Journal of Modern Physics
Vol.08 No.04(2017), Article ID:74995,15 pages
10.4236/jmp.2017.84041
Electronic Band Structure of Graphene Based on the Rectangular 4-Atom Unit Cell
Akira Suzuki1, Masashi Tanabe1, Shigeji Fujita2
1Department of Physics, Faculty of Science, Tokyo University of Science, Tokyo, Japan
2Department of Physics, University at Buffalo, State University of New York, Buffalo, NY, USA

Copyright © 2017 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY 4.0).
http://creativecommons.org/licenses/by/4.0/



Received: February 13, 2017; Accepted: March 27, 2017; Published: March 30, 2017
ABSTRACT
The Wigner-Seitz unit cell (rhombus) for a honeycomb lattice fails to establish a 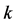 -vector in the 2D space, which is required for the Bloch electron dynamics. Phonon motion cannot be discussed in the triangular coordinates, either. In this paper, we propose a rectangular 4-atom unit cell model, which allows us to discuss the electron and phonon (wave packets) motion in the
-vector in the 2D space, which is required for the Bloch electron dynamics. Phonon motion cannot be discussed in the triangular coordinates, either. In this paper, we propose a rectangular 4-atom unit cell model, which allows us to discuss the electron and phonon (wave packets) motion in the 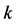 -space. The present paper discusses the band structure of graphene based on the rectangular 4-atom unit cell model to establish an appropriate
-space. The present paper discusses the band structure of graphene based on the rectangular 4-atom unit cell model to establish an appropriate 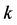 -vector
-vector 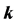 for the Bloch electron dynamics. To obtain the band energy of a Bloch electron in graphene, we extend the tight-binding calculations for the Wigner-Seitz (2- atom unit cell) model of Reich et al. (Physical Review B, 66, Article ID: 035412 (2002)) to the rectangular 4-atom unit cell model. It is shown that the graphene band structure based on the rectangular 4-atom unit cell model reveals the same band structure of the graphene based on the Wigner-Seitz 2-atom unit cell model; the
for the Bloch electron dynamics. To obtain the band energy of a Bloch electron in graphene, we extend the tight-binding calculations for the Wigner-Seitz (2- atom unit cell) model of Reich et al. (Physical Review B, 66, Article ID: 035412 (2002)) to the rectangular 4-atom unit cell model. It is shown that the graphene band structure based on the rectangular 4-atom unit cell model reveals the same band structure of the graphene based on the Wigner-Seitz 2-atom unit cell model; the 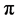 -band energy holds a linear dispersion (
-band energy holds a linear dispersion (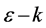 ) relations near the Fermi energy (crossing points of the valence and the conduction bands) in the first Brillouin zone of the rectangular reciprocal lattice. We then confirm the suitability of the proposed rectangular (orthogonal) unit cell model for graphene in order to establish a 2D
) relations near the Fermi energy (crossing points of the valence and the conduction bands) in the first Brillouin zone of the rectangular reciprocal lattice. We then confirm the suitability of the proposed rectangular (orthogonal) unit cell model for graphene in order to establish a 2D 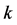 -vector responsible for the Bloch electron (wave packet) dynamics in graphene.
-vector responsible for the Bloch electron (wave packet) dynamics in graphene.
Keywords:
Graphene, Rectangular 4-Atom Unit Cell Model, Primitive Orthogonal Basis Vector, Bloch Electron (Wave Packet) Dynamics, 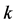 -Vector, Dirac Points, Linear Dispersion Relation
-Vector, Dirac Points, Linear Dispersion Relation

1. Introduction
The electronic band structure of graphene plays an important role for under- standing its unique properties [1] [2] [3] [4] . Graphene is a perfect two- dimensional crystal consisting of a single layer of carbon atoms arranged in a honeycomb lattice. A carbon atom contains four valence electrons, one 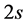 - electron, and three
- electron, and three 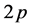 -electrons. They are
-electrons. They are 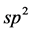 -hybridized, that is, one
-hybridized, that is, one 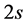 - electron and two
- electron and two 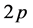 -electrons form strong
-electrons form strong 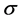 -bonds between carbon atoms leading to the honeycomb structure with the carbon-carbon distance of 0.142 nm. The remaining
-bonds between carbon atoms leading to the honeycomb structure with the carbon-carbon distance of 0.142 nm. The remaining 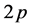 -electron occurs as a
-electron occurs as a 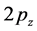 -orbital, which is oriented perpendicularly to the planar structure, and forms a
-orbital, which is oriented perpendicularly to the planar structure, and forms a 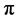 -bond with the neigh- bouring carbon atoms. The
-bond with the neigh- bouring carbon atoms. The 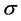 -bonds are completely filled and form a deep valence band [2] . The smallest gap between the bonding and the anti-bonding
-bonds are completely filled and form a deep valence band [2] . The smallest gap between the bonding and the anti-bonding  -bands is approximately 11 eV. Therefore, the majority of low-energy physical effects is determined by the
-bands is approximately 11 eV. Therefore, the majority of low-energy physical effects is determined by the  -bands. Since the overlap with other orbitals (
-bands. Since the overlap with other orbitals ( ) is strictly zero by symmetry,
) is strictly zero by symmetry,  -electrons forming the
-electrons forming the  -bonds can be treated independently from other valence electrons [5] .
-bonds can be treated independently from other valence electrons [5] .
