Journal of Diabetes Mellitus
Vol.05 No.04(2015), Article ID:61217,18 pages
10.4236/jdm.2015.54037
Genetic Engineering of Surrogate β Cells for Treatment of Type 1 Diabetes Mellitus
Andrew M. Handorf, Hans W. Sollinger*, Tausif Alam
Department of Surgery, Division of Transplantation, University of Wisconsin-Madison, Madison, USA

Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 10 October 2015; accepted 15 November 2015; published 18 November 2015
ABSTRACT
Type 1 diabetes mellitus (T1DM) is an autoimmune disease resulting from the destruction of the insulin-producing β cells of the pancreas. While treatment options like daily insulin injections or transplantation of whole-pancreas exist, they are associated with significant drawbacks. As a result, there has been great interest in engineering surrogate β cells, both ex vivo and in situ, to replace the function of those cells lost during the progression of the disease. However, the β cell is highly specialized and extraordinarily adept at synthesizing and rapidly secreting the appropriate amount of insulin in response to even small increases in blood glucose levels. Thus, genetic engineering of the “perfect” β cell may prove impossible. In this review, we will detail the features of β cells that make them so proficient at regulating blood glucose and highlight the key features that absolutely must be met by surrogate β cells if they are to be suitable for treatment of T1DM. Then, we will summarize the current approaches used to genetically engineer surrogate β cells, including the overexpression of β cell-specific transcription factors and insulin gene therapy. Along the way, we will discuss the advantages and disadvantages of each approach and review important studies in the field. Lastly, we will discuss important future directions necessary to genetically engineer surrogate β cells with the potential to treat T1DM.
Keywords:
Gene Therapy, Cell Therapy, Insulin, Type 1 Diabetes Mellitus, Hepatocytes

1. Introduction
Type 1 diabetes mellitus (T1DM) is an autoimmune disorder whereby the insulin-secreting β cells of the pancreas are destroyed. Under physiological conditions, β cells sense and respond to ever-changing blood glucose levels by secreting insulin, which acts on other tissues to promote glucose uptake from the blood and thus lower blood glucose levels. Without sufficient numbers of functional β cells, insulin production becomes inadequate and hyperglycemia ensues. Over time, chronic or poorly controlled hyperglycemia leads to numerous secondary complications, ultimately causing widespread tissue/organ damage and increased mortality. In fact, diabetes is the leading cause of end-stage kidney failure, retinopathy and associated blindness, non-traumatic amputations, and a variety of debilitating neuropathies. Despite active research, there is currently no cure for T1DM.
While there is currently no cure, several therapies exist to control blood glucose levels and limit adverse systemic damages resulting from sustained hyperglycemia. The most common form of treatment involves injection of synthetic insulin. Unfortunately, this option is cumbersome and unable to precisely control blood glucose levels. As a result, exogenous insulin therapy delays the onset and reduces the severity of secondary complications but is unable to prevent them [1] . In addition, exogenous insulin therapy is associated with hypoglycemia, which can lead to seizures, coma, or death. To better imitate normal physiology, specialized insulin pumps, dubbed artificial pancreata, have been developed that use a computer algorithm to deliver insulin [2] [3] or both insulin and glucagon [4] when needed by continuously monitoring blood glucose levels. Using this technology, better glycemic control can be attained compared to traditional insulin therapies. However, a variation in sensitivity with time and longevity of the implanted glucose sensor in vivo remain hurdles, as the glucose sensor will invariably accumulate serum or extracellular matrix proteins that compromise glucose measurements and subsequently affect the amount of insulin delivered. Ultimately, exogenous insulin therapy provides a suboptimal therapy for T1DM. In an effort to establish tighter control of blood glucose levels, whole-pancreas [5] or, to a lesser degree, pancreatic islet transplantation [6] surgeries have proven effective. However, pancreas transplantation therapy is severely hampered by a shortage of donor organs and further complicated by the need for lifelong immunosuppression. Transplantation of pancreatic islets was anticipated to minimize the impact of donor shortage, as islets from one donor could be expanded ex vivo to a quantity sufficient for multiple recipients. Unfortunately, equivalent successes like those observed with whole-pancreas transplantation have yet to be obtained [7] . Hence, there is clearly a need for alternative, state-of-the-art therapies. To that end, there have been considerable efforts to genetically engineer surrogate β cells to replace the function of those cells that were lost during the progression of T1DM.
In this review, we will begin by highlighting the unique features of β cells that make them so adept at controlling hyperglycemia. We will then discuss which of these features are absolutely critical to engineer a surrogate β cell capable of adequately controlling glycemia and describe the current efforts to genetically engineer surrogate β cells. Discussion on efforts to genetically engineer surrogate β cells will include insulin gene therapy―en- compassing various strategies to force the expression of insulin in non-β cells―and generation of β cells from both somatic and stem cells through expression of β cell-specific transcription factors. Along the way, we aim to point out advantages and disadvantages of each approach and ultimately propose the minimum requirements necessary for genetically engineering surrogate β cells with the potential to treat T1DM.
2. Features of the β Cell
In order to engineer a surrogate β cell, it is important to understand the unique features of β cells that make them so adept at controlling glycemia and determine which of these features are absolutely critical to adequately perform its physiological function. The β cell possesses several remarkable features that together allow it to precisely control blood glucose levels. Specifically, the β cell has the ability to regulate insulin production at the transcriptional, post-transcriptional, translational, and post-translational levels, as well as the ability to store and secrete insulin in a highly regulated fashion in response to a variety of different stimuli, most notably glucose. While no individual feature is sufficient in itself to control blood glucose levels, nor specific to β cells, the combination of features makes it a remarkably sophisticated and competent cell for its task―one that has proven quite difficult to engineer identically.
First and foremost, the β cell is capable of sensing and responding to small changes in circulating glucose levels over a broad range of physiological concentrations (2 - 20 mM) through concentration-dependent entry and metabolism of glucose. They do so through expression glucose transporter-2 (GLUT2) and glucokinase. GLUT2 is a transmembrane carrier protein that enables bidirectional glucose transport across cell membranes, whereas glucokinase is an intracellular enzyme that initiates metabolism of glucose into glucose-6-phosphate. The critical feature of GLUT2 and glucokinase is their relatively high Km for glucose (~17 and 8 mM, respectively), which allows their output to vary substantially across a broad range of glucose concentrations and thus serve as “glucose sensors” for β cells [8] . Without them, insulin secretion from β cells would be more like an “on-off switch” instead of a finely tuned trigger.
More remarkably, β cells have the ability to secrete insulin in a precisely regulated fashion in response to elevations in plasma glucose levels, as well as other secretagogues, to maintain euglycemia. β cells do so through their capacity to: 1) synthesize, process and store large quantities of insulin within secretory vesicles and 2) generate secondary stimulus-secretion coupling signals to activate rapid insulin vesicle exocytosis. It has been shown in rat β cells that >99% of all newly synthesized proinsulin molecules are packaged into secretory vesicles (see Figure 1), creating an enormous depot of insulin for rapid, on-demand secretion, which can occur within a minute after exposure to elevated glucose levels [9] . Further, β cells possess a unique combination of metabolic enzymes that ultimately leads to the generation of signals that adjust insulin secretion in response to glucose metabolism. β cells, but not other mammalian cell types, have negligible lactate dehydrogenase activity, so the pyruvate produced from glycolysis is not metabolized to lactate [10] . To compensate, β cells display increased pyruvate carboxylase activity to direct pyruvate almost entirely toward the mitochondria, where it is metabolized via the tricarboxylic acid cycle and oxidative phosphorylation. In so doing, β cells increase their ATP/ADP ratio and subsequently activate ATP-sensitive K+ channels, a key stimulus leading to insulin vesicle exocytosis. There is also a marked increase in mitochondrial metabolic shuttle activities to reoxidize cytosolic NADH to NAD+, a requirement normally provided by lactate dehydrogenase [11] . Increases in NADPH levels serve as another glucose-induced stimulus leading to insulin vesicle exocytosis [12] [13] . Together, the β cell has a unique balance of metabolic enzymes, structures, and mechanisms to generate the secondary stimulus-sec- retion coupling signals necessary for the almost instantaneous secretion of insulin upon exposure to secretagogues (see Figure 2 for schematic).
In addition to glucose, other circulating metabolites greatly influence β cell function. First, the gut produces peptide hormones, termed incretins, which serve as a line of communication between the gut and endocrine pancreas to augment post-prandial insulin production from β cells. These incretins, which include glucose-de- pendent insulinotropic polypeptide (GIP) and glucagon-like peptide-1 (GLP1), bind specific receptors found predominantly on β cells to bolster insulin production [14] . Second, specific amino acids and free fatty acids can serve as insulin secretagogues. Although some amino acids and free fatty acids can, in certain combinations, actively promote insulin secretion, they have a more prominent role in amplifying the stimulatory effects of glucose [15] . Third, neuronal input can influence insulin secretion. In particular, metabolic stress can induce neuronal signals that affect insulin output [16] . Together, these inputs attenuate insulin output from β cells to more precisely regulate glucose levels.
 (a) (b)
(a) (b)
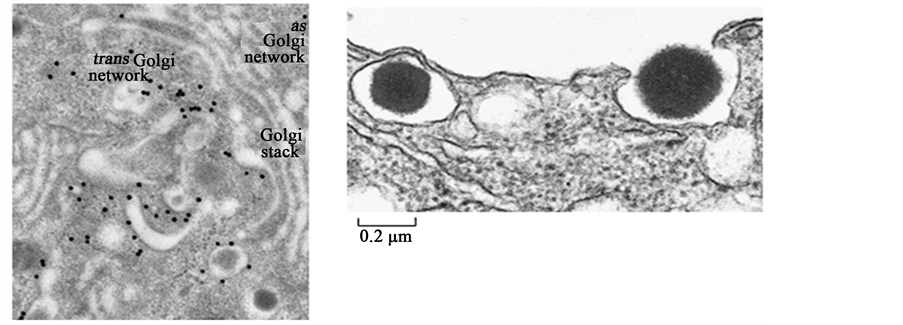
Figure 1. (a) Electron micrograph showing secretory vesicles forming within a β cell. An antibody conjugated to gold particles (black dots) was used to locate clathrin molecules, which are only found on immature secretory vesicles containing proinsulin. Upon maturation, secretory vesicles rapidly shed the clathrin coat. (b) Electron micrograph showing insulin being released from a β cell. The secretory vesicle on the left is about to fuse with the β cell membrane, while the granule on the right is being released into the extracellular space. Each secretory vesicle holds roughly 800,000 insulin molecules on average. Reproduced with modifications from Sci. Am. 256: 85-94, 1988.
