Applied Mathematics
Vol.4 No.8A(2013), Article ID:35183,7 pages DOI:10.4236/am.2013.48A014
Numerical Modeling of Three-Phase Mass Transition with an Application in Atmospheric Chemistry
1Department of Analytical Chemistry and Computer Chemistry, University of Plovdiv, Plovdiv, Bulgaria
2Department of Informatics and Statistics, University of Food Technology, Plovdiv, Bulgaria
Email: atanas@uni-plovdiv.net
Copyright © 2013 Nikolay Kochev et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received May 29, 2013; revised June 29, 2013; accepted July 6, 2013
Keywords: Atmospheric Processes Modeling; Diffusion Equation; Numerical Simulation; Software Library
ABSTRACT
This work presents a software tool for modeling of mass transfer physicochemical processes occurring in the atmosphere. The implemented algorithms provide an efficient theoretical frame for the interpretation of the results obtained from Coated Wall Flow Tube (CWFT) reactor experiments, which is one of the most adequate techniques to study heterogeneous kinetics. The numerical simulations are based on the fundamental Langmuir adsorption theory by ordinary differential equations and the second Fick’s law described by partial differential equations. The main application of the system is to estimate the basic parameters that characterize the processes. The best parameter estimation is found by minimizing the difference between experimental signals from the CWFT reactors and the obtained numerical simulations. A numerical example for an experimental data fit is given.
1. Introduction
Climate changes in the last decades and the expansion of ozone hole have become an important problem for researchers from different fields of natural sciences. A major point of atmospheric processes studies is the understanding of the heterogeneous mass transfer occurring in different altitudes and specifically the role of ice as adsorbent. Atmospheric trace gases interact with the ice in a complicated manner including various physicochemical processes such as adsorption, desorption and diffusion. One of the most efficient ways to study these processes in laboratory are experiments performed in Coated Wall Flow Tube (CWFT) reactors [1-3] in order to match the atmospheric conditions in high altitude such as temperature, pressure, trace gas concentration etc. Usually the thermodynamic and kinetic parameters that characterize the atmospheric processes cannot be derived directly from the raw measurements; hence the mass detector signal must be analyzed with various numerical methods. The calculations can be done in different levels: quantum mechanics (QM), molecular dynamics (MD) and macroscopic kinetic simulations customized for specific experiments. QM and MD typically characterize theoretically (on a microscopic level) small system of several molecules for QM or thousands of molecules for MD (for a very small time interval). However the linking of theoretical data with the experiment still remains quite speculative in many occasions since the theory takes into account localized effects while the experimental data is taken from the whole reactor. The efficient interpretation of the mass signal requires a third numerical approach where the processes in the entire reactor are simulated (the number considered molecules is 1017 for few minutes of simulation/measurement). CWFT reactor consists of a tube cooled down at low and constant temperatures in accordance with atmospheric conditions. A movable injector that ends with a tiny nozzle injects the measured trace gas along the reactor tube while it is moving (see Figure 1). The carrier gas flows laminarly toward the mass spectrometer (detector). Initially ice film is generated by deposition of gaseous water upon injection of water vapor through the sliding injector at low temperatures of the reactor wall. The combined effect of all processes adsorption/desorption with diffusion/segregation kinetics and thermodynamics can be observed as measured mass spectrometer signal Gexp(t). The best estimation of the studied parameters optimizes the mean square error (MSE), which measures the difference between the experimental and simulated (G(l, t)) signal.
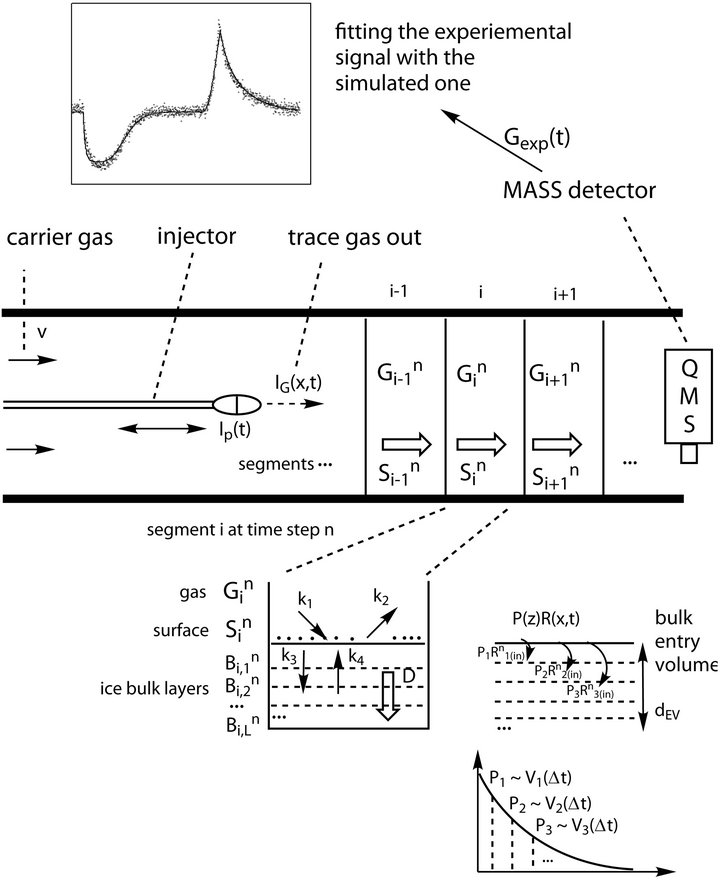
Figure 1. CWFT reactor schematic presentation and formalization of the simulated processes.
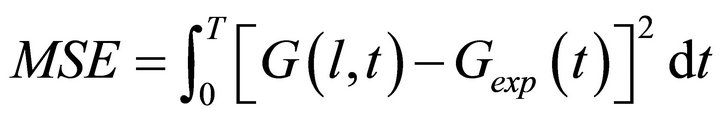 (1)
(1)
In this paper we present in detail developed by us numerical algorithms for simulation of mass transfer physicochemical processes occurring in the atmosphere. Also we give an example application for real measurements of acetic acid interacting with ice surfaces at temperature 190 K.
2. Mathematical Model with a PDE System
The CWFT reactor state is formalized by the following functions:
G(x, t)—gas concentrationS(x, t)—ice surface concentrationB(x, z, t)—ice bulk concentrationwhere the argument 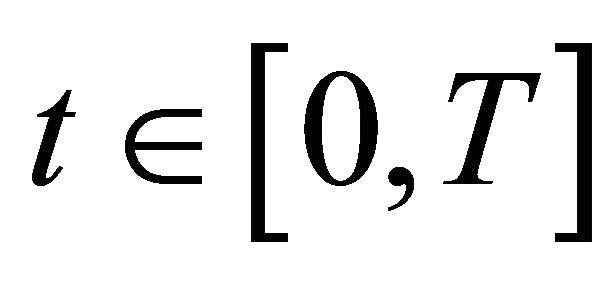 is the time,
is the time, 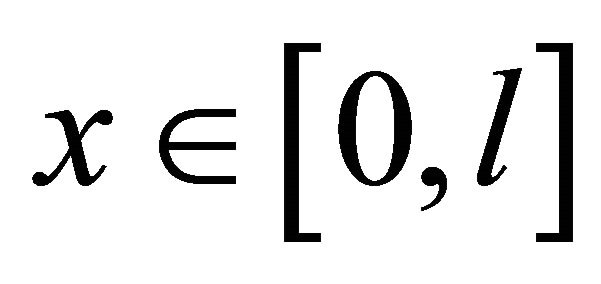 is the position in the reactor and
is the position in the reactor and 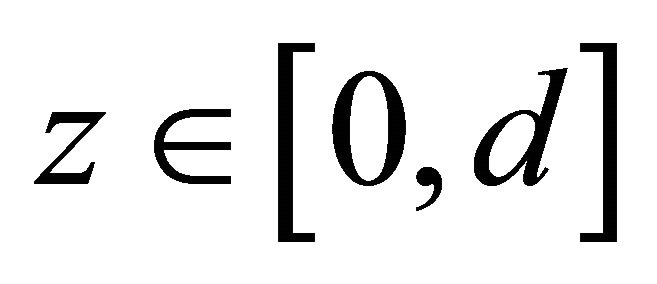 is the position in the ice bulk. G(l, t) is the resulting mathematical model which is used to fit the reactor experimental output (see Equation (1)). G(l, t) is the gas concentration at the end of the CWFT reactor (x = l), where also the mass detected signal, Gexp(t) is obtained (see Figure 1).
is the position in the ice bulk. G(l, t) is the resulting mathematical model which is used to fit the reactor experimental output (see Equation (1)). G(l, t) is the gas concentration at the end of the CWFT reactor (x = l), where also the mass detected signal, Gexp(t) is obtained (see Figure 1).