Indeed, the band structure of graphene can be seen as a triangular lattice with a basis of two atoms per unit cell. This 2-atom unit cell (Wigner-Seitz (WS) cell) model has customarily been used to obtain the graphene band structure for the  -electrons. The band structure provides useful information about the energy dispersion relation of electrons at zero temperature. At finite temperatures those thermally excited “electrons” and “holes” play an essential role for carrier transport properties. Following Ashcroft and Mermin (AM) [6] , if we adopt the semiclassical model of electron dynamics in solids, it is necessary to introduce a
-electrons. The band structure provides useful information about the energy dispersion relation of electrons at zero temperature. At finite temperatures those thermally excited “electrons” and “holes” play an essential role for carrier transport properties. Following Ashcroft and Mermin (AM) [6] , if we adopt the semiclassical model of electron dynamics in solids, it is necessary to introduce a  -vector:
-vector:
 (1)
(1)
where  are the Cartesian orthogonal unit vectors since the
are the Cartesian orthogonal unit vectors since the  -vector,
-vector,  , is involved in the semiclassical equation of (wave packet) motion:
, is involved in the semiclassical equation of (wave packet) motion:
 (2)
(2)
where  and
and  are the electric and magnetic fields, respectively, and the vector
are the electric and magnetic fields, respectively, and the vector 
 (3)
(3)
is the electron velocity where  is the electron energy. In the semiclassical theory electrons are treated as a wave packet. The 2D crystals such as graphene can also be treated similarly, only the
is the electron energy. In the semiclassical theory electrons are treated as a wave packet. The 2D crystals such as graphene can also be treated similarly, only the  -component being dropped. Graphene forms a 2D honeycomb lattice. The WS unit cell is a rhombus (dotted lines) as shown in Figure 1(a).
-component being dropped. Graphene forms a 2D honeycomb lattice. The WS unit cell is a rhombus (dotted lines) as shown in Figure 1(a).
The potential energy  is lattice-periodic:
is lattice-periodic:
 (4)
(4)
where  are Bravais vectors with the primitive non-orthogonal vectors
are Bravais vectors with the primitive non-orthogonal vectors  and integers
and integers . In the field theoretical formulation, the field point
. In the field theoretical formulation, the field point  is given by
is given by , where
, where  is the point defined within the standard WS unit cell. Equation (4) describes the 2D lattice periodicity but
is the point defined within the standard WS unit cell. Equation (4) describes the 2D lattice periodicity but
 (a) (b)
(a) (b)
Figure 1. (a) Lattice structure of graphene. Carbon atoms at vertices. Each honeycomb lattice consists of equivalent carbon (C+) ions labeled by A and B. The Wigner-Seitz 2-atom unit cell (dotted lines) spanned by the basis (lattice unit) vectors  and
and ; (b) Reciprocal lattice showing the first Brillouin zone (BZ) of graphene including the high-symmetry points
; (b) Reciprocal lattice showing the first Brillouin zone (BZ) of graphene including the high-symmetry points , K, and M. The BZ is spanned by two reciprocal lattice vectors
, K, and M. The BZ is spanned by two reciprocal lattice vectors  and
and  constructed from the basis vectors
constructed from the basis vectors  and
and .
.
does not establish the  -space for the Bloch electrons in graphene, which will be explained in Section 4, and this fact has motivated us to study the band structure for a Bloch electron based on the rectangular 4-atom unit cell model for graphene (see Figure 2(a)), which defines the
-space for the Bloch electrons in graphene, which will be explained in Section 4, and this fact has motivated us to study the band structure for a Bloch electron based on the rectangular 4-atom unit cell model for graphene (see Figure 2(a)), which defines the  -vectors playing an important roll for a Bloch electron dynamics.
-vectors playing an important roll for a Bloch electron dynamics.
Our purpose of this paper is to explore the suitability of the rectangular (orthogonal) unit cell model for the Bloch electron band structure and to discuss the  -vector defined in the 2D space for Bloch electrons in graphene. Based on the rectangular 4-atom unit cell model of graphene, we obtain the band structure for the
-vector defined in the 2D space for Bloch electrons in graphene. Based on the rectangular 4-atom unit cell model of graphene, we obtain the band structure for the  -electrons of graphene by extending the prevalent tight-binding calculations for the Wigner-Seitz (WS) 2-atom unit cell model to the rectangular (orthogonal) 4-atom unit cell model introduced by the present authors [7] .
-electrons of graphene by extending the prevalent tight-binding calculations for the Wigner-Seitz (WS) 2-atom unit cell model to the rectangular (orthogonal) 4-atom unit cell model introduced by the present authors [7] .
In Section 2, the rectangular 4-atom unit cell of graphene is introduced. Section 3 presents our tight-binding calculations of the energy band of a Bloch electron in graphene, based on the rectangular 4-atom unit cell model described in Section 2. Section 4 illustrates why we have to utilize the rectangular (orthogonal) unit cell rather than the WS unit cell when considering the electron dynamics of graphene. Results and discussion are given in Section 5. Finally conclusions and some remarks are given in Section 6.
2. The Rectangular 4-Atom Unit Cell Model for Graphene
Graphene is made out of honeycomb lattice (carbon atoms arranged in hexagonal structure) and the 2D honeycomb lattice has a reflection symmetry relative to the  - and
- and  -axis (cp. Figure 2(a)). Then the band structure for a Bloch electron in graphene can be constructed from a rectangular lattice with a basis of four atoms per unit cell, which we call the rectangular 4-atom unit cell.
