1055/s-0031-1291537
- E. L. Wang, Z. R. Qian, M. Nakasono, T. Tanahashi, K. Yoshimoto, Y. Bando, E. Kudo, M. Shimada and T. Sano, “High Expression of Toll-Like Receptor 4/Myeloid Differentiation Factor 88 Signals Correlates with Poor Prognosis in Colorectal Cancer,” British Journal of Cancer Vol. 102, No. 5, 2010, pp. 908-915. doi:10.1038/sj.bjc.6605558
- M. L. Slattery, J. S. Herrick, K. L. Bondurant and R. K. Wolff, “Toll-Like Receptor Genes and Their Association with Colon and Rectal Cancer Development and Prognosis,” International Journal of Cancer, Vol. 130, No. 12, 2012, pp. 2974-2980.
- J. H. Kauppila, H. Takala, K. S. Selander, P. P. Lehenkari, J. Saarnio and T. J. Karttunen, “Increased Toll-Like Receptor 9 Expression Indicates Adverse Prognosis in Oesophageal Adenocarcinoma,” Histopathology, Vol. 59, No. 4, 2011, pp. 643-649. doi:10.1111/j.1365-2559.2011.03991.x
- L. Moise, S. F. Moss and A. S. De Groot, “Moving Helicobacter pylori Vaccine Development Forward with Bioinformatics and Immunomics,” Expert Review of Vaccines, Vol. 11, No. 9, 2012, pp. 1031-1033. doi:10.1586/erv.12.80
- P. Malfertheiner, V. Schultze, B. Rosenkranz, S. H. Kaufmann, T. Ulrichs, D. Novicki, F. Norelli, M. Contorni, S. Peppoloni, D. Berti, et al., “Safety and Immunogenicity of an Intramuscular Helicobacter pylori Vaccine in Noninfected Volunteers: A Phase I Study,” Gastroenterology, Vol. 135, No. 3, 2008, pp. 787-795. doi:10.1053/j.gastro.2008.05.054
- K. Otani, T. Tanigawa, T. Watanabe, Y. Nadatani, M. Sogawa, H. Yamagami, M. Shiba, K. Watanabe, K. Tominaga, Y. Fujiwara, et al., “Toll-Like Receptor 9 Signaling Has Anti-Inflammatory Effects on the Early Phase of Helicobacter pylori-Induced Gastritis,” Biochemical and Biophysical Research Communications, Vol. 426, No. 3, 2012, pp. 342-349. doi:10.1016/j.bbrc.2012.08.080