All processes in the reactor are mathematically described by means of the PDE system (2). The first two equations are based on the Langmuir law [1] plus correction for the gas injection IG(x, t) and transfer from the bulk R(x, t), the third equation is the classical diffusion PDE (also known as the second Fick’s law in physical chemistry) and the last equation describes the gas flow in the reactor.
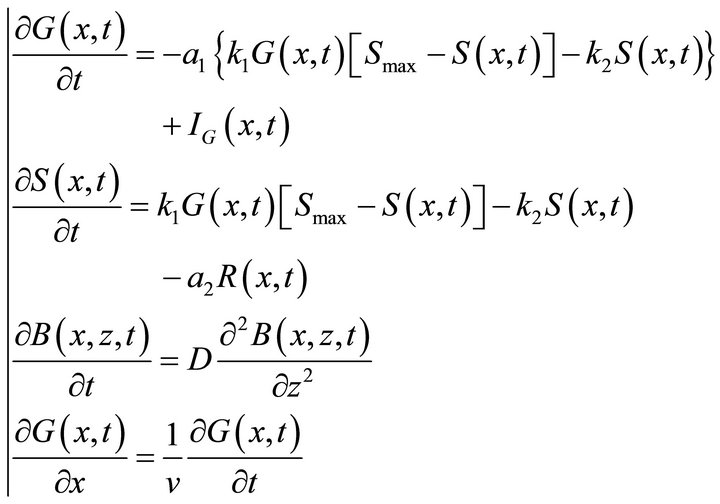 (2)
(2)
The constants k1, k2, Smax and D (also k3, k4 and Bmaxsee Equation (5)) characterize the kinetics of the processes. The constants k1 and k2 respectively describe adsorption and desorption rate coefficients. Smax is the number of the active surface sites and Bmax is the maximum bulk concentration at the given conditions. k3 and k4 represent the solution into the bulk and segregation from the bulk rate constants respectively. The constants a1 and a2 describe the reactor geometry and v is the flow velocity constant.
The function:
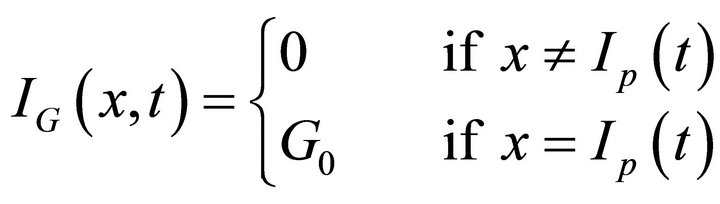 (3)
(3)
represents the injected gas from the movable injector where Ip(t) is the injector position expressed as a function of time:
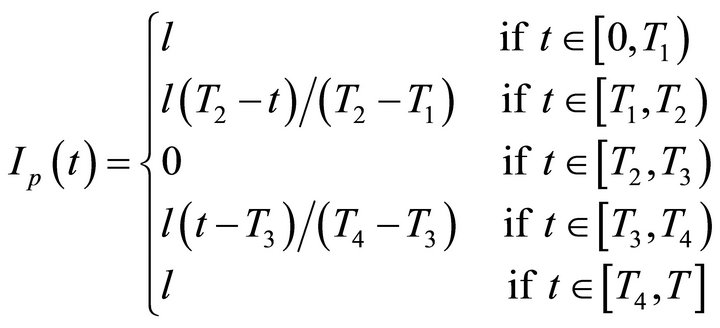 (4)
(4)
Equation (4) describes the injector position as it is changed during the experimental procedures. The injector moves with a constant speed from the end of the CWFT reactor (Ip(t) = l) to the beginning of the reactor (Ip(t) = 0) and backwards to the end. Respectively T1, T2, T3, T4 are the times for the different stages of the injector trajectory (profile).
The rate penetration function 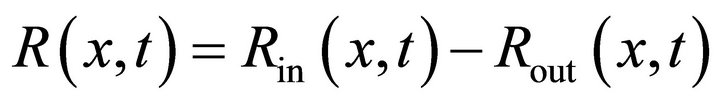 is defined in Equation (5).
is defined in Equation (5).
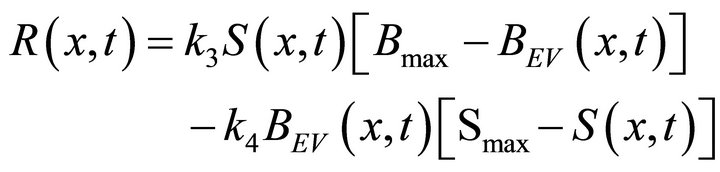 (5)
(5)
where the entry bulk volume concentration is defined for a depth dEV:
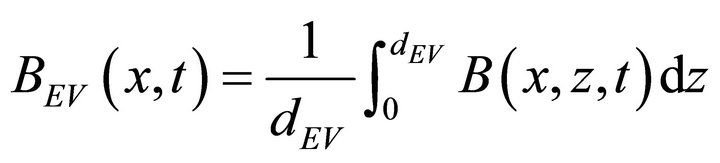 (6)
(6)
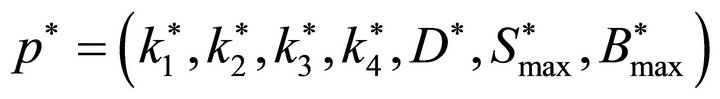 are the optimal values for
are the optimal values for 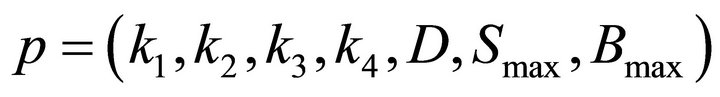 and p* are found by searching a multidimensional parallelepiped
and p* are found by searching a multidimensional parallelepiped 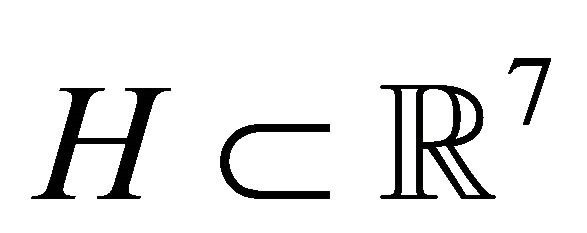 for the minimal value of MSE.
for the minimal value of MSE.
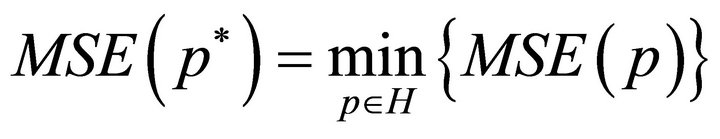 (7)
(7)
MSE from Equation (1) is regarded as a function of the parameters p, MSE = MSE(p). Hyper parallelepiped H is defined as follows:
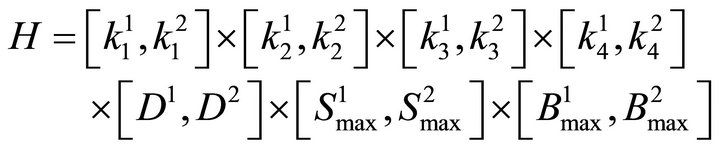 (8)
(8)
The dimensionality of H may change in different occasions. For example, if a simulation is performed without considering the diffusion, only three parameters are used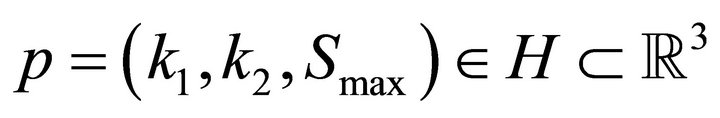 .