-axis (cp. Figure 2(a)). Then the band structure for a Bloch electron in graphene can be constructed from a rectangular lattice with a basis of four atoms per unit cell, which we call the rectangular 4-atom unit cell.
 (a) (b)
(a) (b)
Figure 2. (a) Lattice structure of graphene, made out of the rectangular 4-atom unit cell (a square dotted line) spanned by the basis vectors  and
and . The
. The  are the nearest-neighbor vectors with the constant carbon-carbon distance of
are the nearest-neighbor vectors with the constant carbon-carbon distance of  ; (b) The first Brillouin zone of the rectangular 4-atom unit cell. The
; (b) The first Brillouin zone of the rectangular 4-atom unit cell. The  and
and  are the reciprocal lattice vectors corresponding to the lattice unit vectors. The high-symmetric points within the rectangular unit cell are indicated by
are the reciprocal lattice vectors corresponding to the lattice unit vectors. The high-symmetric points within the rectangular unit cell are indicated by , X, Y, and W within the reciprocal lattice plane.
, X, Y, and W within the reciprocal lattice plane.
The reason why we choose a rectangular (orthogonal) unit cell rather than a triangular (WS) unit cell for graphene will be explained later.
The lattice basis (unit) vectors  of graphene (cp. Figure 2(a)) are defined as
of graphene (cp. Figure 2(a)) are defined as
 (5)
(5)
The lattice constants for the rectangular (orthogonal) unit cell in the Cartesian coordinates are  and
and , respectively, where
, respectively, where  is the nearest-neighbor distance between two carbon atoms. The rectangular 4-atom unit cell of graphene contains four carbon atoms, say A, B, C, and D, as shown in Figure 2(a). The three nearest-neighbor vectors
is the nearest-neighbor distance between two carbon atoms. The rectangular 4-atom unit cell of graphene contains four carbon atoms, say A, B, C, and D, as shown in Figure 2(a). The three nearest-neighbor vectors  in real space are defined (cp. Figure 2(a)) by
in real space are defined (cp. Figure 2(a)) by
 (6)
(6)
The reciprocal-lattice vectors for the rectangular unit cell are given (from Equation (5)) by
 (7)
(7)
The first Brillouin zone is a rectangle as shown in Figure 2(b) with sides of length,  and
and , and area equal to
, and area equal to . There are four key locations of high symmetry in the first Brillouin zone (FBZ) of the rectangular 4-atom unit cell. In Figure 2(b), these locations are identified as the
. There are four key locations of high symmetry in the first Brillouin zone (FBZ) of the rectangular 4-atom unit cell. In Figure 2(b), these locations are identified as the  -point, the X-point, the Y-point, and the W-point. The
-point, the X-point, the Y-point, and the W-point. The  -point is at the center of the Brillouin zone. The locations of these points in the 2D reciprocal space are:
-point is at the center of the Brillouin zone. The locations of these points in the 2D reciprocal space are:
 (8)
(8)
3. Tight-Binding Approach
The single-particle band structure of graphene can be analytically calculated within the tight-binding approximation assuming that electrons are tightly bound to their C+ ion [8] [9] [10] [11] . In order to obtain the band structure of graphene based on the 4-atom rectangular unit cell model [7] for graphene, we follow the tight-binding approach employed by Reich et al. [8] .
The Schrödinger equation for a single electron in the lattice potential field  is expressed by
is expressed by
 (9)
(9)
where the Hamiltonian  is given by
is given by
 (10)
(10)
and the lattice potential  has a lattice periodicity characteristic for graphene. The total wave function
has a lattice periodicity characteristic for graphene. The total wave function  for the electron in the
for the electron in the  -orbital (Bloch electron) may be obtained from the linear combination of the Bloch wave
-orbital (Bloch electron) may be obtained from the linear combination of the Bloch wave  of the form:
of the form:
 (11)
(11)
where  is the number of unit lattices and the index
is the number of unit lattices and the index  refers to the respective carbon atoms. In the above expressions, the phase
refers to the respective carbon atoms. In the above expressions, the phase  are introduced in order to satisfy the Bloch theorem in each conduction channel. Thus the wave function for a Bloch electron in graphene is then expressed by the linear combinations of these Bloch wave functions:
are introduced in order to satisfy the Bloch theorem in each conduction channel. Thus the wave function for a Bloch electron in graphene is then expressed by the linear combinations of these Bloch wave functions:
 (12)
(12)
where ’s are given by Equation (11) and the wave function
’s are given by Equation (11) and the wave function  for the
for the  -orbital is normalized. The energy eigenvalues of Equation (9) for the
-orbital is normalized. The energy eigenvalues of Equation (9) for the  -electron can be obtained in a usual manner by using the graphene Hamil- tonian (10) and the graphene wave function (12) along with Equation (11): Inserting Equation (12) into Equation (9) and multiplying
-electron can be obtained in a usual manner by using the graphene Hamil- tonian (10) and the graphene wave function (12) along with Equation (11): Inserting Equation (12) into Equation (9) and multiplying  from the left to Equation (9), we obtain four separate equations, which are simply expressed in the matrix form as
from the left to Equation (9), we obtain four separate equations, which are simply expressed in the matrix form as
 (13)
(13)
where the matrices,  ,
,  and
and , are respectively expressed by
, are respectively expressed by
 (14)
(14)
 (15)
(15)
 (16)
(16)
Here  are the matrix elements of the Hamiltonian
are the matrix elements of the Hamiltonian , which we call hopping (or transfer) integral, and are the units of energy.
