Journal of Cancer Therapy
Vol.2 No.4(2011), Article ID:7796,12 pages DOI:10.4236/jct.2011.24072
Methylthioadenosine (MTA) Rescues Methylthioadenosine Phosphorylase (MTAP)-Deficient Tumors from Purine Synthesis Inhibition in Vivo via Non-Autonomous Adenine Supply
![]()
1Amgen Inc., South San Francisco, USA; 2Amgen Inc., Seattle, USA.
Email: *astridr@amgen.com
Received June 29th, 2011; revised July 25th, 2011; accepted August 4th, 2011.
Keywords: Methylthioadenosine Phosphorylase, Purine Metabolism, L-Alanosine, Adenine Salvage
ABSTRACT
Methylthioadenosine phosphorylase, (MTAP) is a key enzyme in the adenine and methionine salvage pathways. MTAP is encoded on human chromosome 9p21 in close proximity to the p16INK4a and p14ARF tumor suppressor genes and is frequently co-deleted with p16INK4a in many cancers. Deletion of MTAP has been reported to create a reliance of MTAP–/– tumors on de novo purine synthesis to maintain adequate pools of AMP, leading to increased sensitivity to purine synthesis inhibitors, such as L-alanosine. The ‘Achilles heel’ created by the loss of MTAP in cancer cells provides a unique therapeutic opportunity whereby MTAP–/– tumors could be selectively targeted with purine synthesis inhibitors and normal tissues could be preferentially rescued with MTAP substrates, such as MTA. We demonstrate that, in contrast to published literature, MTAP–/– cells are not more sensitive to inhibition of de novo purine synthesis than MTAP+/+ cells. Although MTA can preferentially rescue MTAP+/+ cells from purine-synthesis inhibitor toxicity in vitro, MTA protects cells of both genotypes from L-alanosine equivalently in vivo. Our data demonstrate that in vivo, adenine salvaged from plasma and adjacent tissues is sufficient to protect MTAP–/– tumors from the effects of purine synthesis inhibitors. These results suggest targeting MTAP–/– tumors with de novo purine synthesis inhibitors is unlikely to provide significant benefit over other therapeutic strategies and may explain, at least in part, the lack of efficacy of Lalanosine in clinical trials.
1. Introduction
Methylthioadenosine phosphorylase (MTAP) is an important metabolic enzyme required for the salvage of adenine and methionine. Methylthioadenosine (MTA) is a product of polyamine synthesis and is cleaved by MTAP to produce adenine and methylthioribose-1- phosphate which is further recycled to methionine [1-3] . MTAP is highly conserved across all Kingdoms, with nearly all organisms expressing either MTAP or 5’-Methylthioadenosine/S-adenosylhomocysteine nucleosidase (MTAN), both of which perform the conversion of MTA to adenine and methionine. Since mammals can only generate AMP via de novo purine synthesis or via salvage of adenosine and adenine, the conversion of MTA to adenine has been thought to provide an important source of adenine for AMP synthesis [4]. Accumulation of MTA is toxic to cells due to feedback inhibition of spermidine synthase; hence MTAP also plays an important function in eliminating this toxic by-product of polyamine synthesis [1,5,6] . Located on chromosome 9 directly adjacent to the tumor suppressor, CDKN2a, MTAP is frequently co-deleted in many human tumors, including leukemia, lymphoma, mesothelioma, lung, pancreatic, glioblastoma, and osteosarcoma [7-9] . Deletion of MTAP creates a unique situation in which the tumor cell is dependent on de novo purine synthesis of AMP to support proliferation, growth and survival. Indeed, previous studies have demonstrated that MTAP deficient tumors are more susceptible to inhibitors of de novo purine synthesis, such as L-alanosine, methotrexate and azaserine [10,11] . These data suggest that targeting MTAP deficient tumors with purine synthesis inhibitors in combination with MTAP substrates, such as MTA or 9-β-D-erythrofuranosyladenine (EFA) would provide greater efficacy and a greater therapeutic window over chemotherapeutic regimens currently in clinical use [12] . In this study, we sought to test the sensitivity of isogenic MTAP–/– and MTAP+/+ cell lines to purine synthesis inhibitors in vitro and in vivo. Using mouse models, we evaluated the contribution of purine nucleosides in plasma and surrounding tissues to the growth and proliferation of MTAP–/– tumors and measured the ability of MTA to preferentially rescue MTAP+/+ cells and tissues from L-alanosine toxicity.
2. Materials and Methods
2.1. Cell Lines
MiaPaCa2, CEM-CCRF and MOLT4 cells were obtained from American Tissue Type Collection and maintained under the recommended culture conditions. Isogenic MTAP+/+ and MTAP–/– HCT116 cells were obtained from the Vogelstein Lab [13]. MTAP expression was restored in MiaPaCa2 cells by retroviral infection of virus generated by pLPC-MTAP transiently transfected in Phoenix A cells [14]. MTAP protein expression was confirmed by Western Blot analysis. Cells were lysed in RIPA lysis buffer (25 mM Tris-HCL, pH 7.6, 150 mM NaCl, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, and 1 X Complete Protease inhibitors (Roche)). Protein concentrations were determined by BCA and 30 ug of protein were loaded on 4% - 12% Bis Tris gels (NuPage Novex), transferred on to PVDF membranes. Blots were probed with rabbit polyclonal anti-MTAP antibodies (0.33 μg/ml) purified from sera of rabbits immunized with full-length MTAP protein and anti-actin monoclonal antibodies (Sigma).
2.2. Viability and Co-Culture Assays
For viability assays, cells were cultured in their normal media (MiaPaCa2 in DMEM CEM-CCRF and HCT116 in RPMI-1640) supplemented with 2 mM L-glutamine and horse serum in place of FBS. Horse serum was used in place of FBS to avoid potential MTAP activity and high levels of hypoxanthine frequently detected in FBS. Five thousand cells/well were seeded in 96 well plates. Cells were incubated for 72 hours with 0 - 10 μM methotrexate (MTX) (Sigma M8407) and 20 μM thymidine (Thy) (Sigma T1895), 0 - 200 μM D, L-Alanosine (Toronto Research Chemicals A510800), or 0 - 100 μM 6- Mercaptopurine (6-MP) (Sigma 852678). In some cases 20 μM MTA (Sigma D5011) or Adenine (Sigma A8751) were added to the wells. Cells were assessed for viability using Cell Titer Glo (Promega G7572) according to the manufacturer’s recommendations. Cell Titer Glo viability results were confirmed in duplicate experiments using CyQuant (Invitrogen C35011) to quantify live cell nuclei (data not shown). Percent of control (POC) was calculated by dividing each sample by the mean of the vehicle controls and the standard deviation (SD) calculated from triplicate samples. To compare the effects of serum from various species, isogenic MiaPaCa2 cells were cultured for 72 hours in media supplemented with 10% human, mouse, horse or fetal bovine serum and treated with 50 μM alanosine and 0 - 200 uM MTA. Viability was measured using Cell Titer Glo.