.
3. Initial and Boundary Conditions of the PDE System
The initial and boundary conditions for PDE system (2) are defined as follows:
 (9)
(9)
P(z) is a probability density function used for the definition of the Neumann boundary conditions in the fourth equation of the PDE system (9). P(z) may be defined in different ways in order to describe the physicochemical processes occurring on the boundary. One of the approaches used by us to define P(z) is the PDE system (10) that is a diffusion process for a time interval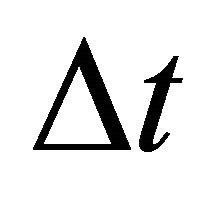 .
.
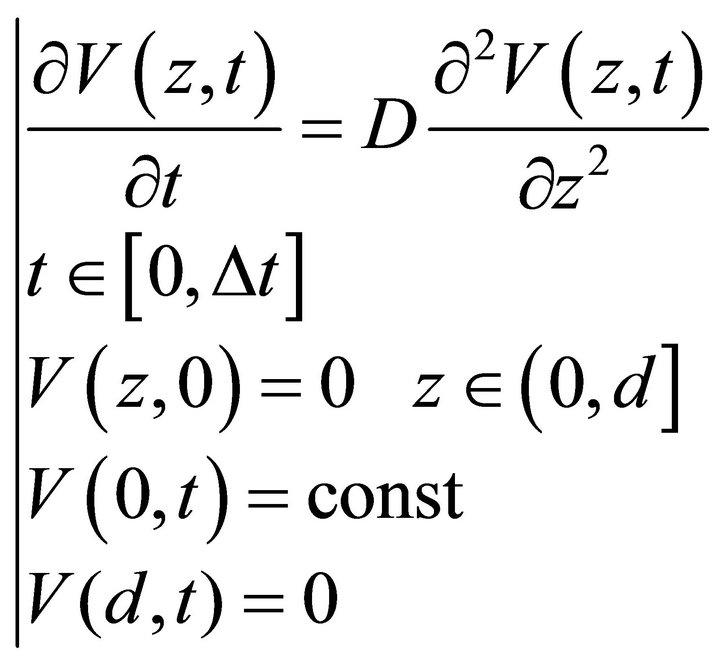 (10)
(10)
The solution V(z, ∆t) is used to define P(z) in the interval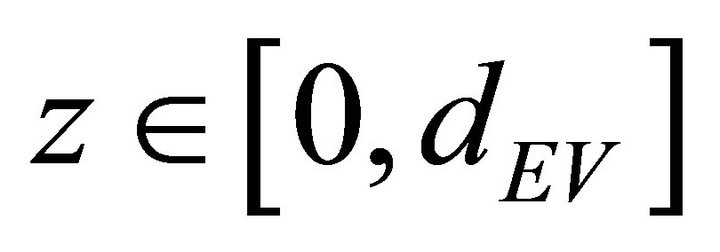 :
:
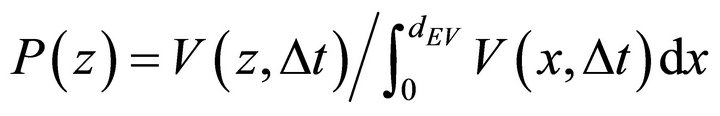 (11)
(11)
System (10) is numerically solved preliminary, before the simulations with the PDE system (2).
4. Numerical Procedures
A discretization is performed for the time:
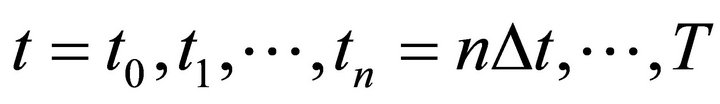 .
.
The reactor is divided into a number of segments K with length ∆x and ice bulk is separated into a number of layers with thickness of ∆z. The functions are represented in discrete form as follows:
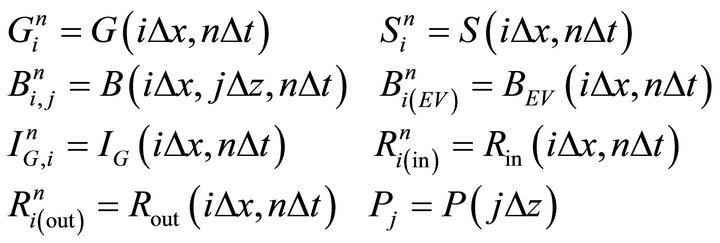
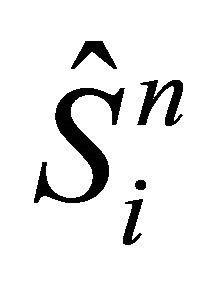 is an intermediate value for
is an intermediate value for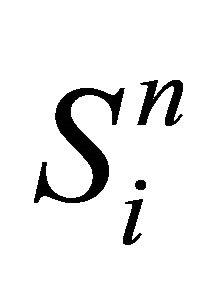 , where the transfer function R(x, t) is not taken into account.
, where the transfer function R(x, t) is not taken into account.
The iterations for a simulation with particular parameters 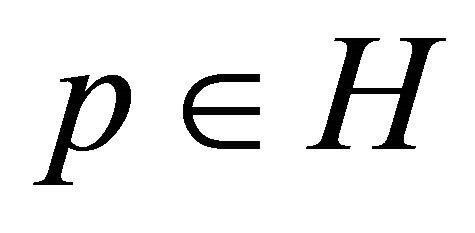 are performed by means of nested cycles over time (index n), reactor segments (index i) and ice bulk layers (index j). The entire simulation process is summarized as follows:
are performed by means of nested cycles over time (index n), reactor segments (index i) and ice bulk layers (index j). The entire simulation process is summarized as follows:
for n = 1,2,…,N
{
for i = 0,1,2,…,K-1
take gas state from previous iteration: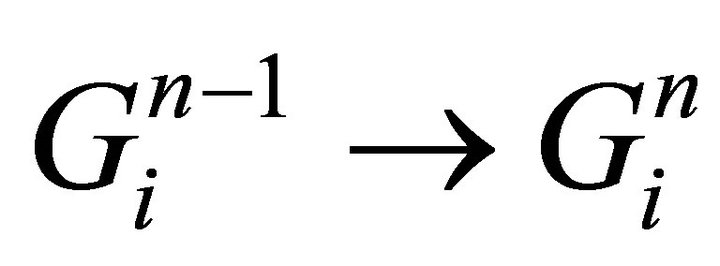
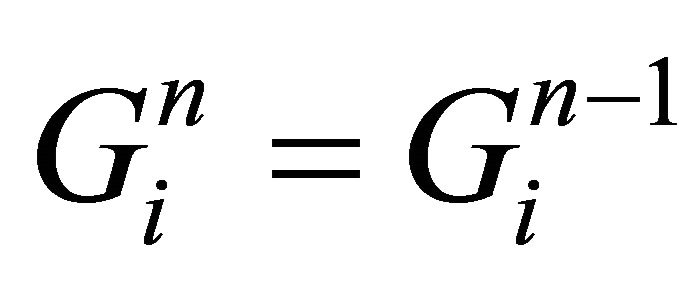
for i = 0,1,2,…,K-1
{
step(i) adsorption/desorption for segment i: 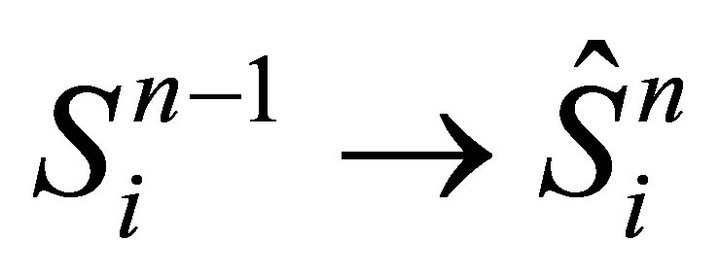 Equation (16)
Equation (16)
step(ii) gas flow from segment i to segment i+1: 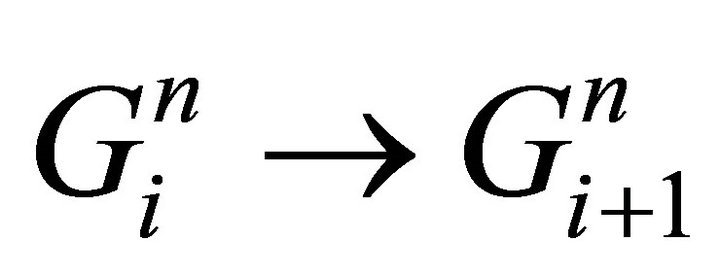 Equation (20)
Equation (20)
step(iii) interface-in: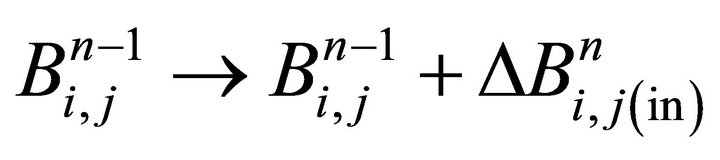 ;
;
transfer to the bulk, j=1,2, …, nEL, Equation (23)
step(iv) diffusion: Equation (25) for j = 1,2,…,L:
update of each layer : 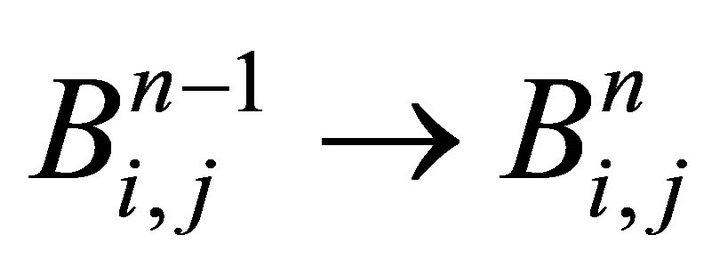
step(v) interface-out: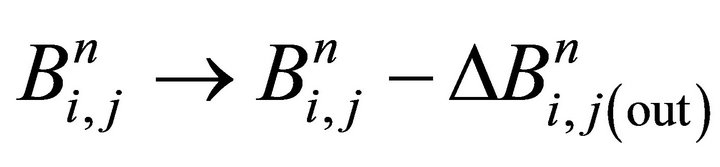 ;
;
transfer out of the bulk to the surfacej=1,2, …, nEL, Equation (24)
step(vi) correction of the surface 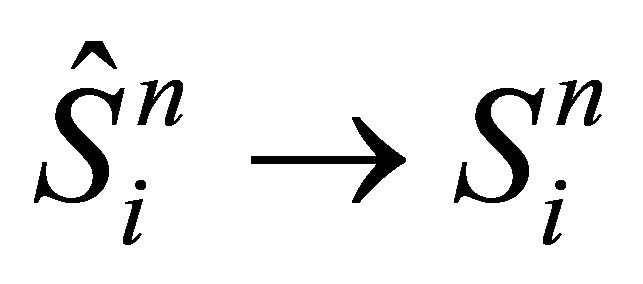
see Equation (27)
}
}
In step (i), the first and second PDEs from (2) are solved without taking into account the terms IG(x, t) and −a2R(x, t):
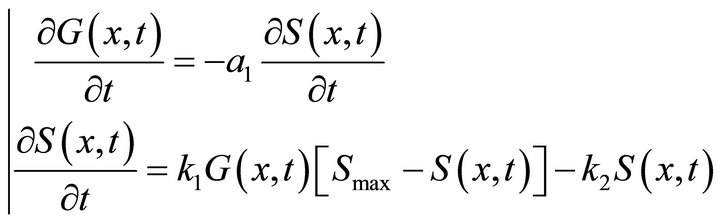 (12)
(12)
the omitted terms are calculated in steps (ii) and (vi) respectively. The system (12) is solved semi-analytically where for a small time interval, G(x, t) is assumed to be constant. After substitution 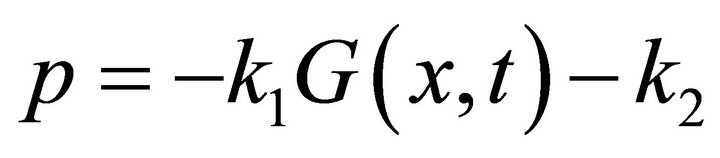 and q = k1G(x, t)Smax in the second equation of system (12) we obtain the following equation:
and q = k1G(x, t)Smax in the second equation of system (12) we obtain the following equation:
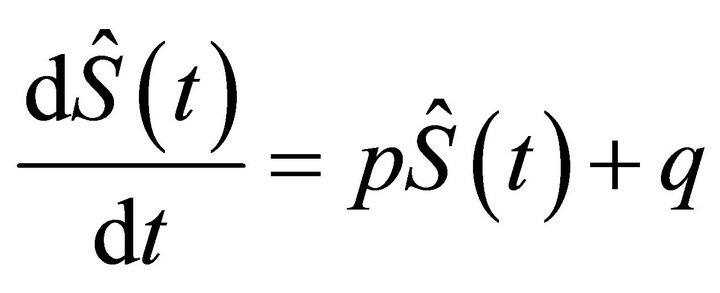 (13)
(13)
It is with separable variables and its integration is trivial as x is fixed.