, which we call hopping (or transfer) integral, and are the units of energy.  are the overlap matrix elements, which are given by the overlap integral between Bloch functions, and are unitless. Equation (13) expresses the simultaneous equations for
are the overlap matrix elements, which are given by the overlap integral between Bloch functions, and are unitless. Equation (13) expresses the simultaneous equations for ,
,  ,
,  and
and . In order to obtain the nontrivial solutions for
. In order to obtain the nontrivial solutions for , the secular equation determi- nant
, the secular equation determi- nant  of their simultaneous equations must be zero:
of their simultaneous equations must be zero:
 (17)
(17)
Since the atomic wave functions are well localized around the carbon atoms, only the nearest-neighbor hopping of Bloch electrons are taken into considera- tion of the following calculations. In other words, as for the electron in atom A orbital, it can hop to a nearest orbital of atom B or atom D. Similarly, the electron in the orbital of atom B can hop to the nearest orbital of atom A or atom C, the electron in the orbital of atom C to the nearest orbital of atom B or atom D, and the electron in the orbital of atom D to the nearest orbital of atom A or atom C.
Let us consider the nearest-neighbor hopping between the orbital of atom A and atom B. The matrix elements  is expressed by the hopping integral:
is expressed by the hopping integral:
 (18)
(18)
Here we only take into account the nearest-neighbor hopping between carbon atoms. From the symmetry of the lattice stracture of graphene, the adjacent hoppings are all the same. We introduce the parametric parameter  for the adjacent hopping between the orbital of atom A and B:
for the adjacent hopping between the orbital of atom A and B:
 (19)
(19)
Since the nearest-neighbor vector  is given by
is given by  and
and  (cp. Equation (6)), Equation (18) can be expressed by
(cp. Equation (6)), Equation (18) can be expressed by
 (20)
(20)
Similarly, the nearest-neighbor hopping integrals are given by
 (21)
(21)
We note that the hopping parameter  is the nearest-neighbor hopping energy (hopping between adjacent carbon atoms).
is the nearest-neighbor hopping energy (hopping between adjacent carbon atoms).
The matrix element  (on-site hopping) is evaluated as
(on-site hopping) is evaluated as
 (22)
(22)
where  is the number of the basic unit cells, and
is the number of the basic unit cells, and  is the parametric parameter for
is the parametric parameter for . Similar evaluation leads to
. Similar evaluation leads to  .
.
Next we evaluate the matrix elements , which are given by the overlap integral
, which are given by the overlap integral . Thus the matrix element
. Thus the matrix element  is evaluated from
is evaluated from
 (23)
(23)
Introducing the parametric parameter  for the overlap integral:
for the overlap integral:
 (24)
(24)
the matrix element  is given by
is given by
 (25)
(25)
Similarly we obtain
 (26)
(26)
We note that the matrix element  is evaluated as
is evaluated as
 (27)
(27)
In a similar manner, we obtain
 (28)
(28)
We can solve Equation (17) for  by using Equations (18)-(28) for the matrix elements, and obtain the four energy eigenvalues of Equation (9) for the graphene
by using Equations (18)-(28) for the matrix elements, and obtain the four energy eigenvalues of Equation (9) for the graphene  -electron bands:
-electron bands:
 (29)
(29)
 (30)
(30)
where  and
and  are the conduction-and the valence-band energies, respec- tively.
are the conduction-and the valence-band energies, respec- tively.
4. Electron Dynamics of Graphene
In semiclassical (wave packet) theory for electron dynamics, it is necessary to introduce a wave vector  (namely,
(namely,  -vector) [6] [12] since the
-vector) [6] [12] since the  -vectors are involved in the semiclassical equation of motion (see Equation (2)). Here, we explain why we employed the rectangular (orthogonal) unit cell for graphene in order to calculate one electron energy band for graphene.
-vectors are involved in the semiclassical equation of motion (see Equation (2)). Here, we explain why we employed the rectangular (orthogonal) unit cell for graphene in order to calculate one electron energy band for graphene.
Graphene forms a 2D honeycomb lattice. Let us first consider the Wigner- Seitz (WS) unit cell (rhombus, dotted lines shown in Figure 1(a)). The potential energy  is lattice-periodic:
is lattice-periodic:
 (31)
(31)
where the position vector  in the potential field
in the potential field  can be represented by Bravais vectors with the primitive (non-orthogonal) basis vectors
can be represented by Bravais vectors with the primitive (non-orthogonal) basis vectors  and integers
and integers :
:
 (32)
(32)
In the field theoretical formulation, the field point  is given by
is given by
 (33)
(33)
where  is the point defined within the standard WS unit cell. Equation (31) describes the (2D) lattice periodicity but does not establish the 2D
is the point defined within the standard WS unit cell. Equation (31) describes the (2D) lattice periodicity but does not establish the 2D  -vector of a Bloch electron in graphene if we choose the non-Cartesian coordinates system. Thus the WS unit cell is not appropriate when one deals with graphene transport problem, which is explained below.