Co-culture experiments were performed in 96-well multiwell insert plates (Becton Dickinson 353938). Five thousand MTAP+/+ or MTAP–/– MiaPaCa2 cells were seeded in triplicate in the bottom and top chambers in media containing either vehicle or 125 nM MTX and 20 μM Thy. In some cases 20 μM MTA was added to the top and bottom chambers. After 72 hours incubation at 37˚C and 5% CO2, cell viability in the bottom or top chambers was measured by Cell Titer Glo. POC was calculated as described above. To test the effects of purines present in whole blood on sensitivity to purine synthesis inhibitors, 5 × 103 MTAP+/+ or MTAP–/– MiaPaCa2 cells were seeded in triplicate in the bottom chamber of multiwell insert plates in media containing vehicle, 125 nM MTX and 20 μM Thy, or 50 μM Lalanosine. 37.5 μl of freshly isolated whole blood in acid citrate dextrose (ACD) was plated in the top chamber in media containing the same treatments as the bottom chambers. After 48 hours incubation at 37˚C and 5% CO2 viability of cells in the bottom chambers was measured by Cell Titer Glo. All p-values were calculated using GraphPad Prism 5.0 1-way analysis of variance (ANOVA).
2.3. In Vivo Studies
Female NCR1-nude mice (model# NCRnu, Taconic Farms) were used for all in vivo studies. To test the effects of L-alanosine on bone marrow proliferation, mice were dosed intraperitonealy (i.p.) with 250 or 500 mg/kg L-alanosine and mice were sacrificed and femoral bone marrow was harvested at 6, 24 or 48 hours. Mice were dosed i.p. with 50 mg/kg BrdU 4 hours prior to sacrifice to label DNA in proliferating tissues. To establish the dose of MTA and adenine that could protect bone marrow from L-alanosine-induced toxicity, mice (n = 4 - 8 per group) were dosed i.p. with 500 mg/kg L-alanosine and a dose range of 50 - 200 mg/kg MTA or adenine. Four hours prior to sacrifice mice were given 50 mg/kg BrdU i.p. Six hours post-L-alanosine treatment mice were sacrificed and femoral bone marrow was collected for FACS analysis. To test the efficacy of L-alanosine in combination with MTA or adenine in CEM xenografts, mice were inoculated subcutaneously (s.c.) with CEM tumor cells (2 million cells per mouse). When tumors reached a volume of 200 mm3 mice were randomly assigned to groups (n = 8 per group). All mice were dosed i.p. with vehicle or 500 mg/kg L-alanosine. Some groups were also dosed i.p. with either 200 mg/kg MTA or Adenine 1 hr before (T – 1) or 1 hr after (T + 1) receiving Lalansoine. All groups were dosed i.p. with 50 mg/kg BrdU two hours after receiving L-alanosine and mice were sacrificed four hours later and single cell suspensions of femoral bone marrow (RBC Lysis Buffer Roche 11814389001) and CEM tumors (Current Protocols in Cytometry 5.2.4) were prepared. P-values were calculated using GraphPad Prism 5.0 1-way analysis of variance (ANOVA).
2.4. BrdU Incorporation by Flow Cytometry
Bone marrow and tumor cells were fixed, permeabilized and processed for FACS analysis of BrdU incorporation using the APC BrdU Flow Kit from BD Pharmingen (559619) according the manufacturer’s instructions. CEM tumors were also stained with FITC-anti-human HLAABC (BD Pharmingen 555552) to distinguish human cells from mouse cells. FACS data were collected on BD Biosciences LSRII flow cytometer using FACS-Diva software and analyzed with FCS Express (De Novo Software). Percent BrdU incorporation is calculated as the mean and SD from 4 - 8 samples per group. P-values were calculated using GraphPad Prism 5.0 1-way analysis of variance (ANOVA).
2.5. Quantitative Analysis of MTA and Endogenous Adenine
Mice were dosed i.p. with 150 - 200 mg/kg MTA. Some mice were also treated with 1.5 U/g bovine xanthine oxidase (XO) (Sigma) intravenously (i.v.) Plasma was collected at various time points from 5 minutes to 6 hours (n = 3). Quantitative analysis of MTA and endogenous adenine levels in plasma was performed on an API 4000 Q-trap mass spectrometer (Applied Biosystems, Foster City, CA) coupled with an Agilent 12200 binary pump (Agilent Technologies, Palo Alto, CA) and a CTC HTS PAL autosampler (CTC Analytics, Switzerland). LC separation was achieved using a Shiseido Capcell PAK AQ C-18 column with a binary gradient of 5 mM ammonium acetate aqueous solution and methanol, over a total LC run time of 10 minutes. Negative ionization mode was used, and selective reaction monitoring was applied for quantification of adenine, MTA, and internal standard (hypoxanthine) with following transitions, respectively: 133.98 to 106.88, 296.0 to 133.80, and 139.88 to 96.0.
The LC/Rad system consisted of an Agilent 1100 series binary pump, an autosampler, a Diode-Array detector, and a beta-RAM radioactive detector (IN/US Systems, Inc.) LC separation was achieved using a Shiseido Capcell PAK AQ C-18 column with a binary gradient of 40 mM Ammonium Acetate aqueous solution (pH 5.5) and methaolo/acetonitrile (80/20), with a total LC run time of 60 minutes.
3. Results
3.1. MTA Preferentially Rescues MTAP+/+ Cell Lines from Purine Synthesis Inhibition in Vitro
Previous studies demonstrated that MTAP deficient cells are more sensitive than cells expressing wildtype MTAP to purine synthesis inhibition induced by Methotrexate (MTX) or L-alanosine treatment. These studies used either isogenic cell line pairs generated by restoring MTAP activity in cell lines with homozygous deletion [11] , or non-isogenic MTAP+/+ or MTAP–/– T-ALL cells [7] . To test the relative sensitivity of MTAP+/+ and MTAP–/– cell lines to purine synthesis inhibition we used three cell systems. We generated isogenic cell line pairs by restoring MTAP expression in the pancreatic adenocarcinoma cell line, MiaPaCa2 which harbors a homozygous deletion in MTAP [11] . In addition, we used isogenic cells generated by deleting endogenous MTAP by homologous recombination in colon adenocarcinoma cell line HCT116 [13]. Finally, we compared the MTAP deficient T-ALL cell line CEM-CCRF to the MTAP positive TALL cell line MOLT4. MTAP protein expression levels were quantified in the various cell lines by Western blot analysis with a polyclonal MTAP antibody (Figure 1(b)). Expression of functional MTAP was confirmed testing conversion of 14C-MTA to adenine (data not shown).
We tested the effects of three different purine synthesis inhibitors with varying mechanisms of action (Figure 1(a)). L-alanosine inhibits adenylosuccinate synthase (ADSS) and adenylosuccinate lyase (ADSL) which catalyze the final two steps in the conversion of IMP to AMP (Figure 2(a)) [15,16] . MTX inhibits de novo purine synthesis at multiple steps in the purine biosynthesis pathway by pre-

Figure 1(a). De novo purine synthesis and adenine salvage are the two main pathways used to generate AMP in mammalian cells. L-alanosine inhibits ADSS and Methotrexate (MTX) inhibits the production of tetrahydrofolate, a cofactor required for the enzyme 5-phosphoribosylglycinamide formyltransferase (GARFT). Hypoxanthine supplements de novo purine synthesis via conversion to inosine monophosphate (IMP) via hypoxanthine phosphoribosyltransferase (HPRT).