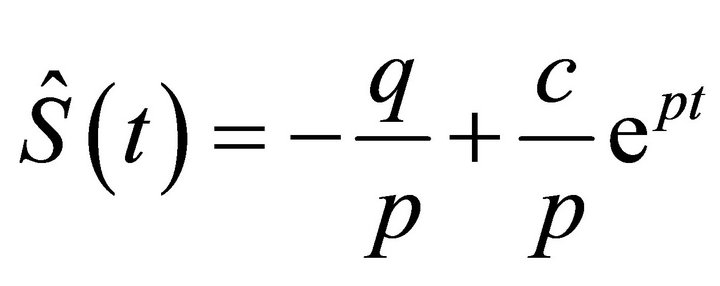 (14)
(14)
In order to calculate the iteration 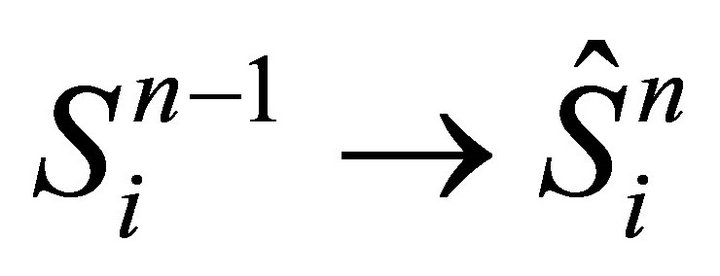 we use (14) together with the following initial condition:
we use (14) together with the following initial condition: 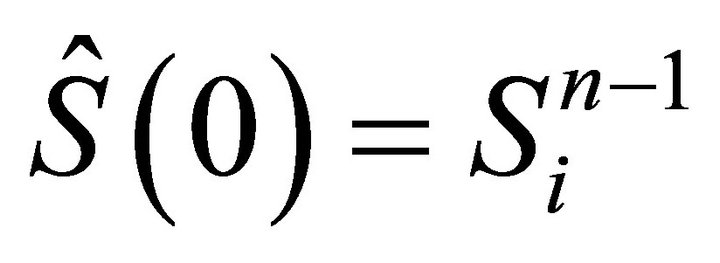 and we obtain constant
and we obtain constant 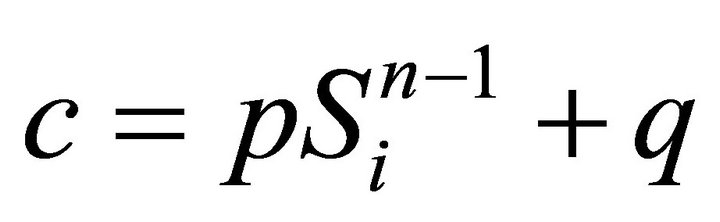 and
and
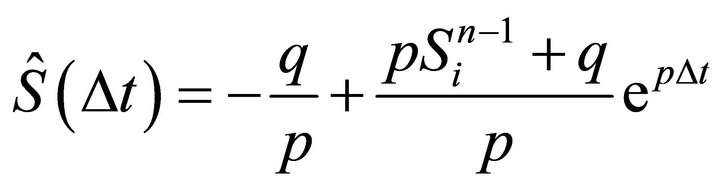 (15)
(15)
After substitution of p and q in Equations (14) and (15), we obtain the following discrete form of numerical step (i):
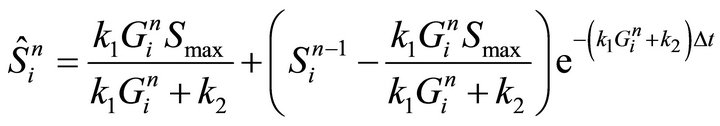 (16)
(16)
In step (ii) the gas flow in the reactor is numerically expressed by the consequent change of the concentrations (iteration ). Taking into account the gas injector function, the first equation of the PDE system (12) is rewritten as follows:
). Taking into account the gas injector function, the first equation of the PDE system (12) is rewritten as follows:
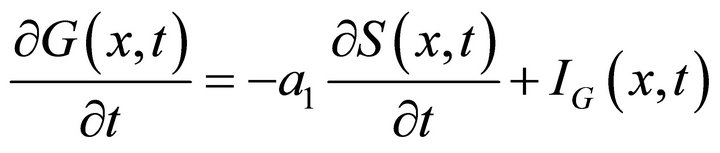 (17)
(17)
We substitute 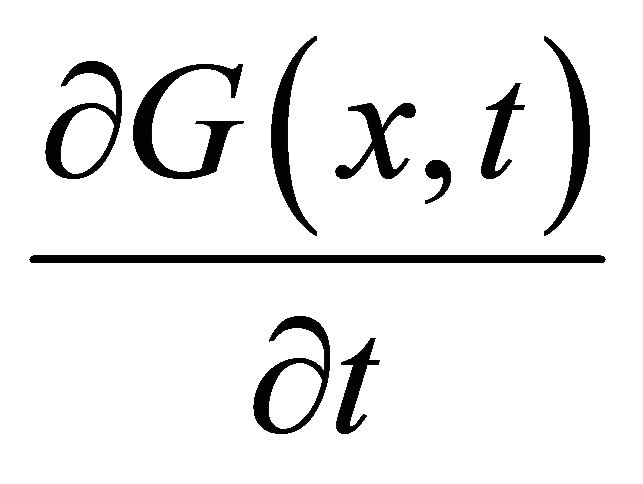 from (17) in the last equation of the PDE system (2) which is then transformed into:
from (17) in the last equation of the PDE system (2) which is then transformed into:
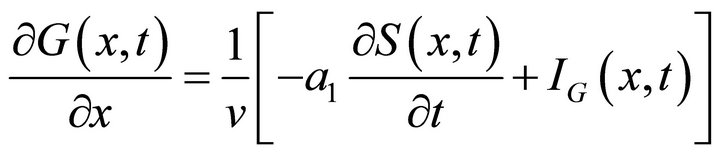 (18)
(18)
The last Equation (18) is resolved numerically with a standard finite difference scheme that is two point derivative approximation and the substitution of velocity expression :
:
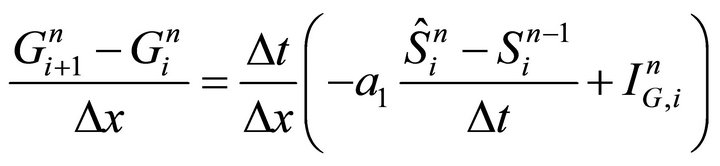 (19)
(19)
Then it follows the iteration of step (ii):
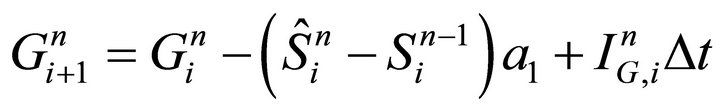 (20)
(20)
Numerical steps (iii) and (v) are performed by the following discrete equations (see also Equation (5) for R(x, t)):
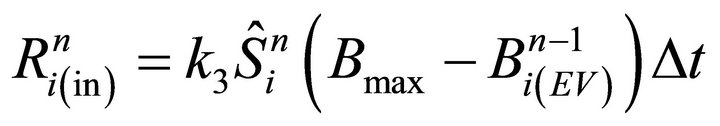 (21)
(21)
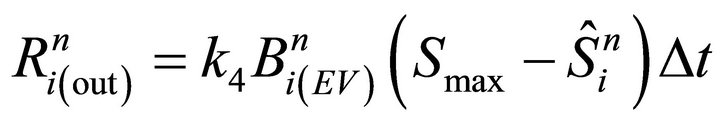 (22)
(22)
In formula (21), member 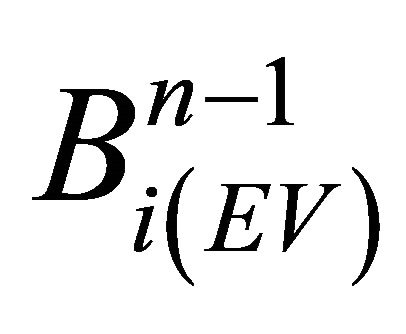 has time index (n − 1) since diffusion step (iv) is not performed yet. The changes for each bulk entry volume layer are obtained from the probability function P(z) (see the fourth equation from the boundary conditions (9)):
has time index (n − 1) since diffusion step (iv) is not performed yet. The changes for each bulk entry volume layer are obtained from the probability function P(z) (see the fourth equation from the boundary conditions (9)):
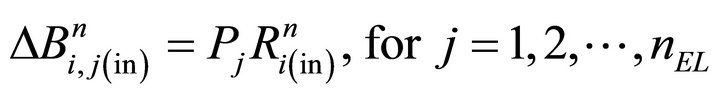 (23)
(23)
where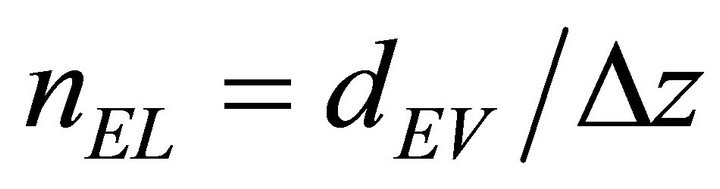 . Analogously, numerical step (v) is calculated:
. Analogously, numerical step (v) is calculated:
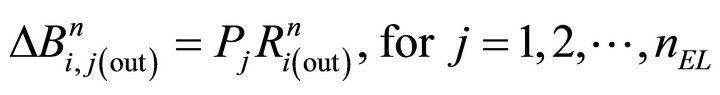 (24)
(24)
that represents molecules that exit from each layer into the surface.