-vector of a Bloch electron in graphene if we choose the non-Cartesian coordinates system. Thus the WS unit cell is not appropriate when one deals with graphene transport problem, which is explained below.
We assume that the wave packet is composed of superposable plane-waves characterized by the  -vectors. The superposability is the basic property of the Schrödinger wave equation. The Schrödinger wave equation for a Bloch electron (wave packet) is
-vectors. The superposability is the basic property of the Schrödinger wave equation. The Schrödinger wave equation for a Bloch electron (wave packet) is
 (34)
(34)
where  is the effective electron mass. The Bravais vectors for the rhombic lattice,
is the effective electron mass. The Bravais vectors for the rhombic lattice,  , are given by Equation (32) and the system is lattice-periodic, cp. Equation (31). The Bloch theorem can be expressed in the form:
, are given by Equation (32) and the system is lattice-periodic, cp. Equation (31). The Bloch theorem can be expressed in the form:
 (35)
(35)
for all  in the Bravais lattice. The theorem in 1D can be proved elementarily [6] . If an oblique lattice is considered and a set of nonorthogonal basis vectors
in the Bravais lattice. The theorem in 1D can be proved elementarily [6] . If an oblique lattice is considered and a set of nonorthogonal basis vectors  are introduced for a 2D Bravais vector, cp. Equation (32), there are no 2D Fourier transform available since there is no 2D
are introduced for a 2D Bravais vector, cp. Equation (32), there are no 2D Fourier transform available since there is no 2D  -vector to satisfy the periodicity condition for the Bloch wave, which must be of the form (35). Thus, the WS unit cell model for graphene is not suitable when one considers the electron dynamics in graphene.
-vector to satisfy the periodicity condition for the Bloch wave, which must be of the form (35). Thus, the WS unit cell model for graphene is not suitable when one considers the electron dynamics in graphene.
The wave function must be Fourier-analyzable. In the rhombic system, however, if we choose the  -axis, say along
-axis, say along , then the potential energy field
, then the potential energy field  is periodic in the
is periodic in the  -direction, but it is aperiodic in the
-direction, but it is aperiodic in the  -direction (cp. Figure 1(a)) since these basis vectors
-direction (cp. Figure 1(a)) since these basis vectors  are not orthogonal each other. Thus, if we choose the rhombic unit cell for graphene, we cannot express
are not orthogonal each other. Thus, if we choose the rhombic unit cell for graphene, we cannot express  - vectors satisfying the condition for the Bloch wave (35). Then, there is no 2D
- vectors satisfying the condition for the Bloch wave (35). Then, there is no 2D  space spanned by 2D
space spanned by 2D  -vectors of Bloch waves if we choose the rhombic unit cell. If we omit the kinetic energy term, then we can still use Equation (31) and obtain the ground state energy (except the zero-point energy).
-vectors of Bloch waves if we choose the rhombic unit cell. If we omit the kinetic energy term, then we can still use Equation (31) and obtain the ground state energy (except the zero-point energy).
Ashcroft and Mermin (AM) [6] introduced a translation operator  such that acting on any field
such that acting on any field  it shifts its argument by
it shifts its argument by :
:
 (36)
(36)
They used
 (37)
(37)
to establish Equation (35). The translation operator  can be expressed in terms of a differential operator:
can be expressed in terms of a differential operator:
 (38)
(38)
as seen from . If the Bravais vector
. If the Bravais vector  is given in
is given in
terms of the orthogonal basis vectors  in the Cartesian coordinates and choose the rectangular unit cell (cp. Figure 2(a)) as in Equation (34), then Equation (37) is satisfied, meaning that the Bloch wave for a rectangular unit cell can be expressed in terms of the Bloch wave (35).
in the Cartesian coordinates and choose the rectangular unit cell (cp. Figure 2(a)) as in Equation (34), then Equation (37) is satisfied, meaning that the Bloch wave for a rectangular unit cell can be expressed in terms of the Bloch wave (35).
If  is written in terms of nonorthogonal
is written in terms of nonorthogonal , then
, then  is a product of differential operators
is a product of differential operators  and
and , which does not commute with the Laplacian
, which does not commute with the Laplacian  in the Hamiltonian operator
in the Hamiltonian operator . Hence, we can not establish Fourier-analyzable 2D
. Hence, we can not establish Fourier-analyzable 2D  -vectors when we choose the rhombic unit cell for graphene. In actual calculations, Equation (37) is valid for the Bloch wave (35), where the Bravais vectors are expressed by the orthogonal basis vectors in the Cartesian coordinates. It should be noted that for an infinite lattice the periodic boundary is the only acceptable boundary condition for the Fourier transformation. AM’s second proof [6] also fails in 2D if the Bravais vector
-vectors when we choose the rhombic unit cell for graphene. In actual calculations, Equation (37) is valid for the Bloch wave (35), where the Bravais vectors are expressed by the orthogonal basis vectors in the Cartesian coordinates. It should be noted that for an infinite lattice the periodic boundary is the only acceptable boundary condition for the Fourier transformation. AM’s second proof [6] also fails in 2D if the Bravais vector  is expressed in nonorthogonal base vectors. This is so because Laplacian terms cannot be handled in non-Cartesian (oblique) coordinates. AM's Equation (8.36) does not hold. We must choose the rectangular unit cell in order to establish the Bloch plane waves [13] [14] for the “electron” in 2D.
is expressed in nonorthogonal base vectors. This is so because Laplacian terms cannot be handled in non-Cartesian (oblique) coordinates. AM's Equation (8.36) does not hold. We must choose the rectangular unit cell in order to establish the Bloch plane waves [13] [14] for the “electron” in 2D.