Figure 1(b). MTAP protein expression in isogenic cell line pairs. Western blots were probed with anti-MTAP or anti-actin antibodies as described in “Materials and Methods”.
venting the production of folate, a required co-factor for several enzymes in the pathway [17]. Since MTX also inhibits thymidylate synthase, 20 μM exogenous thymidine (Thy) was added to MTX to prevent effects on thymidine synthesis and ensure specific inhibition of purine synthesis (Figure 2(b)). Finally, we used 6-mercaptopurine (6-MP) which is converted by HGPRT to a toxic metabolite which interferes with de novo purine
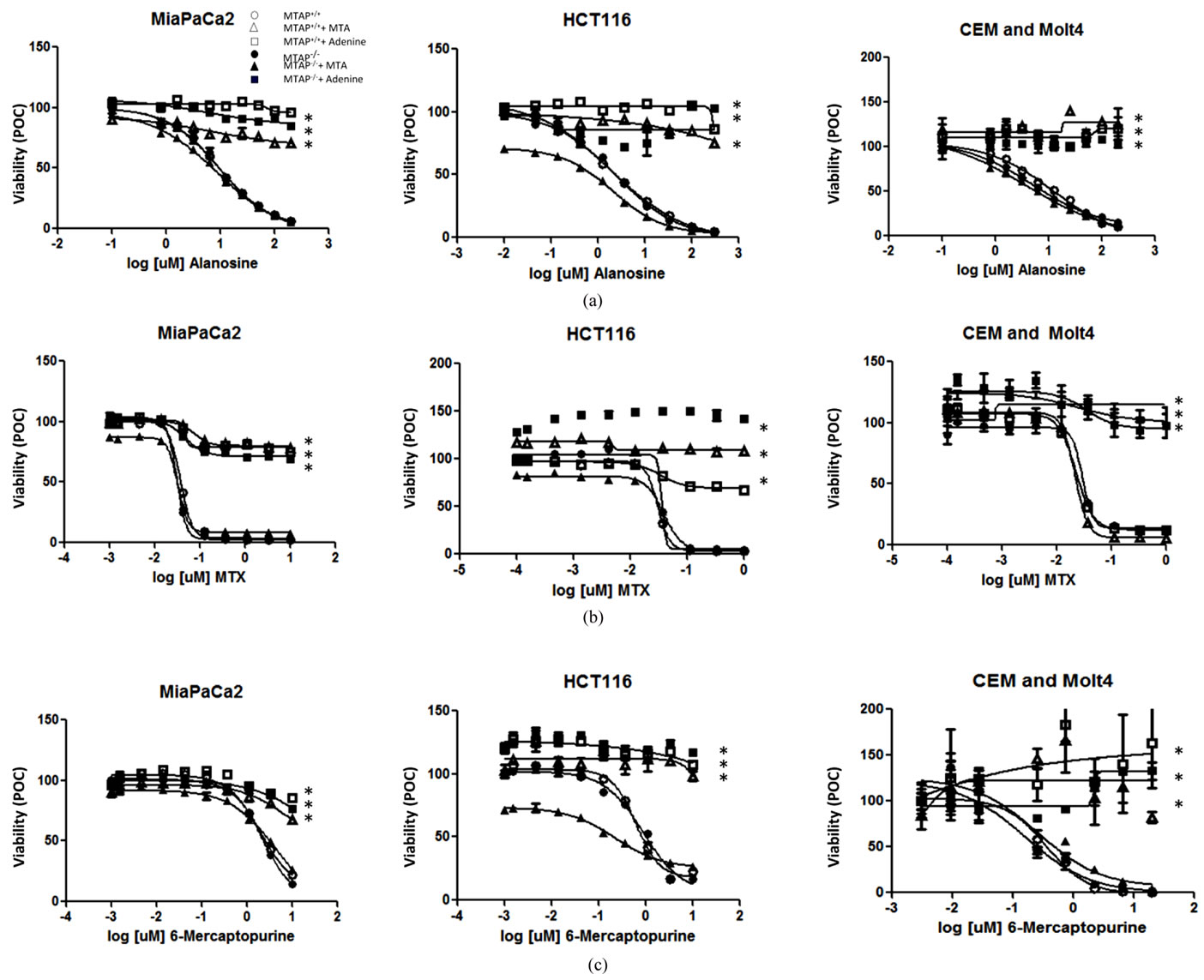
Figure 2. MTA preferentially rescues MTAP+/+ cell lines from purine synthesis inhibitors. (a) Viability of isogenic MTAP+/+ and MTAP–/– MiaPaCa2 , HCT116 cells and CEM and Molt cells after treatment with increasing doses of L-alanosine with or without MTA or Adenine rescue. (b) Same as (a), but cells were treated with increasing doses of Methotrexate. (c) Same as (a) but cells were treated with increasing doses of 6-mercaptopurine. Data is represented as the mean of triplicate samples +/– SD. Each graph is representative of at least duplicate experiments. Symbols: MTAP–/– ˜, MTAP+/+ ™, MTAP–/– + Adenine ¢, MTAP+/+ + Adenine £, MTAP–/– + MTA p, MTAP+/+ + MTA r. * denotes p < 0.001 compared to purine synthesis inhibitor alone.
synthesis (Figure 2(c)) [18]. The doses of thymidine, MTA, adenine and hypoxanthine used for rescue were determined empirically through titration studies (data not shown). In contrast to previous studies [11,19] , there were no apparent differences in IC50 of these drugs comparing MTAP+/+ to MTAP–/– cells of the same tissue type, although addition of MTA completely protected MTAP+/+ cells from toxicity induced by L-alanosine, MTX and 6- MP. Interestingly, MTAP–/– HCT116 were particularly sensitive to toxicity induced by MTA alone, compared to the parental MTAP+/+ HCT116 cells (Figures 2(a)-(c), middle panels). Addition of adenine protected both MTAP+/+ and MTAP–/– cells from purine synthesis inhibitor toxicity.
We also tested the ability of other purines to rescue cells from purine synthesis inhibitor toxicity. Addition of hypoxanthine protected both genotypes from MTX + Thyand 6-MP-induced toxicity, but not from L-alanosine-induced toxicity, consistent with the fact that Lalanosine inhibits ADSS, an enzyme required for both de novo purine synthesis and salvage of hypoxanthine [16] (data not shown). S-adenosylmethionine (SAM), the precursor to MTA, and adenosine were also tested as rescue reagents. SAM protected both genotypes from MTX + Thy, L-alanosine and 6-MP, whereas adenosine was not effective in protecting either genotype from purine synthesis inhibitors(data not shown).
3.2. Purines Are Salvaged from Whole Blood and Rescue MTAP+/+ and MTAP–/– Cells from MTX+Thy, but Not L-Alanosine Induced Toxicity
Since MTAP+/+ cells were able to use MTA as a source of adenine, we tested if MTAP+/+ cells could provide protection to MTAP–/– cells by metabolizing MTA intracellularly and releasing it to surrounding media. MTAP+/+ and MTAP–/– MiaPaCa2 cells were co-cultured in chambers separated by a permeable membrane in the presence or absence of MTX+Thy and MTA. As expected, MTAP+/+ cells were completely rescued by the addition of MTA, regardless of whether they were cocultured with MTAP+/+ or MTAP–/– cells (Figure 3(a)). MTAP–/– cells, however, were not rescued by the addition of MTA regardless of the genotype of cells in coculture, suggesting that MTAP+/+ cells do not release adenine or other purines capable of rescue into the media, even in the presence of excess MTA (Figure 3(a)).