Numerical step (iv) is the diffusion equation (the third PDE from the system (2)). It is numerically solved by means of Euler method with two and three point approximations respectively for the first and second derivatives. The following discrete form of the diffusion equation describes the backward-time centered-spaced (BTCS) method:
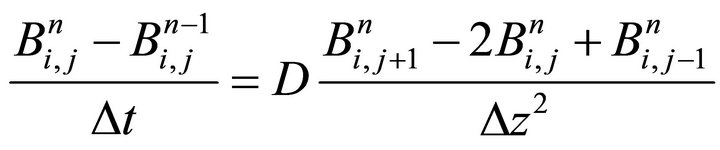 (25)
(25)
The application of the recurrence Equation (25) leads to a 3-diagonal system with known numerical solution in the literature [4,5]. We use time-step restriction (26) which is the Courant-Friedrichs-Lewy stability condition [6]:
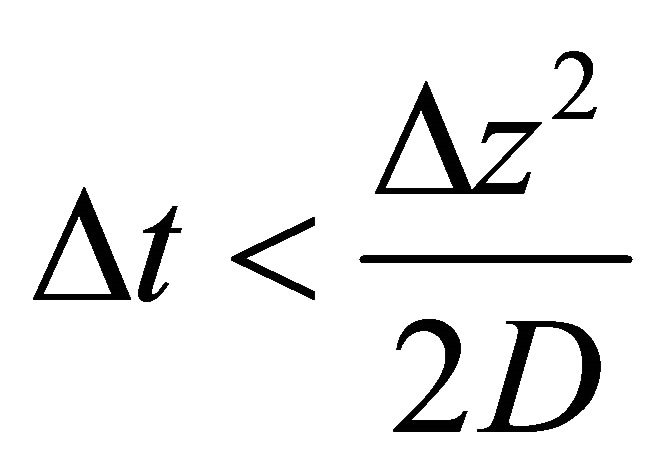 (26)
(26)
The stability restriction (26) of the explicit scheme could be overcome via standard super-time-stepping acceleration procedure, see [6,7]. Thus, explicit schemes become competitive with the efficient implicit scheme with positivity preserving and smoothing properties [8,9] that have also stability restrictions in order to satisfy some practical requirements of the respective problem. For partial differential equations nonstandard finite difference schemes could be invented using the proposed ideas and methodology of Strikwerda [10].
Numerical step (vi) is performed by means of Equation (27) where the transfer from bulk into the surface is taken into account.
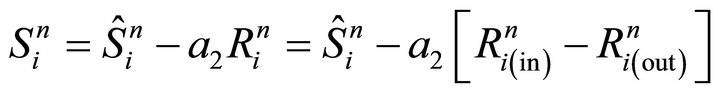 (27)
(27)
Finally the optimal parameters p* (see Equation (7)) are found by a grid search in the hyper parallelepiped, H, with the following discretization:
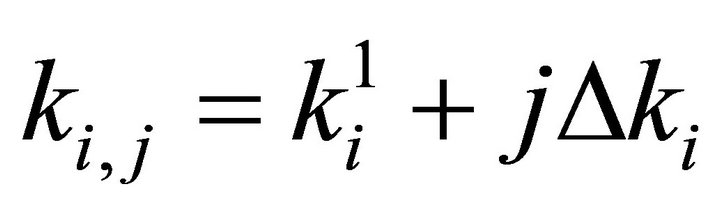 where
where 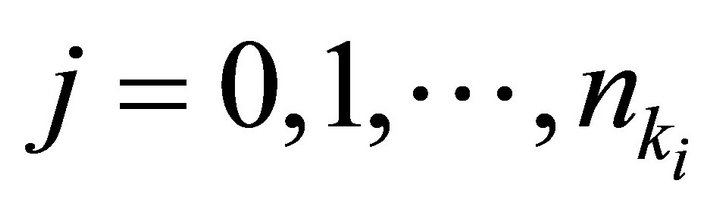
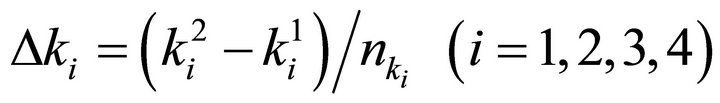
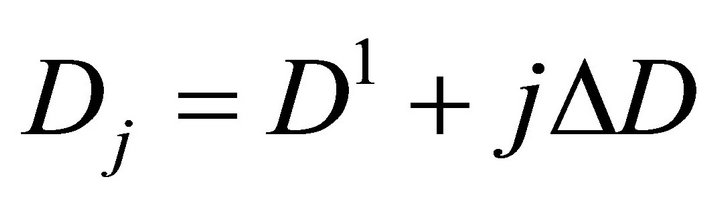 where
where 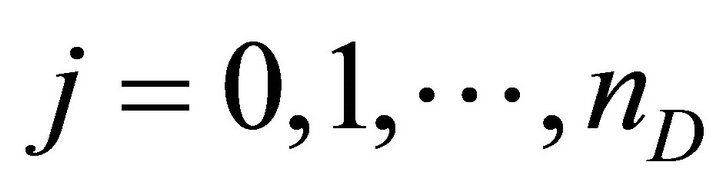
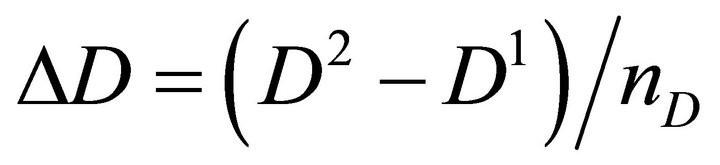
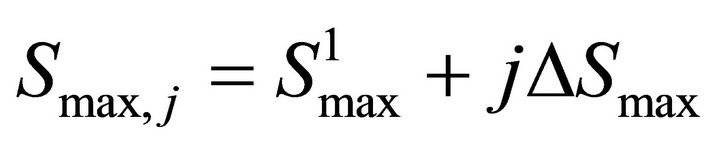 where
where 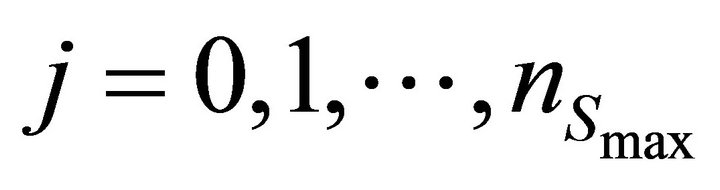
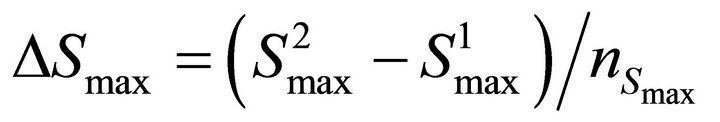
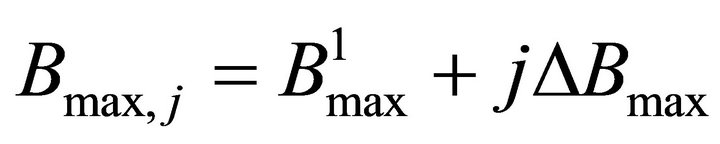 where
where 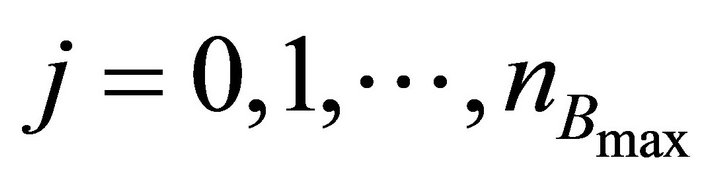
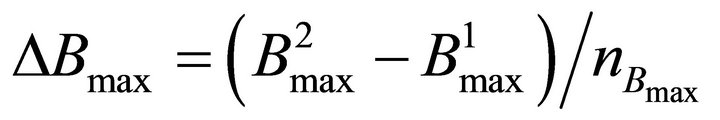
5. Software Development
The first working prototype of the software system (version 1.0) was developed within MS Excel platform as VBA script [1]. Second major milestone (version 2.0) is the implementation of Core Simulation Library (CSL) developed in object oriented language Java. CSL version 2.0 facilitates simulations of the fundamental Langmuir adsorption and desorption processes. CSL library is used with the multi-layer distributed system ADDESSA designed for performing simulations of mass transfer processes in a large scale [11].