We assume that the “electron” (“hole”') (wave packet) has the charge  and a size of the rectangular unit cell, generated above (below) the Fermi energy
and a size of the rectangular unit cell, generated above (below) the Fermi energy . It was shown [7] [15] that (a) the “electron” and the “hole” have different charge distribution and different effective masses, (b) that the “electron” and “hole” move in different easy channels, (c) that the “electrons” and “holes” are thermally excited with different activation energies, and (d) that the “electron” activation energy
. It was shown [7] [15] that (a) the “electron” and the “hole” have different charge distribution and different effective masses, (b) that the “electron” and “hole” move in different easy channels, (c) that the “electrons” and “holes” are thermally excited with different activation energies, and (d) that the “electron” activation energy  is smaller than the “hole” activation energy
is smaller than the “hole” activation energy :
:
 (39)
(39)
Thus, “electrons” are the majority carriers in graphene. The thermally activated electron densities are then given by
 (40)
(40)
where  and 2 represent the “electron” and “hole”, respectively. The pre- factor
and 2 represent the “electron” and “hole”, respectively. The pre- factor  is the density at the high-temperature limit. Magnetotransport ex- periments by Zhang et al. [16] indicate that the “electrons” are the majority carriers in graphene. Thus, our theory based on the rectangular unit cell model is agreement with experiments.
is the density at the high-temperature limit. Magnetotransport ex- periments by Zhang et al. [16] indicate that the “electrons” are the majority carriers in graphene. Thus, our theory based on the rectangular unit cell model is agreement with experiments.
At finite temperature phonons are present in the system. The excitation of phonons can be discussed based on the same rectangular unit cell introduced for the conduction electrons. We note that phonons can be discussed naturally based on the orthogonal unit cells. It is difficult to describe phonons in the WS cell model.
5. Results and Discussion
Based on the rectangular 4-atom unit cell model, we obtained the band disper- sion of graphene by applying the tight-binding theory of Reich et al. [8] . The obtained ( -) band structure based on the rectangular 4-atom unit cell model for graphene computed from Equations (29) and (30) is shown in Figure 3, where the nearest-neighbor hopping parameters
-) band structure based on the rectangular 4-atom unit cell model for graphene computed from Equations (29) and (30) is shown in Figure 3, where the nearest-neighbor hopping parameters  and
and  are taken from the values obtained by the ab initio calculation [8] [17] :
are taken from the values obtained by the ab initio calculation [8] [17] : ,
,  ,
, . This 3D figure illustrates the conduction and the valence band within the first Brillouin zone. The two crossing points P and Q indicated in the figure are of particular importance for the physics of graphene.
. This 3D figure illustrates the conduction and the valence band within the first Brillouin zone. The two crossing points P and Q indicated in the figure are of particular importance for the physics of graphene.
Figure 4 shows the energy band profile along the high symmetric points (indicated by , X, Y, and W) of the rectangular 4-atom unit cell in reciprocal
, X, Y, and W) of the rectangular 4-atom unit cell in reciprocal

Figure 3. (Color online) Band structure of graphene based on the rectangular 4-atom unit cell model. The energy is maximal at the  -point in the conduction band, while there is a saddle-point at the
-point in the conduction band, while there is a saddle-point at the  -point in the valence band in the first BZ. The display shows the linear dispersion structure around the crossing-points P and Q, at which
-point in the valence band in the first BZ. The display shows the linear dispersion structure around the crossing-points P and Q, at which .
.

Figure 4. (Color online) Band profile along the symmetric points of graphene for the rectangular 4-atom unit cell model. One of the cross in points (so-called Dirac points) is indicated by P. Inset is the 1st Brillouin zone of the rectangular unit cell model. Conduction bands (red and blue) are computed from Equation (29) while valence bands (black and green) are computed from Equation (30). Note that the band profile is plotted within the one quarter of the first Brillouin zone.
space. There are two crossing points P and Q in the first BZ (cp. Figure 3), at which the band energy is crossing (i.e., ), suggesting that graphene is a zero gap semiconductor.
), suggesting that graphene is a zero gap semiconductor.
Let us take a close look at the behavior of the band energy close to the crossing points, at which the band energy equals to zero. The conduction and valence bands are degenerate at this point. The dispersion relation for small momenta  near the crossing points
near the crossing points  and
and  is given by Taylor expanding the band energy around the points P and Q, resulting in a unique linear energy dispersion:
is given by Taylor expanding the band energy around the points P and Q, resulting in a unique linear energy dispersion:
 (41)
(41)
where  is now in spherical coordinates (their origin is now at the points P and Q),
is now in spherical coordinates (their origin is now at the points P and Q),  the Fermi velocity given by
the Fermi velocity given by 
with ,
, . This particular band structure re- sembles the physics of massless Dirac fermions with a velocity approximately 300 times smaller than the speed of light. Near the crossing point P in the first Brillouin zone, the linear dispersion (Equation (41)) is clearly seen in the plot in Figure 5 for the rectangular 4-atom unit cell model for graphene. The linear dispersion for small
. This particular band structure re- sembles the physics of massless Dirac fermions with a velocity approximately 300 times smaller than the speed of light. Near the crossing point P in the first Brillouin zone, the linear dispersion (Equation (41)) is clearly seen in the plot in Figure 5 for the rectangular 4-atom unit cell model for graphene. The linear dispersion for small  also holds for the crossing point Q as well.
also holds for the crossing point Q as well.