To test the influence of purines and metabolites in blood on purine synthesis inhibition and resulting toxicity in vitro, co-culture systems were established using mouse whole blood. MTAP–/– or MTAP+/+ MiaPaCa2 cells were co-cultured with freshly isolated mouse whole blood with or without MTX + Thy or L-alanosine. In some conditions, MTA was added to the culture media. Addition of MTA to co-culture with mouse whole blood rescued both genotypes from MTX + Thy (Figure 3(b)), but not L-alanosine induced toxicity (Figure 3(c)). Similar results were obtained with human whole blood (data not shown). Red blood cells lack ADSS and therefore cannot utilize hypoxanthine as a salvage source for purines; hence, hypoxanthine is released from red blood cells as a waste product [4]. Since L-alanosine toxicity cannot be overcome by hypoxanthine, we hypothesized that hypoxanthine was the purine released from cells in whole blood, thereby protecting MTAP+/+ and MTAP–/– cells from MTX + Thy-induced toxicity.
3.3. MTA and Adenine Rescue Mice from Bone-Marrow Toxicity after Treatment with L-Alanosine in Vivo
Our co-culture experiments with whole blood indicated that MTAP–/– cells could be rescued from MTX+Thy induced toxicity with the addition of MTA so we chose to use L-alanosine in our in vivo proof of concept studies to avoid potential cross-feeding and rescue of MTAP–/– tumors. The dose limiting toxicities of L-alanosine in
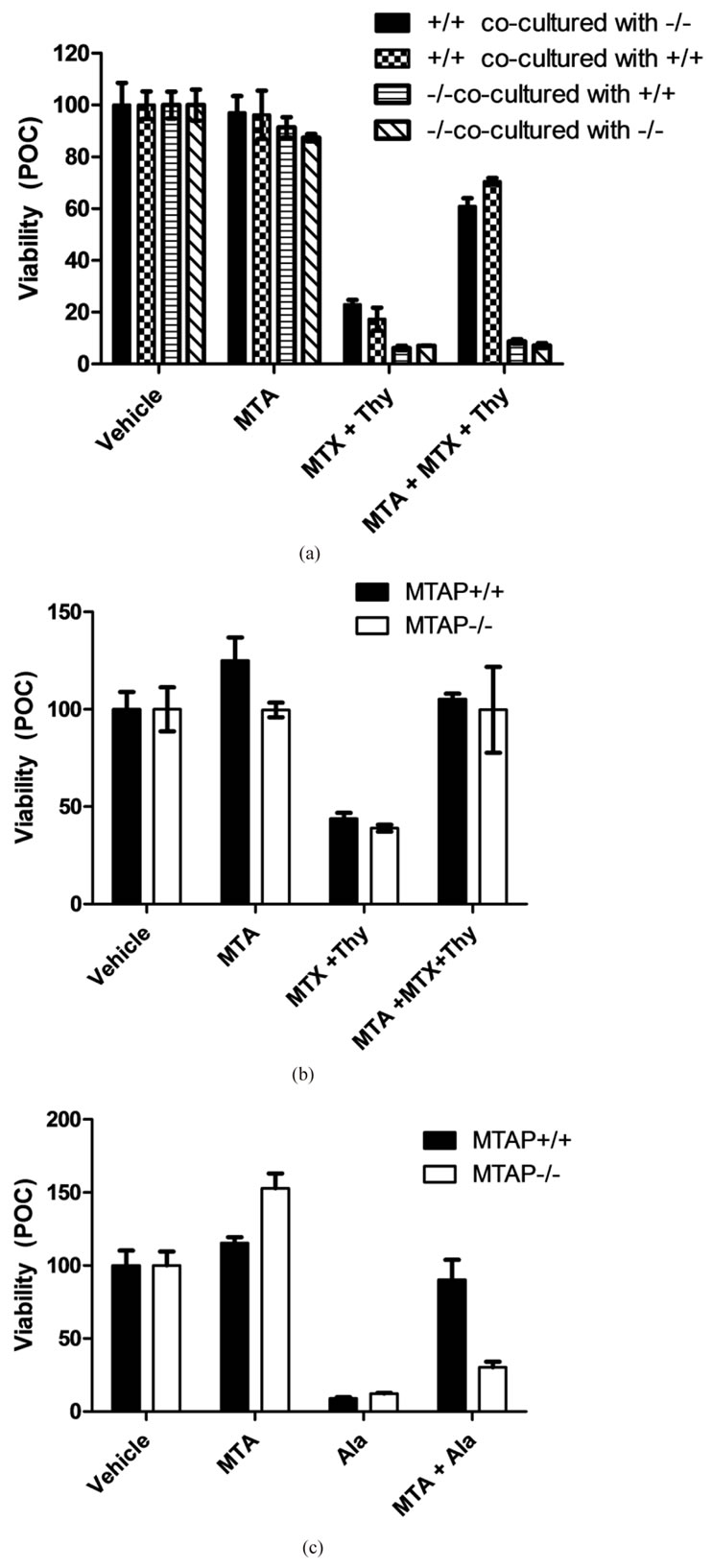
Figure 3. Purines from whole blood rescue MTAP–/– cells from MTX+Thy, but not L-alanosine-induced toxicity. (a) Viability of MTAP+/+ and MTAP–/– cells co-cultured and treated with MTX+Thy with or without MTA rescue. (b) Viability of MTAP+/+ and MTAP–/– cells co-cultured with mouse whole blood and treated with MTX+Thy or (c) Lalanosine with or without MTA rescue. Data is represented as the mean of triplicate samples +/– SD. Each graph is representative of at least duplicate experiments. * denotes p < 0.001 compared to vehicle alone; † denotes p < 0.001 compared to MTX+Thy treatment; ‡ denotes p < 0.001 compared to Ala treatment.
clinical trials are reported to be myelosuppression and mucositis [20,21] . Therefore, as a biomarker of toxicity and measure of proliferation, we measured the effects of L-alanosine on bone marrow proliferation in vivo using BrdU incorporation. BrdU is incorporated into DNA during S-phase. Therefore, the percentage of cells stained with BrdU is equal to the percentage cells which have moved through S-phase and duplicated their DNA during the 4 hour pulse with BrdU.
Mice were given a single i.p. dose of vehicle control or 250 mg/kg or 500 mg/kg L-alanosine and proliferation of bone marrow was measured 6, 24 and 48 hours later. Both doses of L-alanosine completely repressed bone marrow proliferation at 6 and 24 hours, but by 48 hours the mice given 250 mg/kg began to recover (Figure 4(a)). To test the ability of MTA and adenine to restore bone marrow proliferation, mice were given a single i.p. dose of 500 mg/kg L-alanosine, with or without a dose range of adenine or MTA, and bone marrow proliferation was measured 6 hours later. Both MTA and adenine restored proliferation in a dose-dependent manner with 200 mg/kg completely restoring proliferation (Figure 4(b)). It should be noted, however, that mice dosed with >100 mg/kg MTA showed signs of toxicity, such as decreased blood pressure and body temperature within minutes of receiving MTA, but recovered after 30 - 60 minutes, consistent with previous studies [22,23] .