The currently presented CSL version 3.0 implements the algorithms (Equations (2)-(27)) for a combined simulation of all three mass transfer processes: adsoption, desorption and diffusion.
CSL is organized in several Java packages (see Figure 2) where the main functionality is concentrated in package simulate. CSL is implemented complying ObjectOriented Programming representing each component of the experimental reactor by one or more classes. The main input to the program is handled by Simulation Loader class as configuration files which describe the basic parameters of experimental process as well as the simulation parameters. The main program utilizes an object of type Flow Reactor. The reactor contains an array of objects of the type Reactor Segment. Reactor segment is used to store the state of a particular part of the reactor. Additionally Reactor Segment class solves numerically the equations of the Second Fick’s law and stores the state of all ice bulk layers for this segment. Reactor Manager GUI is a Graphical User Interface class which can be used to control the simulation process for a single simulation. The basic parameters inputted from the configuration file can be varied in this module as well. The GUI can be used for a preliminary approximation of the simulation parameters which can help further to chose

Figure 2. Core simulation library—CSL.
more precise intervals for an automatic parameter search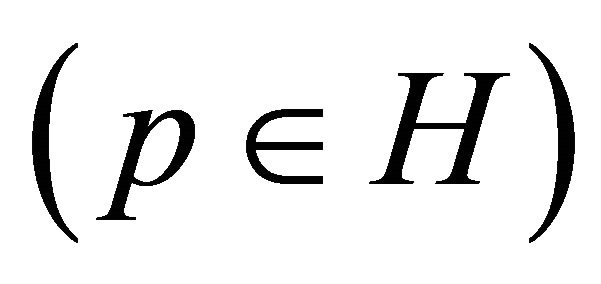 . More information about CSL library and ADDESSA project can be found on the website http://addessa.uni-plovdiv.net.
. More information about CSL library and ADDESSA project can be found on the website http://addessa.uni-plovdiv.net.
6. Example of Numerical Calculations and an Application for Experimental Data
Figure 3 represents the simulation results for a single segment without taking into account the reactor flow. The abscissa on the top graphic is a logarithmic scale representing the time t and the ordinate corresponds to the absolute number of molecules placed on the surface (S(xi, t)) and those remaining in the gas phase (G(xi, t)). The sharp drop of the gas phase until t = 0.1 is due to a rapid adsorption or deposition of molecules on the surface, that is why the surface molecules number is raising. S(xi, t) drops after t = 0.1 because of the concurrent bulk diffusion or mass transfer of molecules to the ice bulk.
The bottom graphics of Figure 3 is illustrating the simultaneous bulk fill (B(xi, zj, t)) as a linear change of the time t. It can be seen that the bulk processes are much slower than the gas/surface interactions. The set of j-plots represent the absolute number of placed molecules in the bulk layers. The system reaches its equilibrium state around t = 900.
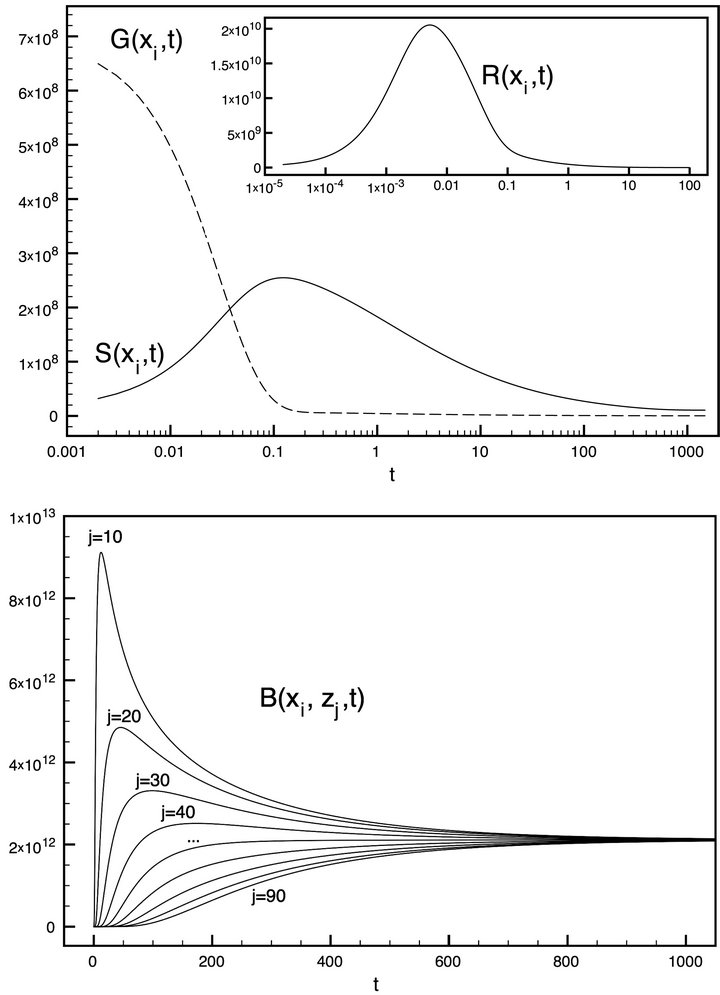
Figure 3. Simulations for G(xi, t), S(xi, t), R(xi, t) and B(xi, zj, t) at different depth j.
Figure 4 shows the simulation result for the entire reactor for three different values of the constant k1. Also the injector position function Ip(t) is plotted in the range of x = 0 (the injector nozzle is far from the detector) to x = l (the injector nozzle is right in front of the mass spectrometer detector).
The three plots of G(l, t) correspond to three different values of the adsorption rate coefficient. The latter is deposition speed of the gas phase molecules on the ice surface. In this case: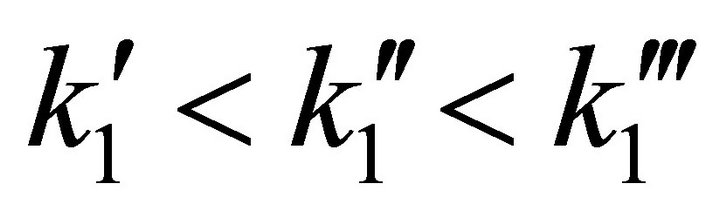 .
.
Figure 5 visualizes the result simulation fit for the experimental measurement of adsorption of acetic acid on ice at temperature 190 K. The graph consists of the raw experimental output (gray dots) and the optimized simulation fit (solid line). The data is measured for about 700 seconds and the output is normalized. The MSE has a minimum value for the found optimal parameters (i.e. kinetic rate constants):
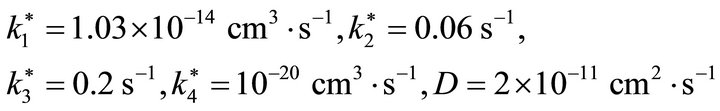
which are in a good agreement with Varotsos and Zellner
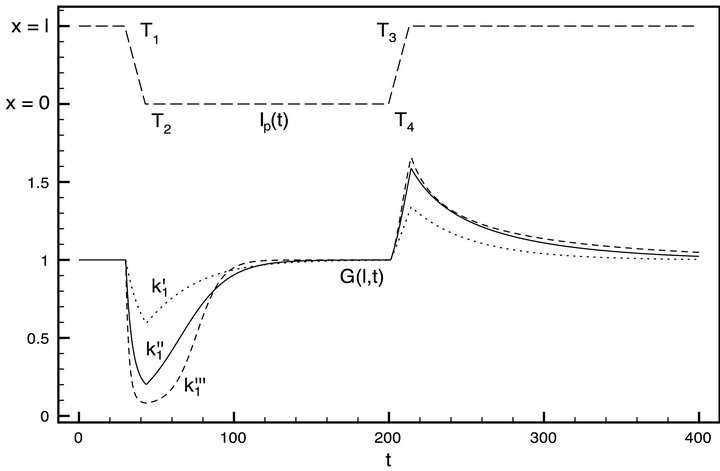
Figure 4. Three numerical solutions G(l, t) of the PDE system (2) and Ip(t) function (visualized above).