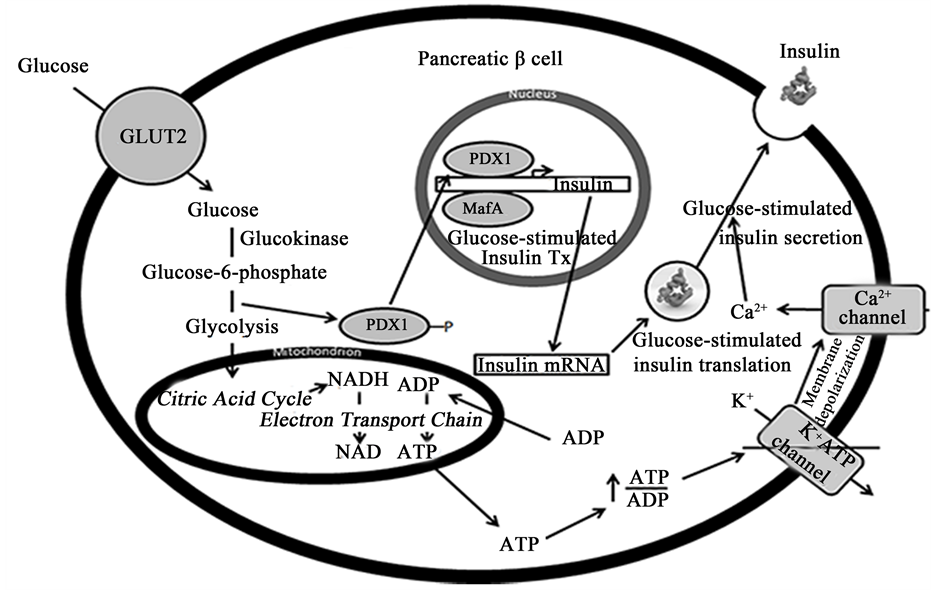
Figure 2. Schematic showing the various glucose-inducible mechanisms within a β cell to promote insulin production and secretion. β cells sense elevated blood glucose levels through GLUT2 and glucokinase. GLUT2 transports glucose into the cell, where it is metabolized into glucose-6-phosphate by glucokinase. Glucose-6-phosphate enters glycolysis, where it is converted to pyruvate. Pyruvate is then converted to acetyl-CoA, which enters the citric acid cycle within mitochondria to produce NADH. NADH is fed into the electron transport chain, where it is oxidized to NAD+, a stimulus leading to insulin vesicle exocytosis. ADP is also converted to ATP in the process, which increases the ratio of ATP/ADP and subsequently closes ATP-sensitive K+ channels. This leads to membrane depolarization and the opening of Ca2+ channels, which promotes insulin secretion. At the same time, increased glucose metabolism leads to the dephosphorylation of PDX1, which causes it to translocate into the nucleus and activate insulin transcription cooperatively with MafA. Glucose metabolism similarly increases insulin mRNA stability and translation to maintain insulin stores during periods of great demand. Abbreviations: GLUT2, glucose transporter-2; PDX1, pancreatic and duodenal homeobox 1; Tx, transcription.
Furthermore, β cells control insulin biosynthesis at the transcriptional, post-transcriptional, translational, and post-translational levels, with each level of control being regulated by glucose availability. β cells do not respond to glucose availability per se but to changes in the rate of glucose metabolism. At the transcriptional level, two transcription factors―PDX1 and MafA―have been identified as crucial regulators that promote insulin gene expression in response to altered glucose metabolism [17] [18] . Namely, it has been reported that PDX1 gets phosphorylated when the concentration of glucose is low, leading to its cytosolic localization [19] [20] . When glucose concentrations become elevated, PDX1 gets dephosphorylated and works cooperatively with MafA and BETA2 to promote insulin transcription [18] .
At the post-transcriptional and translational levels, β cells have glucose-dependent mechanisms to attenuate insulin mRNA stability and the rate of translation, respectively. Insulin mRNA has been estimated to be ~30% of the total mRNA within β cells, and its half-life can vary from anywhere between 29 to 77 hours depending on glucose availability [21] [22] . Association of an RNA-binding protein known as polypyrimidine tract-binding protein (PTB) with a pyrimidine-rich stretch in the 3’ UTR of the insulin mRNA has been shown to be responsible for glucose-dependent changes in its stability [23] [24] . However, overall changes in insulin production increase more dramatically due to glucose-induced changes in insulin mRNA translation. Wicksteed et al. found that glucose-stimulated increases in translation occur through the cooperative activities of a stem-loop at the 5’ UTR and the previously mentioned pyrimidine-rich stretch (UUGAA) in the 3’ UTR [24] . Sequences in the 5’ UTR have similarly been identified as important for glucose-induced increases in insulin mRNA translation [25] [26] , and protein-disulfide isomerase (PDI) has been identified as a binding protein responsible for glucose- stimulated insulin biosynthesis [27] .
Lastly, β cells possess specific post-translational machinery to process proinsulin―a precursor form―into fully active insulin. Proinsulin conversion involves removal of two basic pairs of amino acids―the C-peptide― and is mediated by the β cell endoproteases, PC1/3 and PC2, and the exopeptidase, carboxypeptidase-H [28] [29] . Proinsulin processing occurs within secretory vesicles once they have become acidified in the presence of high concentrations of Ca2+ [30] . Importantly, the glucose-induced increase in proinsulin biosynthesis is matched by a similar increase in the biosynthesis of PC1/3 and PC2 (among other proteins associated with secretory granules), which ensures proper processing of the bulk of proinsulin molecules synthesized [31] .
3. Minimum Requirements of a Surrogate β Cell
Combining all the unique features of the β cell, the end result is a cell with impressive sensitivity to glucose and the ability to modulate insulin secretion almost instantaneously in response to glucose. In addition, the β cell is able to maintain relatively constant stores of insulin through glucose-mediated control of insulin biosynthesis at several levels. So, beyond the ability to express insulin itself (a feat that could conceivably be achieved in any cell type using a strong constitutive promoter), which of these features are absolutely critical in a surrogate β cell and which of these features can be bypassed (see Table 1)?
Given that glucose is the primary stimulus modulating insulin secretion from β cells and the primary effector of insulin action, the impressive glucose sensitivity conferred by GLUT2 and glucokinase is an absolutely critical feature that must be present in an engineered surrogate β cell. Without these proteins, a surrogate β cell would not be able to precisely “sense” glucose concentration and control insulin output over a broad range of circulating glucose concentrations. The response of the surrogate cell would instead be unregulated. Although the expression of GLUT2 and glucokinase is somewhat restricted to β cells, other cells like hepatocytes and certain cells of the gut and hypothalamus also express both proteins. Of course, either or both of these proteins

Table 1. Mechanisms to mimic β cell functionality.
Abbreviations: G6Pase, glucose-6-phosphatase; L-PK, L-pyruvate kinase; GLUT2, glucose transporter-2; IGFBP-1, insulin-like growth factor binding protein-1; PEPCK, phosphoenolpyruvate carboxykinase; ER, endoplasmic reticulum; PC, prohormone convertase; PTB, polypyrimidine tract-binding protein; UTR, untranslated region.
could be co-expressed with insulin to create a surrogate β cell with adequate glucose sensitivity, but this would greatly increase the complexity of genetic manipulation. Nonetheless, several groups have done so in vitro and in vivo. For instance, Callejas et al. enhanced the glucose sensing capacity of skeletal muscle through expression of glucokinase [32] . When co-expressed with insulin using an adeno-associated virus (AAV) in diabetic dogs, they were able to normalize fasting hyperglycemia and accelerate glucose disposal after oral challenge for over four years [33] . Similarly, Faradji et al. co-expressed GLUT2 and glucokinase with insulin in intermediate pituitary cells to confer glucose-sensing capabilities into these cells [34] . While their genetic manipulation conferred a degree of glucose-stimulated insulin secretion (GSIS), they more prominently observed defective glucose metabolism and severe apoptotic cell death. Thus, it is critical to not just express each component but to express them at physiological levels and take into consideration the balance of other metabolic enzymes to obtain a functional β cell [35] . Together, it would be most sensible to target a cell type that already expresses GLUT2 and glucokinase when engineering a surrogate β cell.
In addition to precise glucose sensitivity, a surrogate β cell must also be able to process proinsulin into fully active insulin. This is important because proinsulin has less than 10% of the biologic activity of fully processed insulin [36] . Processing of wild-type proinsulin requires the β cell prohormone convertases, PC1/3 and PC2 [37] . However, these processing enzymes are only found in cells with the regulated secretory pathway, such as pituitary cells and intestinal K cells. An alternative mechanism to process proinsulin without the need for PC1/3 and PC2 is to mutate the preproinsulin sequence so that it can be cleaved by ubiquitously expressed endoproteases like furin [38] [39] . In so doing, any cell of the body could process proinsulin into fully active insulin. Another strategy employed by Lee et al. to bypass the necessity for enzymatic conversion into fully active insulin is to express a single-chain insulin analogue where 35 residues of the C-peptide were replaced by a short turn-forming heptapeptide [40] . Upon delivery of this single-chain insulin analogue using a rAAV, they were able to ameliorate hyperglycemia in streptozotocin (STZ)-induced and autoimmune diabetic rats for over eight months. Thus, while it is critical for a surrogate β cell to have the capacity to process proinsulin into fully active insulin, multiple mechanisms exist to bypass the need for expression of PC1/3 and PC2. In addition, the preproinsulin cDNA sequence can be mutated to increase or decrease the stability of insulin [38] [41] , providing greater flexibility in dosing for gene therapy applications.