3.4. MTA Rescues MTAP–/– CEM Tumors from L-Alanosine-Induced Toxicity in Vivo
We next determined if normal tissues could be protected from L-alanosine toxicity with MTA while still inhibiting MTAP–/– tumor proliferation. We transplanted MTAP–/– CEM tumors subcutaneously in nude mice and allowed tumors to grow to 200 mm3. Mice were then treated with a single dose of 500 mg/kg L-alanosine with or without 200 mg/kg MTA or adenine. MTA and adenine were given either 1 hour before or 1 hour after dosing with Lalanosine. Proliferation of CEM tumors and bone marrow were measured via BrdU incorporation. L-alanosine significantly inhibited proliferation of bone marrow and CEM tumors, though the effects on bone marrow were much more pronounced than on the tumors (Figures 5(a)-(b)). Bone marrow continued to proliferate in mice given either MTA or adenine within 1 hour of dosing L-alanosine (Figure 5(a)). To our surprise, proliferation of MTAP–/– CEM cells was also restored with the addition of either rescue reagent within one hour of Lalanosine treatment (Figure 5(b)).
3.5. Pharmacokinetic Analysis of MTA and Adenine in Nude Mice
To understand why MTA protected MTAP–/– tumors in vivo, we measured the pharmacokinetics of MTA and adenine in plasma over 6 hours after a single i.p. dose of 200 mg/kg MTA. Within five minutes of dosing MTA, adenine appeared in the plasma and displayed a similar pharmacokinetic profile as MTA (Figure 5(c)). Therefore, in contrast to our co-culture studies using isogenic cell lines and whole blood (Figures 3(b)-(c)), MTA was rapidly converted to adenine and was present in mouse plasma in vivo at concentrations high enough (>20 μM) to rescue MTAP–/– CEM cells.
Previous studies have reported the presence of active MTAP in mouse, human and bovine serum and it remained a possibility that MTAP was present and active in mouse serum, causing the conversion of MTA to adenine within the serum [24]. To test this possibility, the activity of MTAP in the serum from various species was measured using two methods. First, we incubated 14C labeled MTA for 0.5 - 24 hours in the presence of fetal bovine serum, mouse serum, human serum and horse serum and analyzed the degradation products by LC with radiometric detection. Incubation of 14C MTA in FBS resulted in the appearance of several degradation products including adenine (Figure 6(a)). In contrast, there was no degradation of MTA when incubated in either horse serum, or freshly isolated mouse or human serum (Figure 6(a)). Second, we tested the ability of MTA to rescue MTAP–/– and MTAP+/+ cells from L-alanosine in vitro in media supplemented with 10% bovine, mouse, horse or human serum (Figure 6(b)). MTA protected MTAP+/+ cells from L-alanosine, but did not protect MTAP–/– cells from toxicity under any media conditions, except for those containing 10% FBS (Figure 6(b)). Together these results demonstrate that there is not sufficient active MTAP in human, mouse or horse serum to convert MTA to adenine extracellularly. Therefore, the adenine observed in mouse plasma likely results from intracellular conversion of MTA to adenine and subsequent transport to the plasma and surrounding tissues. These data further suggested that the rescue of MTAP–/– tumor in vivo was due to the accumulation of excess adenine in plasma.
3.6. Elimination of Plasma Adenine with XOD does Not Prevent Rescue of MTAP–/– Tumors with MTA
Xanthine oxidase (XO) degrades hypoxanthine to uric acid and adenine to 2, 8-dihydroxyadenine, neither of which can be salvaged to purine nucleotides [4]. We hypothesized that treatment with XO in vivo would eliminate excess plasma adenine and cross-feeding of MTAP–/– tumors. We analyzed the pharmacokinetics of MTA and adenine in mouse plasma over six hours after a single dose of either 150 mg/kg MTA with 1.5 U/g XO i.v.
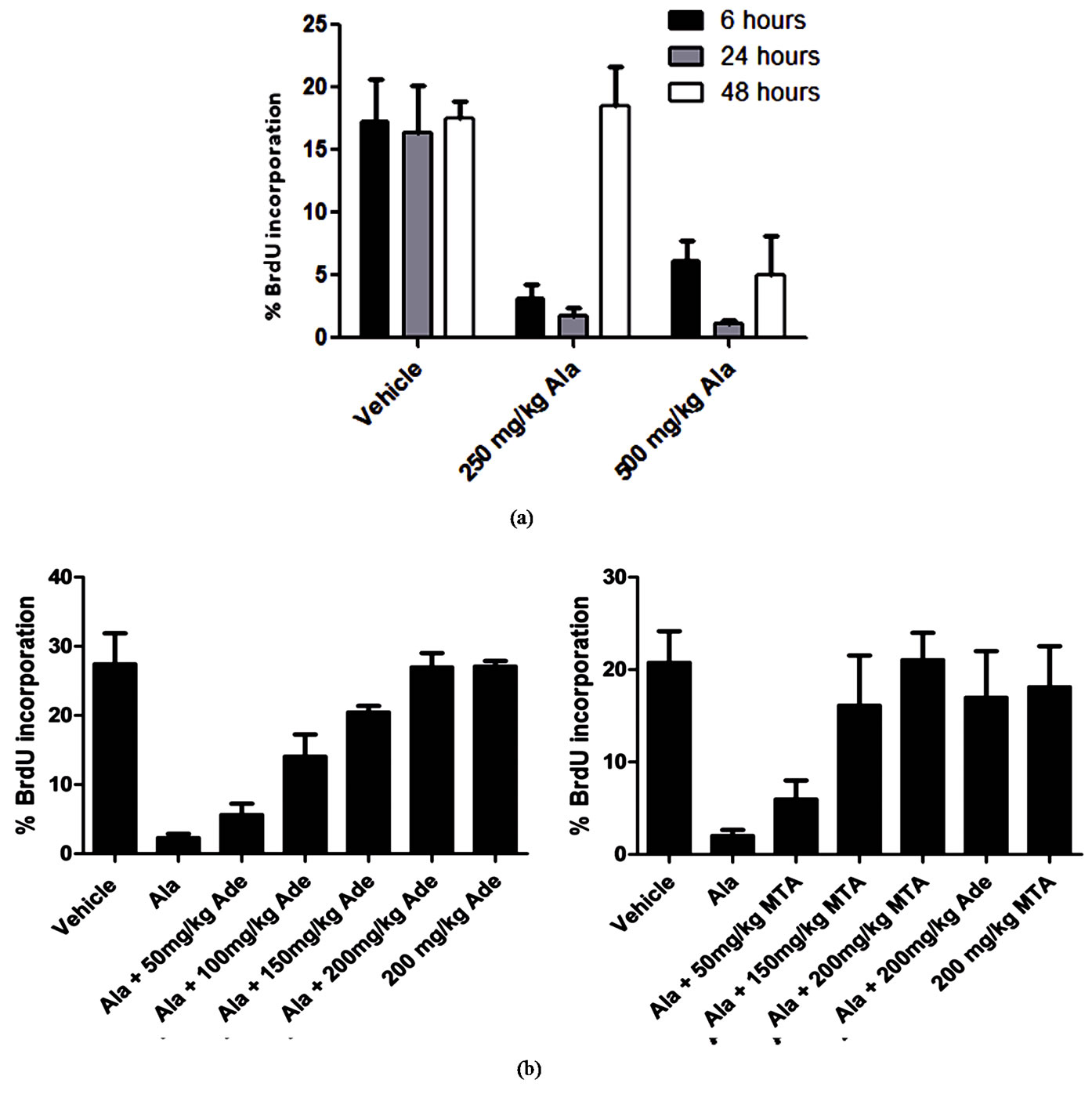
Figure 4. MTA and adenine protect bone marrow from L-alanosine-induced toxicity in vivo. FACS analysis of BrdU incorporation in bone marrow from mice (a), treated with L-alanosine (n = 4 mice per group) (b), treated with L-alanosine and the indicated doses of adenine (left) or MTA (right) (n = 4 - 8 mice per group; * denotes p < 0.001 compared to vehicle alone). Data are calculated as described in “Materials and Methods” and are representative of at least two independent experiments.