The second BZ of the reciprocal lattice for the rectangular 4-atom unit cell is shown by the shaded area in Figure 6. The high symmetric points in the reciprocal lattice of the rectangular 4-atom unit cell are indicated by , X, Y,
, X, Y,

Figure 5. (Color online) The linear dispersion near the point P in the first BZ of the rectangular 4-atom unit cell, where the two bands (valence and conduction) cross each other. The energy is maximal at the  -point, while there is a saddle point at the
-point, while there is a saddle point at the  - point of the first BZ. The linear dispersion relation (red lines) holds for the small
- point of the first BZ. The linear dispersion relation (red lines) holds for the small  near the so-called Dirac point, while the actual bands (black lines) deviate from the linearity near
near the so-called Dirac point, while the actual bands (black lines) deviate from the linearity near .
.

Figure 6. The first Brillouin zone (rectangular) and the second Brillouin (shaded) zone of the rectangular 4-atom unit cell. The region of the first and second BZ’s (i.e., the hexagonal region) of the rectangular 4-atom unit cell is identical to the first BZ of the Wigner-Seitz 2-atom unit cell.
and W, while those in the reciprocal lattice of the 2-atom unit cell are indicated by , M and K. The Dirac points of the Wigner-Seitz 2-atom unit cell are indicated by K and K’ in Figure 6, while the crossing (so-called Dirac) points of the first BZ of the 4-atom unit cell are located at the points P, Q in the first BZ of the 4-atom unit cell. Note that the first Brillouin zone of the Wigner-Seitz 2-atom unit cell is identical to the region of the first and second Brillouin zone of the rectangular 4-atom unit cell (hexagon. cp. Figure 6). The crossing points, P and Q, in the first Brillouin zone of the rectangular 4-atom unit cell correspond to the Dirac points, K and K’ of the Wigner-Seitz 2-atom unit cell since the energy band structure of the rectangular 4-atom unit cell model is identical to the folded band structure of the 2-atom unit cell model along the boundary of the first BZ of the 4-atom unit cell model.
, M and K. The Dirac points of the Wigner-Seitz 2-atom unit cell are indicated by K and K’ in Figure 6, while the crossing (so-called Dirac) points of the first BZ of the 4-atom unit cell are located at the points P, Q in the first BZ of the 4-atom unit cell. Note that the first Brillouin zone of the Wigner-Seitz 2-atom unit cell is identical to the region of the first and second Brillouin zone of the rectangular 4-atom unit cell (hexagon. cp. Figure 6). The crossing points, P and Q, in the first Brillouin zone of the rectangular 4-atom unit cell correspond to the Dirac points, K and K’ of the Wigner-Seitz 2-atom unit cell since the energy band structure of the rectangular 4-atom unit cell model is identical to the folded band structure of the 2-atom unit cell model along the boundary of the first BZ of the 4-atom unit cell model.
The approximate results ( -bands) obtained in this paper are based on the rectangular 4-atom unit cell model for graphene by using tight-binding calculations, where only the nearest-neighbor hopping is taken into account for the transport of Bloch electrons. The horizontal
-bands) obtained in this paper are based on the rectangular 4-atom unit cell model for graphene by using tight-binding calculations, where only the nearest-neighbor hopping is taken into account for the transport of Bloch electrons. The horizontal  line shows the chemical potential. Below the chemical potential, the states are all occupied and the conduction states are above it. The chemical potential is at the points labeled P and Q in the first BZ, where the valence and conduction bands are crossing. The bands with the lowest energy are from the
line shows the chemical potential. Below the chemical potential, the states are all occupied and the conduction states are above it. The chemical potential is at the points labeled P and Q in the first BZ, where the valence and conduction bands are crossing. The bands with the lowest energy are from the  -bands in the planes between the carbon atoms. Electrons in the
-bands in the planes between the carbon atoms. Electrons in the  -bands are tightly bound. The energy bands near the chemical potential are from the
-bands are tightly bound. The energy bands near the chemical potential are from the  -bands. They are formed from the carbon orbital
-bands. They are formed from the carbon orbital , which projects above and below the plane of graphene. These
, which projects above and below the plane of graphene. These  -bands are well described by a nearest-neighbor tight-binding calcula- tions based on the 4-atom unit cell model.
-bands are well described by a nearest-neighbor tight-binding calcula- tions based on the 4-atom unit cell model.
6. Conclusions and Some Remarks
The current authors proposed a rectangular 4-atom unit cell model for graphene [7] . Based on this model, we obtained the band energy by applying the tight- binding approach employed by Reich et al. [8] . We proved the band structure based on the rectangular 4-atom unit cell model for graphene gives the same band structure of graphene based on the prevalent graphene model based on the Wigner-Seitz 2-atom unit cell model.
The rectangular 4-atom unit cell for graphene has the sides perpendicular to each other (see Figure 2). The  -vector
-vector  is defined as in Equation (1) but with the orthogonal unit vector, and the
is defined as in Equation (1) but with the orthogonal unit vector, and the  is obtained as the Fourier conjugate of the position vector
is obtained as the Fourier conjugate of the position vector .