A surrogate β cell must also have some mechanism(s) in place to translate glucose sensitivity into altered insulin production/secretion. As mentioned above, pancreatic β cells have several mechanisms in place to alter insulin production/secretion in response to glucose. Pancreatic β cells: 1) secrete insulin almost instantaneously from storage granules through stimulus-secretion coupling, 2) upregulate insulin transcription, 3) improve preproinsulin mRNA stability, and 4) increase preproinsulin mRNA translation upon increased glucose availability. Together, this enables the β cell to rapidly secrete insulin in response to elevated blood glucose levels and maintain insulin stores at a relatively constant level on a long-term scale. So does a surrogate β cell need to have all of these mechanisms in place to control blood glucose levels, or will a subset of these mechanisms suffice?
The ability to store secreted proteins is not exclusive to β cells but can also be found in all cell types involved in regulated secretion, such as neuroendocrine cells. However, it is debatable whether it is necessary to recapitulate the regulated secretory pathway, which enables β cells to secrete insulin almost immediately upon stimulation with glucose. Keep in mind that a transient hyperglycemia alone is not life-threatening. Diabetes is such a devastating illness because of the secondary complications that arise as a consequence of sustained hyperglycemia. Thus, as long as a surrogate β cell has the ability to produce appropriate amounts of insulin within a reasonable time frame, if perhaps not immediately, in response to elevated glucose concentrations, the kinetics of insulin secretion are not that critical. The quantity of insulin output must simply be large enough to correct hyperglycemia, but not too large as to induce hypoglycemia, a potentially life-threatening consequence. Thus, any glucose-responsive mechanism capable of precisely augmenting insulin production/secretion could be a good starting point for engineering a surrogate β cell, as long as basal, unstimulated insulin output does not induce hypoglycemia under fasting conditions. To endow cells with the regulated secretory pathway, Rivera et al. engineered an insulin protein that aggregates in the endoplasmic reticulum until stimulation with a small molecule that induces protein disaggregation [42] . In so doing, they were able to generate rapid and pulsatile secretion of insulin in vivo and transiently correct diabetic hyperglycemia. However, this manipulation lacks glucose- responsiveness.
Another possible mechanism to control insulin output would be to endow a surrogate β cell with glucose- responsive control of insulin transcription. β cells achieve this through glucose-regulated changes in the intracellular localization and activity of key transcription factors, namely PDX1 and MafA. However, other cells have distinct glucose-responsive mechanisms that could be exploited to alter insulin transcription in a surrogate β cell. For instance, hepatocytes increase the production of enzymes necessary for de novo lipogenesis in response to elevated glucose concentrations [43] [44] . They do so through binding of carbohydrate response elements (ChoREs) by a glucose-regulated transcription factor, known as carbohydrate response element binding protein (ChREBP). ChoREs are composed of two 6-bp motifs with the consensus sequence CACGTG and are found in the promoters of several genes encoding lipogenic enzymes, like L-type pyruvate kinase (L-PK), S14, acetyl-CoA carboxylase, and fatty acid synthase [45] . This mechanism of glucose-regulatable transcriptional control is best understood in hepatocytes but is likely also shared by cells of the small intestine and fat. Importantly, it has been utilized in several instances to control insulin output from surrogate β cells [46] - [51] , as discussed below. Alternatively, Auricchio and colleagues used the dimerizer drug rapamycin to provide pharmacological induction of insulin expression in hepatocytes [52] .
Another mechanism to control insulin output would be to incorporate sequences into the preproinsulin cDNA that lead to glucose-responsive changes in mRNA stability. Namely, it would be advantageous to incorporate sequences that lead to improved proinsulin mRNA stability when blood glucose levels are high and mRNA destabilization at low glucose concentrations. One way to do so would be to incorporate PTB binding sequences into the 3’ UTR of the cDNA construct. Conveniently, PTB is ubiquitously expressed across many (if not all) cell types and represents a common mediator of mRNA stability. An example can be found in hepatocytes, where PTB stabilizes albumin mRNA by binding an identical sequence in the 3’ UTR [53] . This is particularly important for the creation of surrogate β cells, as the half-life of preproinsulin mRNA has been reported to be less than six hours in hepatocytes [24] . The incorporation of PTB binding sites would also endow a surrogate β cell with glucose-responsive control of mRNA translation [24] . Thus, multiple levels of glucose-responsive control could be attained through this mechanism.
Overall, a surrogate β cell should, at minimum, be able to synthesize and secrete fully-processed insulin in a glucose-responsive fashion. This would require the ability to sense small changes in glucose concentration (i.e. express GLUT2 and glucokinase), respond to those changes by altering insulin output (either through altered biosynthesis or secretion), and process proinsulin into fully active insulin. Of utmost importance, constitutive insulin secretion should be limited to avoid the possibility of hypoglycemia during fasting periods, and in theory, a truly glucose-responsive system would be effective at preventing constitutive insulin release that exceeds basal needs.
4. Genetic Engineering Strategies
The process of genetically engineering surrogate β cells begins by delivering suitably designed genes into target cells. Many gene delivery tools exist to accomplish this task and can be broadly grouped into two categories: viral and non-viral. Non-viral methods have the advantage of being less immunogenic. However, non-viral methods typically only drive transgene expression transiently, as most do not have a mechanism to induce chromosomal integration. In addition, they tend to deliver genes inefficiently in vivo. Viral methods, on the other hand, deliver transgenes with greater efficiency but will often elicit some degree of immune response. Several viruses have been used for the purpose of generating surrogate β cells, with each possessing their own unique features.
Adenoviruses were among the first viral vectors used for the generation of surrogate β cells due to their ability to transduce non-dividing cells with high efficiency. In addition, adenoviral vectors can be produced at very high titers relatively easily, enabling the type of production necessary for translating a treatment to the clinic. However, adenoviruses are unable to integrate their genetic cargo into the genome and thus provide only transient gene expression [54] [55] . Studies using adenoviral vectors to deliver the insulin gene into diabetic animals generally noted a correction of hyperglycemia for only a month [56] . Thus, repeated injection of adenoviral vectors would be necessary to sustain normoglycemia. Unfortunately, the immune response elicited by the first treatment precludes effective repeated adenovirus-based treatments. In fact, adenoviruses elicit such a profound immune response that their administration led to the death of a patient in 1999 [57] [58] . Thus, while adenoviruses are extremely efficient gene delivery tools, they are not well suited as a therapeutic tool for treatment of T1DM.
To combat issues regarding immunogenicity and short-term expression, AAVs have been used. AAVs are able to transduce both dividing and non-dividing cells and are able to integrate transgenes into the host genome at a specific site on chromosome 19, enabling long-term transgene expression [59] . The primary disadvantage of using AAVs is that their packaging capacity is less than 5 kb, limiting the use of larger expression cassettes with greater complexity for regulated expression. Furthermore, they cannot be produced in as high a titer as adenoviruses, limiting their utility as a therapeutic tool. Nonetheless, AAVs have been used successfully to generate surrogate β cells. Park et al. used a rAAV to deliver furin-cleavable insulin under control of the CMV promoter into STZ-induced diabetic rats and found improved glucose tolerance (at 2 g/kg) comparable to that of non-dia- betic control rats [60] . Additionally, they observed less inflammation and fewer side effects using rAAV when compared to the same treatment using adenoviral vectors.
Retroviral vectors are perhaps the most widely used gene delivery vector, owing to their natural ability to integrate their genetic cargo into a host’s genome and obtain sustained gene expression. However, retroviral integration has been shown to have some preference to occur in close proximity to the transcriptional regulatory sequences of proto-oncogenes, as observed in 1999 following the treatment of nine severe-combined immunodeficiency patients [61] . Insertional mutagenesis led to the development of leukemia in four of the nine patients, and as cautionary measure, the field of clinical gene therapy was temporarily halted. Retroviral vectors are also limited by their inability to transduce non-dividing cells, a problem that severely hinders their utility for the production of surrogate β cells from quiescent cells. Accordingly, retroviral vectors have only been used sparingly in insulin gene therapy applications, and in these cases, hepatocyte proliferation must first be stimulated before retrovirus infusion. For instance, Muzzin and colleagues performed a partial hepatectomy before using retroviral vectors to deliver furin-cleavable preproinsulin-1 into STZ-induced diabetic rats [62] . Remarkably, they were able to attain near normoglycemia, but the necessity for hepatocyte proliferation precludes the translation of retroviral vectors in humans.
Lentiviral vectors provide a particularly attractive gene delivery vehicle to genetically engineer surrogate β cells, owing to their ability to transduce dividing and non-dividing cells and integrate their cargo into the host’s genome. Lentiviral vectors are a type of retrovirus and thus share some of the same concerns of insertional mutagenesis. However, lentiviral vectors display a more random integration profile with less preference to integrate near regulatory sequences of proto-oncogenes than retroviral vectors, reducing the risk of insertional mutagenesis [63] [64] . Additionally, lentiviral vectors do not elicit a strong immune response. Unfortunately, it remains a challenge to produce lentiviral vectors in high enough titer for clinical application [65] . Nonetheless, lentiviral vectors offer stable, long-term transgene expression with an improved biosafety profile and are thus an excellent candidate as a therapeutic tool for treatment of T1DM. Ren and colleagues used lentiviral vectors to deliver furin-cleavable insulin to the livers of STZ-induced diabetic rats [66] and non-obese diabetic mice [67] . In each case, correction of diabetic hyperglycemia was observed for at least five months post-treatment with no evidence of impaired liver function, intrahepatic inflammation, or recurring autoimmunity against the newly-formed insulin-producing cells.