Treatment with XO reduced plasma adenine to less than 1 mM at the peak concentration but had no effect on plasma MTA levels (Figure 7(a)).
Next we tested the ability of XO to prevent the rescue of MTAP–/– CEM tumors from L-alanosine-induced toxicity. Although XO significantly reduced plasma adenine, proliferation of both bone marrow (Figure 7(b)) and tumors (Figure 7(c)) were restored in mice treated with the combination of XO, MTA and L-alanosine. These data suggest that MTAP–/– tumors can obtain purines from adjacent tissues to circumvent purine synthesis inhibition and maintain proliferation, even when plasma adenine is reduced to levels below those required for rescue in vitro.
4. Conclusions
Our data are the first to demonstrate that MTAP–/– tumors can obtain purines from plasma and surrounding tissues in vivo and imply that inhibition of purine synthesis in combination with rescue therapies, such as MTA, or EFA would not be an effective therapeutic strategy for tumors harboring MTAP deletions. We observed no differential sensitivity to purine synthesis inhibitors based on MTAP status in vitro, unless exogenous MTA was supplied. In vitro co-culture experiments demonstrated
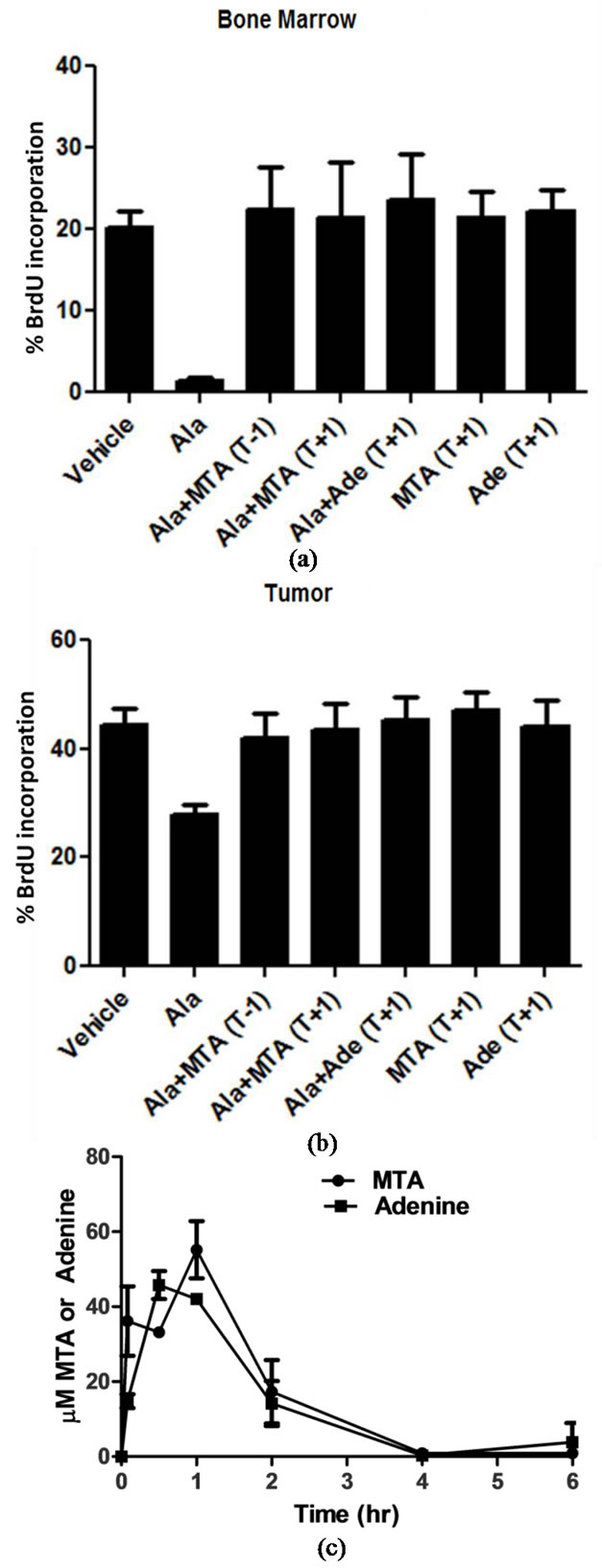
Figure 5. MTA and adenine protect MTAP–/– CEM tumors from L-alanosine-induced toxicity in vivo. (a) FACS analysis of BrdU incorporation in bone marrow from CEM-tumor bearing mice treated with L-alanosine (n = 8 per group). 200 mg/kg MTA or adenine were given either 1 hour prior to L-alanosine (T – 1) or 1 hour post-L-alanosine (T + 1). B, FACS analysis of BrdU incorporation in CEMtumors collected from the same animals described in (a) (* denotes p < 0.05). (c), Plasma MTA and adenine concentrations in mice treated with MTA for the indicated times (n = 3 per group). Data are representative of at least two independent experiments.
that adenine was not released from MTAP+/+ cells or whole blood in the presence of exogenous MTA. In vivo, MTAP–/– tumors were actually less sensitive to Lalanosine treatment than MTAP+/+ bone marrow. In contrast to our co-culture experiments, treatment with MTA caused a rapid increase in plasma adenine and effectively protected bone marrow and MTAP–/– tumors from the anti-proliferative effects of L-alanosine. Treatment with xanthine oxidase significantly reduced plasma adenine, however MTA still rescued proliferation of MTAP–/– tumors.
These results suggest that in vivo metabolism and transport of purines are more complicated than first proposed and support several conclusions. First, endogenous intracellular stores of MTA and salvaged adenine are not sufficient to substitute for de novo synthesis of AMP and support cell growth and survival in MTAP+/+ cells. Therefore, no therapeutic window between normal tissues and MTAP–/– tumors exists without supportive care measures under these conditions. This conclusion is further supported by recent clinical trials with L-alanosine targeting MTAP–/– tumors which failed to achieve significant clinical responses when dosed to MTD [21]. Second, excess intracellular adenine produced by MTA conversion is not retained in MTAP+/+ cells as evidenced by the rapid increase in plasma adenine observed after dosing MTA in vivo. Third, MTAP–/– tumors are adept at salvaging purines from plasma and surrounding tissues to circumvent a block in de novo purine synthesis. Even when plasma adenine levels were drastically reduced by XOD, MTAP–/– tumors were rescued from the anti-proliferative effects of L-alanosine by MTA. This suggests that purines, and perhaps even ADP can pass directly from MTAP+/+ stroma to MTAP–/– tumor to provide protection. Finally, in vitro cell-based systems do not adequately model or predict in vivo metabolism or transport of MTA.