.
The transport electrons in graphene originating from the  -band can be described by the 2D Bloch
-band can be described by the 2D Bloch  -vector defined in the proposed rectangular unit cell. We showed that the energy dispersion relation near the Dirac points (small
-vector defined in the proposed rectangular unit cell. We showed that the energy dispersion relation near the Dirac points (small ) shows the linear dependence of
) shows the linear dependence of  (see Equation (41)).
(see Equation (41)).
A material (density) wave such as a phonon wave can be presented by a traveling wave function of the form:  with C = material density,
with C = material density,  = angular frequency, and
= angular frequency, and  -vector. The direction of
-vector. The direction of  points to the direction of the traveling plane wave.
points to the direction of the traveling plane wave.
In the currently prevailing theory [2] [3] [4] [5] [8] [9] [10] [11] [17] the solid state theory dealing with a hexagonal crystal starts with a primitive non- orthogonal unit cell, the 2-atom unit cell. This theoretical model has difficulties in particular for a superconductor. The ground state of a superconductor must be condensed in a single particle-state in accordance with Nernst’s theorem (the third law of thermodynamics). Many fermions have a distribution in energy and hence a many-fermion system cannot be a superconductor. Only many-boson system can be a superconductor.
In the prevailing theory [2] [3] [4] [5] [8] [9] [10] [11] [17] a 2-atom unit cell model is used to set up  -vectors with non-orthogonal unit vectors
-vectors with non-orthogonal unit vectors  and
and . A Bloch
. A Bloch  -vector is
-vector is  constructed with the Born-von Karman boundary condition [6] . But this vector is not useful in dealing with a superconducting intercalation graphite compound.
constructed with the Born-von Karman boundary condition [6] . But this vector is not useful in dealing with a superconducting intercalation graphite compound.
In the present work we have shown that the non-orthogonal 2-atom and orthogonal 4-atom models give the same band energies in the mean field appro- ximation. In separate publication we report the superconductivity in graphite intercalation compounds C8K, C6Ca and in compound MgB2.
Cite this paper
Suzuki, A., Tanabe, M. and Fujita, S. (2017) Electronic Band Structure of Graphene Based on the Rectangular 4-Atom Unit Cell. Journal of Modern Physics, 8, 607-621. https://doi.org/10.4236/jmp.2017.84041
References
- 1. Geim, A.K. and Noboselov, K.S. (2007) Nature Materials, 6, 183-191.
https://doi.org/10.1038/nmat1849 - 2. Neto, A.H.C., Guinea, F., Peres, N.M.R., Novoselov, K.S. and Geim, A.K. (2009) Reviews of Modern Physics, 81, 109.
https://doi.org/10.1103/RevModPhys.81.109 - 3. Sarma, S.D., Adam, S., Hwang, E. and Rossi, E. (2011) Reviews of Modern Physics, 83, 407.
https://doi.org/10.1103/RevModPhys.83.407 - 4. Katsnelson, M.I. (2012) Graphene: Carbon in Two Dimensions. Cambridge University Press, Cambridge.
https://doi.org/10.1017/CBO9781139031080 - 5. Reich, S., Thomsen, C. and Maultzsch, J. (2004) Carbon Nanotubes: Basic Concepts and Physical Properties. Wiley-VCH Verlag GmbH, Berlin.
- 6. Ashcroft, N.W. and Mermin, N.D. (1976) Solid State Physics. Saunders, Philadelphia, 75, 133-135, 137-139, 228-229.
- 7. Fujita, S. and Suzuki, A. (2010) Journal of Applied Physics, 107, Article ID: 013711.
https://doi.org/10.1063/1.3280035 - 8. Reich, S., Maultzsch, J., Thomsen, C. and Ordejön, P. (2002) Physical Review B, 66, Article ID: 035412.
https://doi.org/10.1103/PhysRevB.66.035412 - 9. Wallace, P.R. (1947) Physical Review, 71, 622.
https://doi.org/10.1103/PhysRev.71.622 - 10. McClure, J.W. (1957) Physical Review, 108, 612.
https://doi.org/10.1103/PhysRev.108.612 - 11. Slonczewski, J.S. and Weiss, P.R. (1958) Physical Review, 109, 272.
https://doi.org/10.1103/PhysRev.109.272 - 12. Fujita, S. and Ito, K. (2007) Quantum Theory of Conducting Matter: Newtonian Equations of Motion for a Bloch Electron. Springer, New York, 120-121, 192-193.
https://doi.org/10.1007/978-0-387-74103-1 - 13. Bloch, F. (1929) Zeitschrift für Physik, 52, 555-600.
https://doi.org/10.1007/BF01339455 - 14. Fujita, S., Jovaini, A., Godoy, S. and Suzuki, A. (2012) Physics Letters A, 376, 2808-2811.
- 15. Fujita, S., Takato, Y. and Suzuki, A. (2011) Modern Physics Letters B, 25, 223.
https://doi.org/10.1142/S0217984911025675 - 16. Zhang, Y., Tan, Y.W., Stormer, H.L. and Kim, P. (2005) Nature, 438, 201-204.
https://doi.org/10.1038/nature04235 - 17. Saito, R., Dresselhaus, G. and Dresselhaus, M.S. (2005) Physical Properties of Carbon Nanotubes. Imperial College Press, London.