5. Engineering of Surrogate β Cells
Given the prevalence and gravity of diabetes, significant research efforts have been put forth to treat the disease. Some of these efforts have been aimed at preventing the autoimmune destruction of native β cells [68] - [71] , whereas a more significant portion of research has been aimed at generating surrogate β cells, either through expansion of remaining β cells in vitro [72] , differentiation of stem cells into β cells [73] - [75] , or genetic modification. Indeed, attempts to differentiate embryonic stem cells (ESCs) into β cells have been quite successful but are outside the scope of this review [76] . Here, we will review the different strategies used to genetically engineer surrogate β cells, beginning with the overexpression of β cell-specific transcription factors and concluding with insulin gene therapy.
5.1. Overexpression of β Cell-Specific Transcription Factors
During development, β cells are derived from the endodermal germ layer. After endodermal specification, developing β cells are first specified to become pancreatic progenitors with the potential to generate ductal, acinar and endocrine cells, and then specified to become endocrine progenitors before becoming β cells. Throughout this process, specific transcription factors control each cell fate decision. Thus, an attractive strategy to produce surrogate β cells would be to overexpress β cell-specific transcription factors in non-β cells. Transcription factors play a critical role in executing differentiation programs during development by driving the expression of cell-specific genes. Further, transcription factors are important during adulthood for maintaining the differentiated status of somatic cells. Thus, groups have overexpressed β cell-specific transcription factors with the hopes of recapitulating key features of development to produce surrogate β cells that possess all the unique features of β cells, including the ability to synthesize, store and secrete insulin in response to glucose.
Pancreatic and duodenal homeobox 1 (PDX1), a transcription factor that regulates early pancreatic development and controls glucose-dependent insulin expression in β cells, has been shown to be a potent inducer of β cell fate when ectopically expressed. For example, Ferber et al. expressed PDX1 in the livers of diabetic mice using an adenovirus and observed insulin expression and secretion, as well as restoration of normoglycemia. However, they also observed the development of hepatitis and an increased likelihood of autoimmune destruction of the newly-formed β cells [77] . Similarly, other investigators have ectopically expressed PDX1 in various cell types of endodermal origin, including mouse pancreas [78] , rat intestinal epithelium-derived cells [79] , and primary duct cells [80] , with similar efficacy. PDX1 has also been used to differentiate stem cells from a variety of sources into surrogate β cells. Both mesenchymal (MSCs) [81] - [84] and embryonic stem cells (ESCs) [85] [86] have been used to generate surrogate β cells through overexpression of PDX1. For example, Raikwar and Zavazava generated surrogate β cells from mouse ESCs by constitutively overexpressing both PDX1 and insulin. These cells were capable of GSIS in vitro and corrected hyperglycemia when implanted into diabetic mice [87] .
In addition to PDX1, several other transcription factors implicated in β cell development have been tested for their ability to generate surrogate β cells. NeuroD1, a transcription factor of the helix-loop-helix family, is important for maintaining the endocrine cell fate during development and induces insulin expression in β cells. Co-expression of NeuroD1 and insulin in a rat liver cell line that does not otherwise express β cell transcription factors led to a surrogate β cell with the capacity to store insulin in secretory granules [88] . When transplanted into non-obese diabetic/SCID mice, this engineered cell line reversed diabetes and regulated insulin secretion in response to increasing concentrations of glucose. Expression of NeuroD1 also stimulated the expression of other β cell-specific transcription factors, including PDX1, Pax6, Nkx2.2, and Nkx6.1, thus demonstrating the promise of transcription factor-mediated generation of surrogate β cells. In addition, NeuroD1 has been shown to be a potent inducer of insulin expression in both primary duct cells and hepatocytes, further demonstrating its potential for production of surrogate β cells [89] [90] .
Neurog3 has also been tested for its ability to produce surrogate β cells, given its role in directing the endocrine pancreas fate. In fact, all hormone-producing cells of the pancreas express Neurog3, and Neurog3 has been shown to activate the persistent expression of NeuroD1. However, the use of Neurog3 in β cell engineering has been met with limited success [91] - [94] . On the other hand, co-delivery of Neurog3 with betacellulin, a growth factor important for β cell differentiation, resulted in the transdifferentiation of hepatic oval cells into insulin-producing cells [95] . Another study showed that co-delivery of Neurog3 with PDX1 and MafA, a transcription factor important for maintenance of β cell function, was successful in converting pancreatic exocrine cells into cells closely resembling β cells in vivo [96] . This suggests that while Neurog3 may be important in the engineering of surrogate β cells, it may not have the ability to efficiently instruct a β cell fate alone.
Once the endocrine cell fate has been determined, Pax4 is necessary for determining β and γ cell fates within the developing pancreas. Thus, researchers have explored the impact of Pax4 expression on the production of surrogate β cells. Overexpression of Pax4 in mouse ESCs was found to drive the production of surrogate β cells capable of maintaining normal blood glucose levels for 14 days after implantation into STZ-induced diabetic mice [97] . Together, these studies implicate Pax4 as another important transcription factor for the production of surrogate β cells.
Given its role in driving β cell differentiation, Nkx6.1 has also been overexpressed in an effort to generate surrogate β cells. However, this approach has been met with limited success. For instance, Gefen-Halevi et al. overexpressed Nkx6.1 but found that it alone was unable to induce insulin expression despite inducing the expression of immature pancreatic markers, like Neurog3 and Isl-1. However, when co-expressed with PDX1, it promoted reprogramming of liver cells to β cells capable of GSIS [98] . Regardless, Nkx6.1 does not appear to be an ideal candidate for production of surrogate β cells.
Overall, the overexpression of β cell-specific transcription factors provides an exciting strategy to engineer surrogate β cells. However, the translation of this strategy will greatly depend on whether the newly-formed surrogate β cells can evade recurring autoimmunity. This is particularly problematic, given the abundance of previously identified autoantigens implicated in leading to the autoimmune destruction of native β cells [99] . Furthermore, success will likely require the simultaneous overexpression of multiple transcription factors, which inevitably increases the complexity of such a therapy. Nonetheless, the overexpression of β cell-specific transcription factors has been met with a great deal of success to this point.
5.2. Insulin Gene Therapy
At the most basic level, β cells are essentially nothing more than extremely sophisticated insulin secretors. Thus, an alternative approach to generate surrogate β cells is to simply force the expression of insulin in non-β cells. In theory, this would greatly limit the threat of recurring autoimmunity, as fewer β cell-specific autoantigens would be present. Remarkably, this strategy was first employed more than 30 years ago when Nicolau and colleagues encapsulated a plasmid encoding the rat preproinsulin-I gene in liposomes and intravenously injected it in rats [100] . Indeed, they found that the liposomes were taken up by the spleen and liver, where it led to insulin production and a modest decrease in blood glucose levels. However, secretion of insulin was constitutive and unresponsive to glucose. Around the same time, Moore and coworkers expressed human proinsulin cDNA in the AtT-20 adenoma cell line derived from the mouse anterior pituitary, a cell endowed with regulated secretory mechanisms that ordinarily secretes adrenocorticotropic hormone [101] . In addition to simply producing insulin in a non-β cell, they showed that it was possible to have it stored in secretory vesicles, proteolytically processed into insulin-like molecules, and secreted upon stimulation with a cell-permeable cAMP analogue. Although these insulin-expressing pituitary cells showed little to no sensitivity to glucose as a secretagogue, implantantation into STZ-induced diabetic mice improved hyperglycemia to a small degree from the constitutive release of insulin [102] [103] . Co-transfection of these insulin-producing AtT-20 cells with GLUT2 conferred them with GSIS, although it was within a subphysiological range [104] . While these research efforts provided a nice proof- of-concept that insulin could be stored and released from non-β cells in a glucose-responsive fashion, implantation of these cells intraperitoneally led to the formation of tumor-like aggregates, emphasizing the importance of choosing the appropriate cell type for clinical translation.
While pituitary cell lines seem attractive for their inherent capacity to produce, sequester, process, and secrete proteins in a regulated fashion, they lack the glucose-sensing capacity of β cells. Therefore, use of such cell lines, besides the need for total isolation in vivo for safety reasons, cannot provide the normal profile of β cell-like glucose-responsiveness. Hepatocytes, on the other hand, express GLUT2 and glucokinase, enabling precise glucose sensitivity analogous to β cells. As a result, a variety of liver cell lines have been used to generate surrogate β cells. For instance, Simpson and colleagues used a human hepatoma cell line (HEP G2) to express the full-length human cDNA. Unexpectedly, their initial attempts resulted in the creation of a hepatoma cell line capable of synthesizing, storing, and secreting insulin in a semi-regulated manner, but the cells were unresponsive to glucose [105] . Co-transfection of these cells with GLUT2 endowed them with regulated, near-physio- logical secretion of (pro)insulin in response to glucose, although insulin was predominantly secreted via the constitutive pathway [106] .
A hepatocyte-derived cellular carcinoma cell line, HUH7, has also been used to genetically engineer surrogate β cells. Tuch and coworkers expressed insulin in these cells under the control of the cytomegalovirus (CMV) promoter and found that these cells synthesized, stored and secreted insulin in response to glucose in a rapid and near-physiological manner [107] . When transplanted into diabetic immunoincompetent mice, the cells normalized blood glucose levels before overcorrecting them due to uncontrolled proliferation. While it is unexpected that both the HEP G2 and HUH7 cells were able to store, process, and secrete insulin in a semi-regulated fashion, examination of the HUH7 cells uncovered the endogenous expression of NeuroD, a β cell-specific transcription factor, that was likely responsible for the presence of storage granules and the ability to process proinsulin to insulin. This feature is perhaps a product of these cells being cancer cell lines and thus possessing features of a common progenitor between β cells and hepatocytes.