Our data are in contrast to previously published evidence that MTAP–/– primary T-ALL cells from patients are more sensitive to L-alanosine than MTAP+/+ primary T-ALL cells in vitro [10] . L-alanosine is a pro-drug that must be converted to its active antimetabolite, L-alanosyl-5-amino-4-imidazolecarboxylic acid ribonucleotide (alanosyl-AICOR), by 5-aminoimidozaole-4-(N-succinylocarboxamide) ribotide (SACAIR) synthase, and is further metabolized and inactivated by ADSL. Thus, the increased sensitivity of MTAP–/– primary patient samples to L-alanosine may not be due to deletion of MTAP, but rather to alteration of other enzymes involved in the activation and catabolism of L-alanosine. Indeed, the activity and expression levels of these enzymes have been shown to mediate the toxicity of L-alanosine in vivo [25].
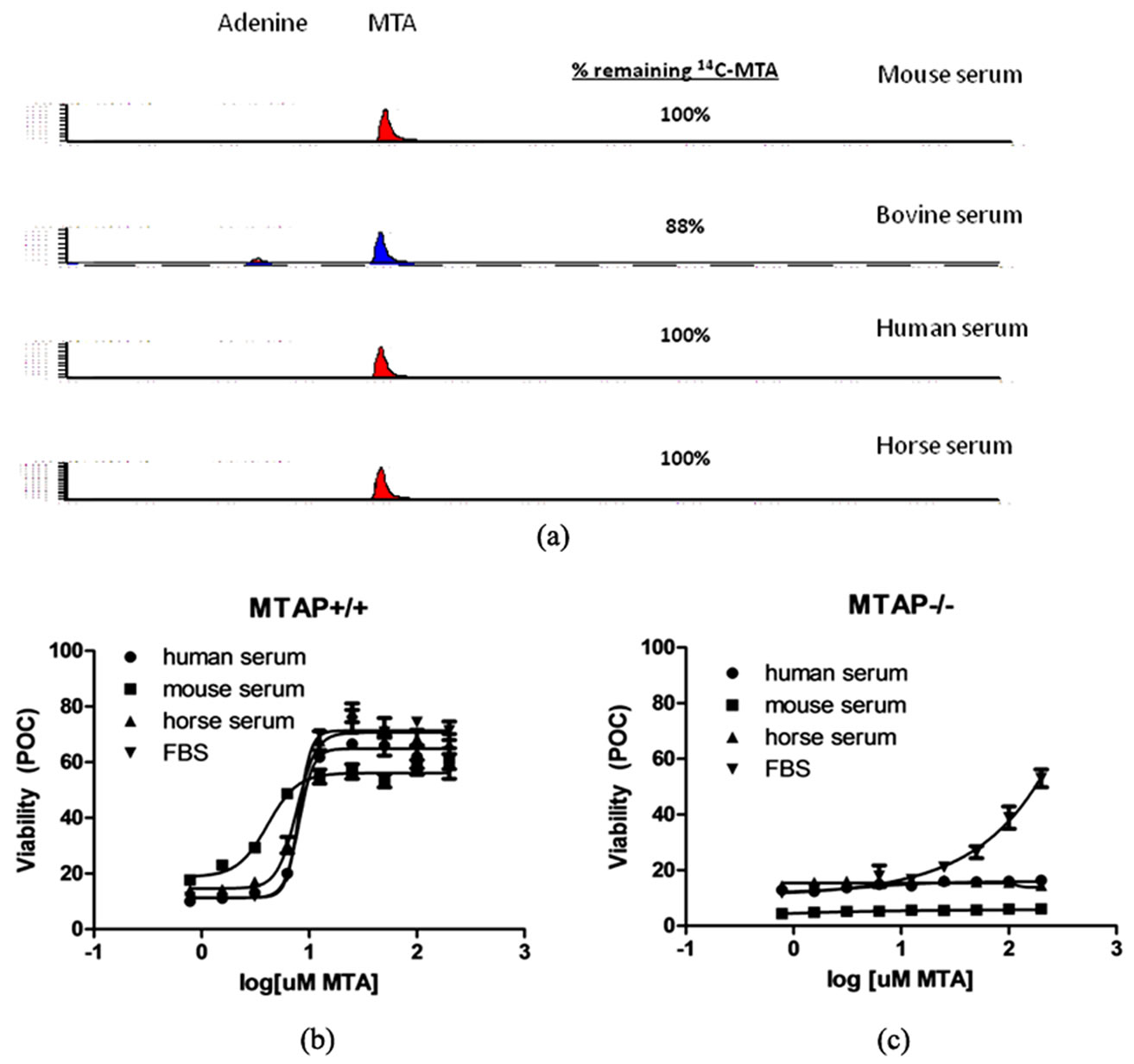
Figure 6. FBS, but not human, mouse or horse serum contains active MTAP. (a) LC/RAD traces of MTA and metabolites in serum incubated with 14CMTA. B, ATP content of MTAP+/+ and MTAP–/– MiaPaCa2 cells cultured in the indicated sera and 50 μM L-alanosine and increasing doses of MTA. Data are calculated as the mean and SD of triplicate samples and are representative of at least two independent experiments. * denotes p < 0.001.
Previous publications have demonstrated that bone marrow stem cells are highly sensitive to MTA alone when cultured ex vivo in colony formation assays [26]. In contrast to these studies, we found no adverse effects on bone marrow proliferation in vivo. However, at concentrations sufficient to protect bone marrow from toxicity induced by inhibition of de novo purine synthesis, MTA induced physiological signs of toxicity potentially associated with adenosine receptor agonism, such as depressed blood pressure and body temperature. Although the mice recovered from these physiological effects within 30 minutes, twice daily doses (BID) of MTA of 100 mg/kg or greater were lethal. For this reason, we limited our studies to short-term single-dose studies. The observed side-effects and toxicity could make the strategy of dosing MTA as a rescue reagent therapeutically challenging. Alternative MTAP substrates, such as EFA have been tested in vitro, but whether these have the same toxicities as MTA in vivo is yet to be studied.
MTAP activity impinges on three metabolic pathways: adenine salvage, methionine recycling and polyamine synthesis. Although our studies suggest that targeting MTAP-deficient tumors by inhibiting de novo purine synthesis in combination with MTA rescue will not be a viable approach, it remains possible that MTAP-deleted tumors may respond to combinations of inhibitors of polyamine synthesis or restriction of methionine. Finally, these studies were limited to evaluation of L-alanosine and rescue with MTA in nude mice. However it remains to be determined if there are differences in the transport and metabolism of purines in humans which might allow for preferential rescue of MTAP+/+ tissues over MTAP–/– tumors.

Figure 7. Xanthine oxidase eliminates plasma adenine, but does not abrogate rescue of MTAP–/– CEM tumors with MTA. (a) Plasma MTA and adenine concentrations in mice treated with MTA and XO for the indicated times (n = 3 per group). FACS analysis of BrdU incorporation in (b), bone marrow and (c), tumors from mice treated with L-alanosine with or without MTA, adenine or XO (n = 8 per group). Data are calculated as described in “Materials and Methods” and are representative of at least two independent experiments.
REFERENCES
- L. Christa, L. Thuillier, A. Munier and J. L. Perignon, “Salvage of 5’-Deoxy-methylthioadenosine into Purines and Methionine by Lymphoid Cells and Inhibition of Cell Proliferation,” Biochimica et Biophysica Acta, Vol. 803, No. 1-2, 1984, pp. 7-10. doi:10.1016/0167-4889(84)90048-X
- A. E. Pegg and H. G. Williams-Ashman, “PhosphateStimulated Breakdown of 5’-Methylthioadenosine by Rat Ventral Prostate,” Biochemical Journal, Vol. 115, 1969, pp. 241-247.