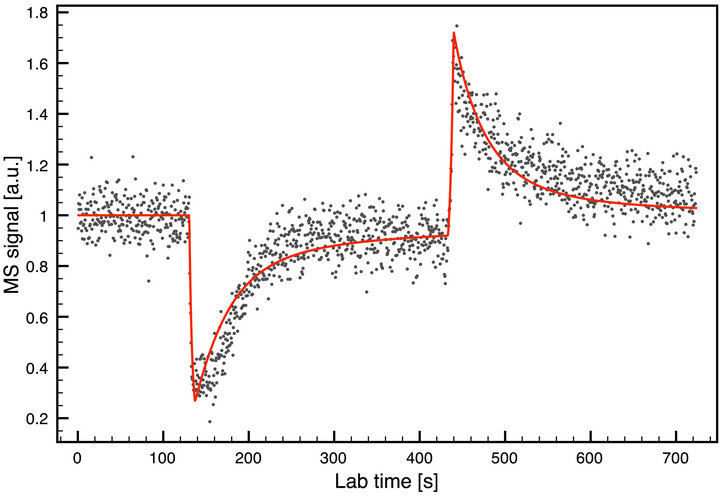
Figure 5. Experimental measurement of acetic acid on ice and the corresponding best theoretical fit.
results [12]. The adsorption capacity of ice was estimated to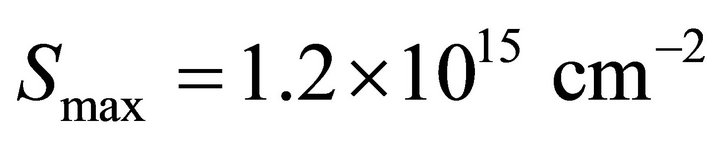 . The thermodynamic values as Henry’s constant
. The thermodynamic values as Henry’s constant 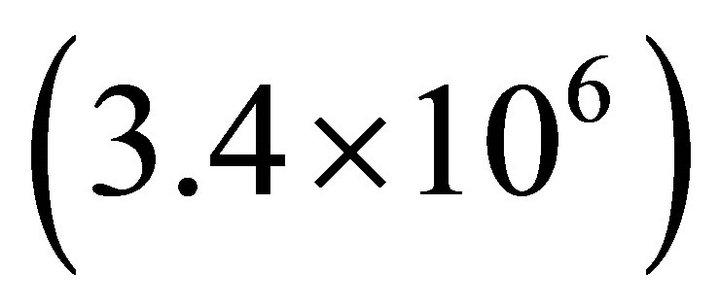 [13] and Langmuir’s constant
[13] and Langmuir’s constant 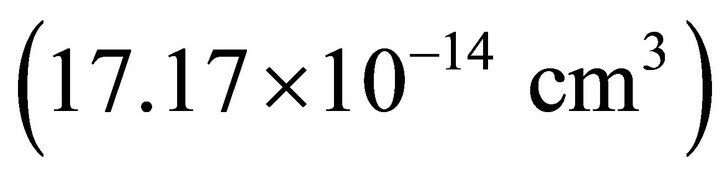 [14] are in a good agreement with the literature for the chosen experimental conditions.
[14] are in a good agreement with the literature for the chosen experimental conditions.
7. Conclusion
The implemented numerical methods for simulation of combined mass transfer can be efficiently applied for study of atmospheric processes. The estimation of basic gas kinetic rate constants is performed by finding the best fit of the measured mass spectrometer signal against the simulated one. The described methods are theoretically proved and practically tested for numerical stability. The procedures have been tested for solving of real gas kinetics problems by performing numerous simulations in various high performance computing systems.
8. Acknowledgements
We would like to thank the Bulgarian National Fund for Scientific Research NFNI (project MU02/12) for supporting this study. This work is also helped by MADARA National Computing Centre (RNF01/0110).
REFERENCES
- P. Behr, A. Terziyski and R. Zellner, “Reversible Gas Adsorption in Coated Wall Flow Tube Reactors. Model Simulations for Langmuir Kinetics,” Zeitschrift für Physikalische Chemie, Vol. 218, No. 11, 2004, pp. 1307-1327. doi:10.1524/zpch.218.11.1307.50806
- P. Behr, A. Terziyski and R. Zellner, “Acetone Adsorption on Ice Surfaces in the Temperature Range T = 190- 220 K: Evidence for Aging Effects Due to Crystallographic Changes of the Adsorption Sites,” The Journal of Physical Chemistry A, Vol. 110, No. 26, 2006, pp. 8098- 8107. doi:10.1021/jp0563742
- R. A. Cox, M. A. Fernandez, A. Symington, M. Ullerstam and J. P. D. Abbatt, “A Kinetic Model for Uptake of HNO3 and HCl on Ice in a Coated Wall Flow System,” Physical Chemistry Chemical Physics, Vol. 7, No. 19, 2005, pp. 3434-3442. doi:10.1039/b506683b
- W. H. Press, S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, “Numerical Recipes in FORTRAN Example Book,” Cambridge University Press, Cambridge, 1992.
- K. Morton and D. Mayers, “Numerical Solution of Partial Differential Equations: An Introduction,” 2nd Edition, Cambridge University Press, Cambridge, 2005. doi:10.1017/CBO9780511812248
- G. Smith, “Numerical Solution of Partial Differential Equations: Finite Difference Methods,” Oxford University Press, Oxford, 1985.
- V. Alexiades, “Overcoming the Stability Restriction of Explicit Schemes via Super-Time-Stepping,” Proceedings of Dynamic Systems and Applications, Vol. 2, Dynamic Publishers, 1996, pp. 39-44.
- M. Milev and A. Tagliani, “Efficient Implicit Scheme with Positivity Preserving and Smoothing Properties,” Journal of Computational and Applied Mathematics, Vol. 243, 2013, pp. 1-9. doi:10.1016/j.cam.2012.09.039
- J. C. Ndogmo and D. B. Ntwiga, “High-Order Accurate Implicit Methods for Barrier Option Pricing,” Applied Mathematics and Computation, Vol. 218, No. 5, 2011, pp. 2210-2224. doi:10.1016/j.amc.2011.07.037
- J. Strikwerda, “Finite Difference Schemes and Partial Differential Equations,” 2nd Edition, SIAM: Society for Industrial and Applied Mathematics, 2004. doi:10.1137/1.9780898717938
- A. Terziyski and N. Kochev, “Distributed Software System for Data Evaluation and Numerical Simulations of Atmospheric Processes,” Numerical Methods and Applications, Lecture Notes in Computer Science, Vol. 6046, 2011, pp. 182-189.
- C. A. Varotsos and R. Zellner, “A New Modeling Tool for the Diffusion of Gases in Ice or Amorphous Binary Mixture in the Polar Stratosphere and the Upper Troposphere,” Atmospheric Chemistry and Physics, Vol. 10, No. 6, 2010, pp. 3099-3105.
- R. Sander, “Compilation of Henry’s Law Constants for Inorganic and Organic Species of Potential Importance in Environmental Chemistry,” Version 3, 1999. http://www.henrys-law.org
- R. Atkinson, D. L. Baulch, R. A. Cox, J. N. Crowley, R. F. Hampson, R. G. Hynes, M. E. Jenkin, M. J. Rossi, J. Troe and T. J. Wallington, “Evaluated Kinetic and Photochemical Data for Atmospheric Chemistry: Volume IV—Gas Phase Reactions of Organic Halogen Species,” Atmospheric Chemistry and Physics, Vol. 8, No. 15, 2008, pp. 4141-4496. doi:10.5194/acp-8-4141-2008