Use of native hepatocytes to generate surrogate β cells offers several advantages. Namely, they: 1) are closely related to β cells developmentally, 2) play a pivotal role in glucose homeostasis, 3) express GLUT2 and glucokinase, 4) possess a robust capacity to synthesize and secrete proteins, and 5) are relatively easy to target for gene therapy applications (Table 2). One of the primary functions of the liver is to serve as a processing center for blood, and it has been shown that in certain cases, greater than 90% of infused viral particles are taken up by the liver within minutes after intravenous injection [108] . As a result, it has been the most commonly targeted organ for in vivo production of surrogate β cells. To improve liver targeting further, a variety of liver-specific
Table 2. Features of target cells for generation of surrogate β cells.
A “+” indicates that the cell type shares that property with β cells, whereas a “−” indicates that the cell type does not. Abbreviations: BMSC, bone marrow stromal cell; GLUT2, glucose transporter-2; PC, prohormone convertase.
promoters have been used to drive transgene expression in vivo. For instance, Chen et al. used a glucose-6- phosphatase (G6Pase) promoter to drive insulin production in cultured hepatoma cells and found that insulin production was modulated in response to alterations in both glucose and insulin levels [109] . Specifically, insulin strongly inhibited G6Pase promoter activity under low glucose conditions, whereas elevated glucose concentrations enhanced promoter activity [110] . The group then delivered the insulin gene to the liver of STZ-induced diabetic rats under the control of the G6Pase promoter and found that non-fasting hyperglycemia was significantly reduced, glucose utilization was accelerated after glucose challenge, and fasting glucose levels were within a normal range without hypoglycemia after a 16-hour fast.
Thule and colleagues used the insulin-like growth factor binding protein-1 (IGFBP1) promoter to direct liver-specific insulin expression. Like the G6Pase promoter, the IGFBP1 promoter is repressed by insulin, creating a feedback inhibition loop on insulin expression. However, IGFBP1 promoter activity is not enhanced by elevated glucose concentrations. To generate glucose-inducible insulin expression, they additionally incorporated glucose-inducible response elements (GIREs) from the L-PK gene and successfully generated a system whereby glucose stimulated and insulin repressed IGFBP1 promoter activity in vitro and in vivo, ultimately resulting in a treatment that enabled diabetic animals to maintain near-normoglycemia and weight gain without significant hypoglycemia [47] [48] [51] . Similarly, Burkhardt et al. employed the GLUT2 promoter to drive hepatic insulin gene expression in a glucose-inducible but insulin-repressive fashion and found a reduction in diabetic hyperglycemia [111] . However, while creating surrogate β cells that direct insulin expression in an insulin-repressive fashion may seem intuitive to prevent hypoglycemia, it should be noted that the activity of the wild-type insulin promoter used by native β cells is actually enhanced by insulin, creating a feed-forward system to quickly amplify insulin expression. Given the elegance and effectiveness with which nature operates, it would be difficult to envision a scenario where doing the opposite would be beneficial.
Accordingly, Hsu and coworkers used the rat insulin-1 promoter to drive hepatic insulin expression, thus producing a system with the potential to be activated by both glucose and insulin, similar to native β cells [112] . In so doing, they were successful at driving insulin secretion from Huh7 hepatoma cells in vitro in response to glucose and pharmacological activators of cAMP signaling. They were also able to augment insulin expression in vivo in response to glucose and theophylline and ameliorate hyperglycemia in STZ-induced diabetic mice. However, they did not test whether insulin activated transgene expression using this promoter. It is also unclear how this promoter was active in hepatocytes, which do not ordinarily express the β cell-specific transcription factors necessary to drive insulin expression. Regardless, hepatocytes do not possess enough of the β cell-spe- cific regulatory mechanisms to safely express insulin in a feed-forward manner with respect to insulin. Thus, for the sake of hepatic insulin gene therapy, it would be ideal to create a system that was unresponsive to insulin altogether.
In our lab, we created a simplified system to control hepatic insulin gene expression by incorporating the GIREs from the S14 promoter upstream of the albumin promoter, which alone is neither glucose- nor insulin- responsive [49] . Indeed, we found that this system was able to produce large amounts of glucose-inducible insulin in vitro and promote euglycemia in STZ-induced diabetic rats when fasted or fed ad libitum. Additionally, weight loss due to uncontrolled hyperglycemia was reversed in treated rats, and a glucose tolerance test revealed glucose-responsive increases in insulin production and accelerated glucose clearance. Lastly, treated rats displayed restoration of healthy liver function without any sign of hepatic inflammation [50] . Together, we created surrogate β cells with exceptional glucose sensitivity (due to the natural expression of GLUT2 and glucokinase from hepatocytes) and glucose responsive insulin expression from a hepatocyte-specific promoter that is capable of fully correcting diabetic hyperglycemia.
Another promising cell type for engineering surrogate β cells is the intestinal K cell. Like hepatocytes, intestinal K cells express glucokinase and GLUT2, giving them enhanced glucose sensitivity. Additionally, K cells express the proinsulin processing enzymes and have the machinery for regulated insulin secretion, similar to the aforementioned pituitary cells. Cheung and colleagues exploited these unique advantages to engineer surrogate β cells from K cells. To do so, they transfected a tumor-derived K cell line with the human insulin gene under the control of the GIP promoter, a K cell-specific promoter believed, albeit controversially, to be regulated by glucose [46] . They found that the cells expressed and processed insulin and secreted it in a glucose-dependent manner. They then generated transgenic mice expressing human insulin under control of the GIP promoter and found that insulin expression was targeted specifically to K cells. Further, these transgenic mice maintained normal fasting glucose levels and disposed of glucose in a normal fashion in response to an oral glucose challenge for up to three months after mice were rendered diabetic with STZ. Thus, K cells provide an exciting target for generation of surrogate β cells. However, the translation of this strategy to the clinic will be severely hampered by the following concerns: 1) The gut is one of the primary hubs for the immune system, and given that insulin itself is an autoantigen that has been shown to lead to the initial destruction of pancreatic β cells [113] , intestinal K cells may be particularly susceptible to recurring autoimmune attack. 2) More importantly, the GIP promoter is not only regulated by glucose but also by other nutrients―most notably fats; thus, meals lacking carbohydrates but rich in fats would lead to the unnecessary secretion of insulin and very real possibility of severe hypoglycemia. Patients receiving this treatment would, at the very least, need to pay extremely close attention to every nutrient ingested to avoid potentially fatal consequences. Further, another study found that glucose alone does not even regulate insulin secretion in these surrogate β cells [114] .
6. Conclusions and Future Directions
All current therapies for T1DM possess significant drawbacks. In addition to effectively correcting hyperglycemia through precise glucose-responsiveness, a treatment for T1DM should be safe, affordable, readily available, and long-lasting without the need for lifelong immunosuppression. The genetic engineering of surrogate β cells provides an exciting alternative to treat T1DM with the potential to meet all of these needs. For instance, by delivering a glucose-responsive insulin expression cassette into a cell type with GLUT2 and glucokinase expression using an integrating viral vector, long-term correction of diabetic hyperglycemia could be attained. However, if such a therapy is to have success in the clinic, several challenges must be addressed.
Lentiviral vectors provide a promising delivery tool for the production of surrogate β cells but are severely hampered by our inability to produce them in high enough titer to affordably translate this technology to the clinic. In addition, while lentiviral vectors have the capacity to transduce non-dividing cells, transduction efficiency has been shown to be greater in cells that are actively dividing [115] . Thus, improvements to lentiviral vector design that enhance transduction efficiency and/or transgene expression would reduce the necessity for large viral doses. This is particularly important for insulin gene therapy because the insulin molecule has a relatively short circulating half-life, estimated at around 4 - 6 minutes [116] . Adenoviral vectors, on the other hand, are able to be produced in extremely high titers and are very efficient at transducing non-dividing cells but are only capable of producing transient transgene expression. As a result, they do not provide a viable option for the production of surrogate β cells. However, recent advances have allowed researchers to endow adenoviruses with the ability to integrate their cargo by incorporating the Sleeping Beauty transposase system into the expression cassette [117] . To overcome or minimize immunity to adenoviral vectors, a newer version of “gutted” adenovirus with minimal wild-type adenovirus sequences is currently being used in a variety of human clinical trials.
An exciting option to generate surrogate β cells that closely mimic native β cells is through forced expression of β cell-specific transcription factors. However, this strategy is greatly limited by concerns of recurring autoimmunity, as the newly-formed β cells will inevitably express a more expansive repertoire of β cell proteins, including autoantigens. To protect against autoimmunity, successful execution of this strategy will ultimately require immunosuppression, especially if the newly-formed β cells are generated ex vivo. An alternative strategy to protect ex vivo-generated β cells from autoimmune destruction would be to implant them in an immunoprotective implantation device. However, no biomaterial is completely inert, and proteins from serum and extracellular matrix will accumulate on the implantation device over time, which will affect diffusion of nutrients and waste products into and out of the device. More importantly, this will affect the precision of glucose sensitivity and insulin secretion over time, as well.
Lastly, it will be essential to get sufficient glucose-induced insulin expression from surrogate β cells to precisely control glucose metabolism. For insulin gene therapy, this requires glucose-regulatable expression of insulin in a cell type that is capable of this task. We have achieved this by restricting insulin expression to hepatocytes by using the albumin promoter and incorporating GIREs from the S14 gene to amplify insulin expression in response to elevated glucose levels. Using the relatively weak albumin promoter has the added advantage of reducing basal insulin expression, thus reducing the risk of hypoglycemic episodes. Similarly, other groups have devised glucose-regulatable strategies to produce surrogate β cells of clinical relevance. Ultimately, the success of such a strategy will hinge on the amplitude of glucose induction and ability to reproducibly generate a therapeutic quantity of surrogate β cells. As the current gene delivery tools continue to advance, these goals will become more attainable for treatment of T1DM.