- P. S. Backlund Jr. and R. A. Smith, “Methionine Synthesis from 5’-Methylthioadenosine in Rat Liver,” Journal of Biological Chemistry, Vol. 256, 1981, pp. 1533-1535.
- T. W. Stone and H. A. Simmonds, “Purines: Basic and Clinical Aspects,” Kluwer Academic Publishers, Dordrecht, 1991. doi:10.1007/978-94-011-3911-3
- N. Kamatani and D. A. Carson, “Dependence of Adenine Production upon Polyamine Synthesis in Cultured Human Lymphoblasts,” Biochim Biophys Acta, Vol. 675, 1981, pp. 344-350.
- A. L. Subhi, P. Diegelman, C. W. Porter, B. Tang, Z. J. Lu, G. D. Markham and W. D. Kruger, “Methylthioadenosine Phosphorylase Regulates Ornithine Decarboxylase by Production of Downstream Metabolites,” Journal of Biological Chemistry, Vol. 278, 2003, pp. 49868-49873. doi:10.1074/jbc.M308451200
- A. Batova, M. B. Diccianni, T. Nobori, T. Vu, J. Yu, L. Bridgeman and A. L. Yu, “Frequent Deletion in the Methylthioadenosine Phosphorylase Gene in T-Cell Acute Lymphoblastic Leukemia: Strategies for Enzyme-Targeted Therapy,” Blood, Vol. 88, 1996, pp. 3083-3090.
- T. Nobori, J. G. Karras, F. Della Ragione, T. A. Waltz, P. P. Chen and D. A. Carson, “Absence of Methylthioadenosine Phosphorylase in Human Gliomas,” Cancer Research, Vol. 51, 1991, pp. 3193-3197.
- T. Nobori, K. Takabayashi, P. Tran, L. Orvis, A. Batova, A. L. Yu and D. A. Carson, “Genomic Cloning of Methylthioadenosine Phosphorylase: A Purine Metabolic Enzyme Deficient in Multiple Different Cancers,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 93, No. 12, 1996, pp. 6203-6208. doi:10.1073/pnas.93.12.6203
- A. Batova, M. B. Diccianni, M. Omura-Minamisawa, J. Yu, C. J. Carrera, L. J. Bridgeman, F. H. Kung, J. Pullen, M. D. Amylon and A. L. Yu, “Use of Alanosine as a Methylthioadenosine Phosphorylase-Selective Therapy for T-Cell Acute Lymphoblastic Leukemia in Vitro,” Cancer Research, Vol. 59, 1999, pp. 1492-1497.
- Z. H. Chen, O. I. Olopade and T. M. Savarese, “Expression of Methylthioadenosine Phosphorylase cDNA in p16-, MTAPMalignant Cells: Restoration of Methylthioadenosine Phosphorylase-Dependent Salvage Pathways and Alterations of Sensitivity to Inhibitors of Purine De Novo Synthesis,” Molecular Pharmacology, Vol. 52, 1997, pp. 903-911.
- A. Batova, H. Cottam, J. Yu, M. B. Diccianni, C. J. Carrera and A. L. Yu, “EFA (9-Beta-D-erythrofuranosyladenine) Is an Effective Salvage Agent for Methylthioadenosine Phosphorylase-Selective Therapy of T-Cell Acute Lymphoblastic Leukemia with L-Alanosine,” Blood, Vol. 107, No. 3, 2006, pp. 898-903. doi:10.1182/blood-2005-06-2430
- C. Rago, B. Vogelstein and F. Bunz, “Genetic Knockouts and Knockins in Human Somatic Cells,” Nature Protocols, Vol. 2, 2007, pp. 2734-2746. doi:10.1038/nprot.2007.408
- S. Swift, J. Lorens, P. Achacoso and G. P. Nolan, “Rapid Production of Retroviruses for Efficient Gene Delivery to Mammalian Cells Using 293T Cell-Based Systems,” Current Protocols in Immunology, 2001, Unit 10.17C.
- P. J. Casey and J. M. Lowenstein, “Inhibition of Adenylosuccinate Lyase by L-Alanosyl-5-aminoimidazole-4-carboxylic Acid Ribonucleotide (Alanosyl-Aicor),” Biochemical Pharmacology, Vol. 36, No. 5, 1987, pp. 705- 709. doi:10.1016/0006-2952(87)90722-2
- A. K. Tyagi and D. A. Cooney, “Identification of the Antimetabolite of L-Alanosine, L-Alanosyl-5-amino-4- imidazole-carboxylic Acid Ribonucleotide, in Tumors and Assessment of Its Inhibition of Adenylosuccinate Synthetase,” Cancer Research, Vol. 40, 1980, pp. 4390- 4397.
- C. H. Takimoto, “New Antifolates: Pharmacology and Clinical Applications,” Oncologist, Vol. 1, 1996, pp. 68- 81.
- S. Coulthard and L. Hogarth, “The Thiopurines: An Update,” Investigational New Drugs, Vol. 23, No. 6, 2005, pp. 523-532. doi:10.1007/s10637-005-4020-8
- N. Kamatani, W. A. Nelson-Rees and D. A. Carson, “Selective Killing of Human Malignant Cell Lines Deficient in Methylthioadenosine Phosphorylase, a Purine Metabolic Enzyme,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 78, No. 2, 1981, pp. 1219-1223. doi:10.1073/pnas.78.2.1219
- G. M. Dosik, D. Stewart, M. Valdivieso, M. A. Burgess and G. P. Bodey, “Phase I Study of L-Alanosine Using a Daily x 3 Schedule,” Cancer Treatment Reports, Vol. 66, 1982, pp. 73-76.
- H. L. Kindler, H. A. Burris, A. B. Sandler and I. A. Oliff, “A Phase II Multicenter Study of L-Alanosine, a Potent Inhibitor of Adenine Biosynthesis, in Patients with MTAPDeficient Cancer,” Investigational New Drugs, Vol. 27, No. 1, 2009, pp. 75-81. doi:10.1007/s10637-008-9160-1
- R. Munshi, A. S. Clanachan and H. P. Baer, “5’-Deoxy-5’-methylthioadenosine: A Nucleoside Which Differentiates between Adenosine Receptor Types,” Biochemical Pharmacology, Vol. 37, No. 10, 1988, pp. 2085- 2089. doi:10.1016/0006-2952(88)90560-6
- R. A. Olsson, E. M. Khouri, J. L. Bedynek Jr. and J. McLean, “Coronary Vasoactivity of Adenosine in the Conscious Dog,” Circulation Research, Vol. 45, 1979, pp. 468-478.
- M. K. Riscoe and A. J. Ferro, “5-Methylthioribose. Its effects and Function in Mammalian Cells,” Journal of Biological Chemistry, Vol. 259, 1984, pp. 5465-5471.
- A. K. Tyagi, D. C. Thake, E. McGee and D. A. Cooney, “Determinants of the Toxicity of L-Alanosine to Various Organs of the Mouse,” Toxicology, Vol. 21, No. 1, 1981, pp. 59-69. doi:10.1016/0300-483X(81)90016-0
- J. Yu, A. Batova, L. Shao, C. J. Carrera and A. L. Yu, “Presence of Methylthioadenosine Phosphorylase (MTAP) in Hematopoietic Stem/Progenitor Cells: Its Therapeutic Implication for MTAP (-) Malignancies,” Clinical Cancer Research, Vol. 3, 1997, pp. 433-438.