Cite this paper
Andrew M. Handorf,Hans W. Sollinger,Tausif Alam, (2015) Genetic Engineering of Surrogate β Cells for Treatment of Type 1 Diabetes Mellitus. Journal of Diabetes Mellitus,05,295-312. doi: 10.4236/jdm.2015.54037
References
- 1. The Diabetes Control and Complications Trial Research Group (1993) The Effect of Intensive Treatment of Diabetes on the Development and Progression of Long-Term Complications in Insulin-Dependent Diabetes Mellitus. The New England Journal of Medicine, 329, 977-986.
http://dx.doi.org/10.1056/NEJM199309303291401 - 2. Hovorka, R., Kumareswaran, K., Harris, J., Allen, J.M. and Elleri, D. (2011) Overnight Closed Loop Insulin Delivery (Artificial Pancreas) in Adults with Type 1 Diabetes: Crossover Randomised Controlled Studies. BMJ, 342, d1855.
http://dx.doi.org/10.1136/bmj.d1855 - 3. Hovorka, R. (2011) Closed-Loop Insulin Delivery: From Bench to Clinical Practice. Nature Reviews Endocrinology, 7, 385-395.
http://dx.doi.org/10.1038/nrendo.2011.32 - 4. El-Khatib, F.H., Russell, S.J., Nathan, D.M., Sutherlin, R.G. and Damiano, E.R. (2010) A Bihormonal Closed-Loop Artificial Pancreas for Type 1 Diabetes. Science Translational Medicine, 2, 27ra27.
http://dx.doi.org/10.1126/scitranslmed.3000619 - 5. Kelly, W.D., Lillehei, R.C., Merkel, F.K., Idezuki, Y. and Goetz, F.C. (1967) Allotransplantation of Pancreas and Duodenum Along with Kidney in Diabetic Nephropathy. Surgery, 61, 827-&.
- 6. Paty, B.W., Ryan, E.A., Shapiro, A.M., Lakey, J.R. and Robertson, R.P. (2002) Intrahepatic Islet Transplantation in Type 1 Diabetic Patients Does Not Restore Hypoglycemic Hormonal Counterregulation or Symptom Recognition after Insulin Independence. Diabetes, 51, 3428-3434.
http://dx.doi.org/10.2337/diabetes.51.12.3428 - 7. Shapiro, A.M., Lakey, J.R., Ryan, E.A., Korbutt, G.S. and Toth, E. (2000) Islet Transplantation in Seven Patients with Type 1 Diabetes Mellitus Using a Glucocorticoid-Free Immunosuppressive Regimen. The New England Journal of Medicine, 343, 230-238.
http://dx.doi.org/10.1056/NEJM200007273430401 - 8. Newgard, C.B. and McGarry, J.D. (1995) Metabolic Coupling Factors in Pancreatic Beta-Cell Signal Transduction. Annual Review of Biochemistry, 64, 689-719.
http://dx.doi.org/10.1146/annurev.bi.64.070195.003353 - 9. Wollheim, C.B. and Sharp, G.W.G. (1981) Regulation of Insulin Release by Calcium. Physiological Reviews, 61, 914-973.
- 10. Ishihara, H., Wang, H., Drewes, L.R. and Wollheim, C.B. (1999) Overexpression of Monocarboxylate Transporter and Lactate Dehydrogenase Alters Insulin Secretory Responses to Pyruvate and Lactate in Beta Cells. The Journal of Clinical Investigation, 104, 1621-1629.
http://dx.doi.org/10.1172/JCI7515 - 11. Schuit, F., De Vos, A., Farfari, S., Moens, K. and Pipeleers, D. (1997) Metabolic Fate of Glucose in Purified Islet Cells. Glucose-Regulated Anaplerosis in Beta Cells. The Journal of Biological Chemistry, 272, 18572-18579.
http://dx.doi.org/10.1074/jbc.272.30.18572 - 12. Ronnebaum, S.M., Ilkayeva, O., Burgess, S.C., Joseph, J.W. and Lu, D. (2006) A Pyruvate Cycling Pathway Involving Cytosolic Nadp-Dependent Isocitrate Dehydrogenase Regulates Glucose-Stimulated Insulin Secretion. The Journal of Biological Chemistry, 281, 30593-30602.
http://dx.doi.org/10.1074/jbc.M511908200 - 13. Ivarsson, R., Quintens, R., Dejonghe, S., Tsukamoto, K., in’t Veld, P., Renstrom, E. and Schuit, F.C. (2005) Redox Control of Exocytosis: Regulatory Role of Nadph, Thioredoxin, and Glutaredoxin. Diabetes, 54, 2132-2142.
http://dx.doi.org/10.2337/diabetes.54.7.2132 - 14. McIntosh, C.H.S., Widenmaier, S. and Kim, S.J. (2009) Glucose-Dependent Insulinotropic Polypeptide (Gastric Inhibitory Polypeptide; GIP). Vitamins & Hormones, 80, 409-471.
http://dx.doi.org/10.1016/S0083-6729(08)00615-8 - 15. Nolan, C.J. and Prentki, M. (2008) The Islet Beta-Cell: Fuel Responsive and Vulnerable. Trends in Endocrinology and Metabolism, 19, 285-291.
http://dx.doi.org/10.1016/j.tem.2008.07.006 - 16. Pongratz, R.L., Kibbey, R.G., Shulman, G.I. and Cline, G.W. (2007) Cytosolic and Mitochondrial Malic Enzyme Isoforms Differentially Control Insulin Secretion. The Journal of Biological Chemistry, 282, 200-207.
http://dx.doi.org/10.1074/jbc.M602954200 - 17. Melloul, D., Marshak, S. and Cerasi, E. (2002) Regulation of Insulin Gene Transcription. Diabetologia, 45, 309-326.
http://dx.doi.org/10.1007/s00125-001-0728-y - 18. Zhao, L., Guo, M., Matsuoka, T.A., Hagman, D.K. and Parazzoli, S.D. (2005) The Islet Beta Cell-Enriched Mafa Activator Is a Key Regulator of Insulin Gene Transcription. The Journal of Biological Chemistry, 280, 11887-11894.
http://dx.doi.org/10.1074/jbc.M409475200 - 19. Macfarlane, W.M., McKinnon, C.M., Felton-Edkins, Z.A., Cragg, H. and James, R.F. (1999) Glucose Stimulates Translocation of the Homeodomain Transcription Factor Pdx1 from the Cytoplasm to the Nucleus in Pancreatic Beta-Cells. The Journal of Biological Chemistry, 274, 1011-1016.
http://dx.doi.org/10.1074/jbc.274.2.1011 - 20. McKinnon, C.M. and Docherty, K. (2001) Pancreatic Duodenal Homeobox-1, Pdx-1, a Major Regulator of Beta Cell Identity and Function. Diabetologia, 44, 1203-1214.
http://dx.doi.org/10.1007/s001250100628 - 21. Welsh, M., Nielsen, D.A., MacKrell, A.J. and Steiner, D.F. (1985) Control of Insulin Gene Expression in Pancreatic Beta-Cells and in an Insulin-Producing Cell Line, Rin-5f Cells. Ii. Regulation of Insulin Mrna Stability. The Journal of Biological Chemistry, 260, 13590-13594.
- 22. Tillmar, L. and Welsh, N. (2002) Hypoxia May Increase Rat Insulin Mrna Levels by Promoting Binding of the Polypyrimidine Tract-Binding Protein (Ptb) to the Pyrimidine-Rich Insulin Mrna 3’-Untranslated Region. Molecular Medicine, 8, 263-272.
- 23. Tillmar, L. and Welsh, N. (2004) Glucose-Induced Binding of the Polypyrimidine Tract-Binding Protein (Ptb) to the 3’-Untranslated Region of the Insulin Mrna (Ins-Prs) Is Inhibited by Rapamycin. Molecular and Cellular Biochemistry, 260, 85-90.
http://dx.doi.org/10.1023/B:MCBI.0000026059.56089.e4 - 24. Wicksteed, B., Herbert, T.P., Alarcon, C., Lingohr, M.K. and Moss, L.G. (2001) Cooperativity between the Preproinsulin Mrna Untranslated Regions Is Necessary for Glucose-Stimulated Translation. Journal of Biological Chemistry, 276, 22553-22558.
http://dx.doi.org/10.1074/jbc.M011214200 - 25. Wicksteed, B., Uchizono, Y., Alarcon, C., McCuaig, J.F. and Shalev, A. (2007) A Cis-Element in the 5’ Untranslated Region of the Preproinsulin Mrna (Ppige) Is Required for Glucose Regulation of Proinsulin Translation. Cell Metabolism, 5, 221-227.
http://dx.doi.org/10.1016/j.cmet.2007.02.007 - 26. Muralidharan, B., Bakthavachalu, B., Pathak, A. and Seshadri, V. (2007) A Minimal Element in 5’utr of Insulin Mrna Mediates Its Translational Regulation by Glucose. FEBS Letters, 581, 4103-4108.
http://dx.doi.org/10.1016/j.febslet.2007.07.050 - 27. Kulkarni, S.D., Muralidharan, B., Panda, A.C., Bakthavachalu, B. and Vindu, A. (2011) Glucose-Stimulated Translation Regulation of Insulin by the 5’ Utr-Binding Proteins. Journal of Biological Chemistry, 286, 14146-14156.
http://dx.doi.org/10.1074/jbc.M110.190553 - 28. Hutton, J.C. (1994) Insulin Secretory Granule Biogenesis and the Proinsulin-Processing Endopeptidases. Diabetologia, 37, S48-S56.
http://dx.doi.org/10.1007/BF00400826 - 29. Steiner, D.F. (1998) The Proprotein Convertases. Current Opinion in Chemical Biology, 2, 31-39.
http://dx.doi.org/10.1016/S1367-5931(98)80033-1 - 30. Davidson, H.W., Rhodes, C.J. and Hutton, J.C. (1988) Intraorganellar Calcium and Ph Control Proinsulin Cleavage in the Pancreatic Beta-Cell Via 2 Distinct Site-Specific Endopeptidases. Nature, 333, 93-96.
http://dx.doi.org/10.1038/333093a0 - 31. Knoch, K.P., Bergert, H., Borgonovo, B., Saeger, H.D. and Altkruger, A. (2004) Polypyrimidine Tract-Binding Protein Promotes Insulin Secretory Granule Biogenesis. Nature Cell Biology, 6, 207-214.
http://dx.doi.org/10.1038/ncb1099 - 32. Mas, A., Montane, J., Anguela, X.M., Munoz, S. and Douar, A.M. (2006) Reversal of Type 1 Diabetes by Engineering a Glucose Sensor in Skeletal Muscle. Diabetes, 55, 1546-1553.
http://dx.doi.org/10.2337/db05-1615 - 33. Callejas, D., Mann, C.J., Ayuso, E., Lage, R. and Grifoll, I. (2013) Treatment of Diabetes and Long-Term Survival after Insulin and Glucokinase Gene Therapy. Diabetes, 62, 1718-1729.
http://dx.doi.org/10.2337/db12-1113 - 34. Faradji, R.N., Alcazar, O., Havari, E., Worku, A.D. and Tornheim, K. (2001) Glucose-Induced Toxicity in Bioengineered Surrogate Beta Cells. Diabetes, 50, A6-A6.
- 35. Halban, P.A., Kahn, S.E., Lernmark, A. and Rhodes, C.J. (2001) Gene and Cell-Replacement Therapy in the Treatment of Type 1 Diabetes—How High Must the Standards Be Set? Diabetes, 50, 2181-2191.
http://dx.doi.org/10.2337/diabetes.50.10.2181 - 36. Revers, R.R., Henry, R., Schmeiser, L., Kolterman, O. and Cohen, R. (1984) The Effects of Biosynthetic Human Proinsulin on Carbohydrate-Metabolism. Diabetes, 33, 762-770.
http://dx.doi.org/10.2337/diab.33.8.762 - 37. Newgard, C.B. (1994) Cellular Engineering and Gene-Therapy Strategies for Insulin Replacement in Diabetes. Diabetes, 43, 341-350.
http://dx.doi.org/10.2337/diab.43.3.341 - 38. Groskreutz, D.J., Sliwkowski, M.X. and Gorman, C.M. (1994) Genetically-Engineered Proinsulin Constitutively Processed and Secreted as Mature, Active Insulin. Journal of Biological Chemistry, 269, 6241-6245.
- 39. Yanagita, M., Hoshino, H., Nakayama, K. and Takeuchi, T. (1993) Processing of Mutated Proinsulin with Tetrabasic Cleavage Sites to Mature Insulin Reflects the Expression of Furin in Nonendocrine Cell-Lines. Endocrinology, 133, 639-644.
- 40. Lee, H.C., Kim, S.J., Kim, K.S., Shin, H.C. and Yoon, J.W. (2000) Remission in Models of Type 1 Diabetes by Gene Therapy Using a Single-Chain Insulin Analogue. Nature, 408, 483-488.
http://dx.doi.org/10.1038/35044106 - 41. Brems, D.N., Alter, L.A., Beckage, M.J., Chance, R.E. and Dimarchi, R.D. (1992) Altering the Association Properties of Insulin by Amino-Acid Replacement. Protein Engineering, 5, 527-533.
http://dx.doi.org/10.1093/protein/5.6.527 - 42. Rivera, V.M., Wang, X., Wardwell, S., Courage, N.L. and Volchuk, A. (2000) Regulation of Protein Secretion through Controlled Aggregation in the Endoplasmic Reticulum. Science, 287, 826-830.
http://dx.doi.org/10.1126/science.287.5454.826 - 43. Towle, H.C., Kaytor, E.N. and Shih, H.M. (1997) Regulation of the Expression of Lipogenic Enzyme Genes by Carbohydrate. Annual Review of Nutrition, 17, 405-433.
http://dx.doi.org/10.1146/annurev.nutr.17.1.405 - 44. Girard, J., Ferre, P. and Foufelle, F. (1997) Mechanisms by Which Carbohydrates Regulate Expression of Genes for Glycolytic and Lipogenic Enzymes. Annual Review of Nutrition, 17, 325-352.
http://dx.doi.org/10.1146/annurev.nutr.17.1.325 - 45. Towle, H.C. (2005) Glucose as a Regulator of Eukaryotic Gene Transcription. Trends in Endocrinology and Metabolism, 16, 489-494.
http://dx.doi.org/10.1016/j.tem.2005.10.003 - 46. Cheung, A.T., Dayanandan, B., Lewis, J.T., Korbutt, G.S. and Rajotte, R.V. (2000) Glucose-Dependent Insulin Release from Genetically Engineered K Cells. Science, 290, 1959-1962.
http://dx.doi.org/10.1126/science.290.5498.1959 - 47. Thule, P.M., Liu, J. and Phillips, L.S. (2000) Glucose Regulated Production of Human Insulin in Rat Hepatocytes. Gene Therapy, 7, 205-214.
http://dx.doi.org/10.1038/sj.gt.3301076 - 48. Thule, P.M. and Liu, J.M. (2000) Regulated Hepatic Insulin Gene Therapy of Stz-Diabetic Rats. Gene Therapy, 7, 1744-1752.
http://dx.doi.org/10.1038/sj.gt.3301297 - 49. Alam, T. and Sollinger, H.W. (2002) Glucose-Regulated Insulin Production in Hepatocytes. Transplantation, 74, 1781-1787.
http://dx.doi.org/10.1097/00007890-200212270-00024 - 50. Alam, T., Wai, P., Held, D., Vakili, S.T. and Forsberg, E. (2013) Correction of Diabetic Hyperglycemia and Amelioration of Metabolic Anomalies by Minicircle DNA Mediated Glucose-Dependent Hepatic Insulin Production. PLoS ONE, 8, e67515.
http://dx.doi.org/10.1371/journal.pone.0067515 - 51. Olson, D.E., Paveglio, S.A., Huey, P.U., Porter, M.H. and Thule, P.M. (2003) Glucose-Responsive Hepatic Insulin Gene Therapy of Spontaneously Diabetic Bb/Wor Rats. Human Gene Therapy, 14, 1401-1413.
http://dx.doi.org/10.1089/104303403769211628 - 52. Auricchio, A., Gao, G.P., Yu, Q.C., Raper, S. and Rivera, V.M. (2002) Constitutive and Regulated Expression of Processed Insulin Following in Vivo Hepatic Gene Transfer. Gene Therapy, 9, 963-971.
http://dx.doi.org/10.1038/sj.gt.3301746 - 53. Kuwahata, M., Kuramoto, Y., Sawai, Y., Amano, S. and Tomoe, Y. (2008) Polypyrimidine Tract-Binding Protein Is Involved in Regulation of Albumin Synthesis in Response to Food Intake. Journal of Nutritional Science and Vitaminology, 54, 142-147.
http://dx.doi.org/10.3177/jnsv.54.142 - 54. Morgan, R.A. and Anderson, W.F. (1993) Human Gene-Therapy. Annual Review of Biochemistry, 62, 191-217.
http://dx.doi.org/10.1146/annurev.bi.62.070193.001203 - 55. Volpers, C. and Kochanek, S. (2004) Adenoviral Vectors for Gene Transfer and Therapy. Journal of Gene Medicine, 6, S164-S171.
http://dx.doi.org/10.1002/jgm.496 - 56. Short, D.K., Okada, S., Yamauchi, K. and Pessin, J.E. (1998) Adenovirus-Mediated Transfer of a Modified Human Proinsulin Gene Reverses Hyperglycemia in Diabetic Mice. American Journal of Physiology-Endocrinology and Metabolism, 275, E748-E756.
- 57. Marshall, E. (1999) Clinical Trials—Gene Therapy Death Prompts Review of Adenovirus Vector. Science, 286, 2244-2245.
http://dx.doi.org/10.1126/science.286.5448.2244 - 58. Mary Ann Liebert, Inc. (2002) Assessment of Adenoviral Vector Safety and Toxicity: Report of the National Institutes of Health Recombinant DNA Advisory Committee. Human Gene Therapy, 13, 3-13.
http://dx.doi.org/10.1089/10430340152712629 - 59. Muzyczka, N. (1992) Use of Adenoassociated Virus as a General Transduction Vector for Mammalian-Cells. Current Topics in Microbiology and Immunology, 158, 97-129.
- 60. Park, Y.M., Woo, S.A., Lee, G.T., Ko, J.Y. and Lee, Y. (2005) Safety and Efficacy of Adeno-Associated Viral Vector-Mediated Insulin Gene Transfer via Portal Vein to the Livers of Streptozotocin-Induced Diabetic Sprague-Dawley Rats. Journal of Gene Medicine, 7, 621-629.
http://dx.doi.org/10.1002/jgm.708 - 61. Cavazzana-Calvo, M., Hacein-Bey, S., Basile, C.D., Gross, F. and Yvon, E. (2000) Gene Therapy of Human Severe Combined Immunodeficiency (Scid)-X1 Disease. Science, 288, 669-672.
http://dx.doi.org/10.1126/science.288.5466.669 - 62. Muzzin, P., Eisensmith, R.C., Copeland, K.C. and Woo, S.L.C. (1997) Hepatic Insulin Gene Expression as Treatment for Type 1 Diabetes Mellitus in Rats. Molecular Endocrinology, 11, 833-837.
http://dx.doi.org/10.1210/mend.11.6.0017 - 63. Ronen, K., Negre, O., Roth, S., Colomb, C. and Malani, N. (2011) Distribution of Lentiviral Vector Integration Sites in Mice Following Therapeutic Gene Transfer to Treat Beta-Thalassemia. Molecular Therapy, 19, 1273-1286.
http://dx.doi.org/10.1038/mt.2011.20 - 64. Papayannakos, C. and Daniel, R. (2013) Understanding Lentiviral Vector Chromatin Targeting: Working to Reduce Insertional Mutagenic Potential for Gene Therapy. Gene Therapy, 20, 581-588.
http://dx.doi.org/10.1038/gt.2012.88 - 65. Schweizer, M. and Merten, O.W. (2010) Large-Scale Production Means for the Manufacturing of Lentiviral Vectors. Current Gene Therapy, 10, 474-486.
http://dx.doi.org/10.2174/156652310793797748 - 66. Ren, B., O’Brien, B.A., Swan, M.A., Koina, M.E. and Nassif, N. (2007) Long-Term Correction of Diabetes in Rats after Lentiviral Hepatic Insulin Gene Therapy. Diabetologia, 50, 1910-1920.
http://dx.doi.org/10.1007/s00125-007-0722-0 - 67. Ren, B.H., O’Brien, B.A., Byrne, M.R., Ch’ng, E. and Gatt, P.N. (2013) Long-Term Reversal of Diabetes in Non-Obese Diabetic Mice by Liver-Directed Gene Therapy. Journal of Gene Medicine, 15, 28-41.
http://dx.doi.org/10.1002/jgm.2692 - 68. Tisch, R., Wang, B., Weaver, D.J., Liu, S. and Bui, T. (2001) Antigen-Specific Mediated Suppression of Beta Cell Autoimmunity by Plasmid DNA Vaccination. Journal of Immunology, 166, 2122-2132.
http://dx.doi.org/10.4049/jimmunol.166.3.2122 - 69. Piccirillo, C.A., Chang, Y.G. and Prud’homme, G.J. (1998) Tgf-Beta 1 Somatic Gene Therapy Prevents Autoimmune Disease in Nonobese Diabetic Mice. Journal of Immunology, 161, 3950-3956.
- 70. Von Herrath, M.G., Efrat, S., Oldstone, M.B.A. and Horwitz, M.S. (1997) Expression of Adenoviral E3 Transgenes in Beta Cells Prevents Autoimmune Diabetes. Proceedings of the National Academy of Sciences of the United States of America, 94, 9808-9813.
http://dx.doi.org/10.1073/pnas.94.18.9808 - 71. Tian, C.R., Bagley, J., Cretin, N., Seth, N. and Wucherpfennig, K.W. (2004) Prevention of Type 1 Diabetes by Gene Therapy. Journal of Clinical Investigation, 114, 969-978.
http://dx.doi.org/10.1172/JCI22103 - 72. Dominguez-Bendala, J., Inverardi, L. and Ricordi, C. (2012) Regeneration of Pancreatic Beta-Cell Mass for the Treatment of Diabetes. Expert Opinion on Biological Therapy, 12, 731-741.
http://dx.doi.org/10.1517/14712598.2012.679654 - 73. Pagliuca, F.W. and Melton, D.A. (2013) How to Make a Functional Beta-Cell. Development, 140, 2472-2483.
http://dx.doi.org/10.1242/dev.093187 - 74. Phillips, B.W., Hentze, H., Rust, W.L., Chen, Q.P. and Chipperfield, H. (2007) Directed Differentiation of Human Embryonic Stem Cells into the Pancreatic Endocrine Lineage. Stem Cells and Development, 16, 561-578.
http://dx.doi.org/10.1089/scd.2007.0029 - 75. Xu, X.F., Browning, V.L. and Odorico, J.S. (2011) Activin, Bmp and Fgf Pathways Cooperate to Promote Endoderm and Pancreatic Lineage Cell Differentiation from Human Embryonic Stem Cells. Mechanisms of Development, 128, 412-427.
http://dx.doi.org/10.1016/j.mod.2011.08.001 - 76. Pagliuca, F.W., Millman, J.R., Gurtler, M., Segel, M. and Van Dervort, A. (2014) Generation of Functional Human Pancreatic Beta Cells in Vitro. Cell, 159, 428-439.
http://dx.doi.org/10.1016/j.cell.2014.09.040 - 77. Ferber, S., Halkin, A., Cohen, H., Ber, I. and Einav, Y. (2000) Pancreatic and Duodenal Homeobox Gene 1 Induces Expression of Insulin Genes in Liver and Ameliorates Streptozotocin-Induced Hyperglycemia. Nature Medicine, 6, 568-572.
http://dx.doi.org/10.1038/75050 - 78. Taniguchi, H., Yamato, E., Tashiro, F., Ikegami, H. and Ogihara, T. (2003) Beta-Cell Neogenesis Induced by Adenovirus-Mediated Gene Delivery of Transcription Factor Pdx-1 into Mouse Pancreas. Gene Therapy, 10, 15-23.
http://dx.doi.org/10.1038/sj.gt.3301846 - 79. Yoshida, S., Kajimoto, Y., Yasuda, T., Watada, H. and Fujitani, Y. (2002) Pdx-1 Induces Differentiation of Intestinal Epithelioid Iec-6 into Insulin-Producing Cells. Diabetes, 51, 2505-2513.
http://dx.doi.org/10.2337/diabetes.51.8.2505 - 80. Noguchi, H., Xu, G., Matsumoto, S., Kaneto, H. and Kobayashi, N. (2006) Induction of Pancreatic Stem/Progenitor Cells into Insulin-Producing Cells by Adenoviral-Mediated Gene Transfer Technology. Cell Transplant, 15, 929-938.
http://dx.doi.org/10.3727/000000006783981431 - 81. Karnieli, O., Izhar-Prato, Y., Bulvik, S. and Efrat, S. (2007) Generation of Insulin-Producing Cells from Human Bone Marrow Mesenchymal Stem Cells by Genetic Manipulation. Stem Cells, 25, 2837-2844.
http://dx.doi.org/10.1634/stemcells.2007-0164 - 82. Li, Y.H., Zhang, R., Qiao, H.F., Zhang, H.P. and Wang, Y.F. (2007) Generation of Insulin-Producing Cells from Pdx-1 Gene-Modified Human Mesenchymal Stem Cells. Journal of Cellular Physiology, 211, 36-44.
http://dx.doi.org/10.1002/jcp.20897 - 83. Lin, G.T., Wang, G.F., Liu, G., Yang, L.J. and Chang, L.J. (2009) Treatment of Type 1 Diabetes with Adipose Tissue-Derived Stem Cells Expressing Pancreatic Duodenal Homeobox 1. Stem Cells and Development, 18, 1399-1406.
http://dx.doi.org/10.1089/scd.2009.0010 - 84. Kajiyama, H., Hamazaki, T.S., Tokuhara, M., Masui, S. and Okabayashi, K. (2010) Pdx1-Transfected Adipose Tissue-Derived Stem Cells Differentiate into Insulin-Producing Cells in Vivo and Reduce Hyperglycemia in Diabetic Mice. The International Journal of Developmental Biology, 54, 699-705.
http://dx.doi.org/10.1387/ijdb.092953hk - 85. Lavon, N., Yanuka, O. and Benvenisty, N. (2006) The Effect of Overexpression of Pdx1 and Foxa2 on the Differentiation of Human Embryonic Stem Cells into Pancreatic Cells. Stem Cells, 24, 1923-1930.
http://dx.doi.org/10.1634/stemcells.2005-0397 - 86. Vincent, R., Treff, N., Budde, M., Kastenberg, Z. and Odorico, J. (2006) Generation and Characterization of Novel Tetracycline-Inducible Pancreatic Transcription Factor-Expressing Murine Embryonic Stem Cell Lines. Stem Cells and Development, 15, 953-962.
http://dx.doi.org/10.1089/scd.2006.15.953 - 87. Raikwar, S.P. and Zavazava, N. (2012) Pdx1-Engineered Embryonic Stem Cell-Derived Insulin Producing Cells Regulate Hyperglycemia in Diabetic Mice. Transplantation Research, 1, 19.
http://dx.doi.org/10.1186/2047-1440-1-19 - 88. Simpson, A.M., Tao, C., Swan, M.A., Ren, B. and O’Brien, B.A. (2007) Glucose Regulated Production of Human Insulin in H4iie Rat Liver Cells. Diabetes, 56, A120-A120.
- 89. Noguchi, H., Xu, G., Matsumoto, S., Kaneto, H. and Kobayashi, N. (2006) Induction of Pancreatic Stem/Progenitor Cells into Insulin-Producing Cells by Adenoviral-Mediated Gene Transfer Technology. Cell Transplant, 15, 929-938.
http://dx.doi.org/10.3727/000000006783981431 - 90. Yatoh, S., Akashi, T., Chan, P.P., Kaneto, H. and Sharma, A. (2007) Neurod and Reaggregation Induce Beta-Cell Specific Gene Expression in Cultured Hepatocytes. Diabetes/Metabolism Research and Reviews, 23, 239-249.
http://dx.doi.org/10.1002/dmrr.678 - 91. Wang, A.Y., Ehrhardt, A., Xu, H. and Kay, M.A. (2007) Adenovirus Transduction Is Required for the Correction of Diabetes Using Pdx-1 or Neurogenin-3 in the Liver. Molecular Therapy, 15, 255-263.
http://dx.doi.org/10.1038/sj.mt.6300032 - 92. Kaneto, H., Nakatani, Y., Miyatsuka, T., Matsuoka, T. and Matsuhisa, M. (2005) Pdx-1/Vp16 Fusion Protein, Together with Neurod or Ngn3, Markedly Induces Insulin Gene Transcription and Ameliorates Glucose Tolerance. Diabetes, 54, 1009-1022.
http://dx.doi.org/10.2337/diabetes.54.4.1009 - 93. Song, Y.D., Lee, E.J., Yashar, P., Pfaff, L.E. and Kim, S.Y. (2007) Islet Cell Differentiation in Liver by Combinatorial Expression of Transcription Factors Neurogenin-3, Beta2, and Ripe3b1. Biochemical and Biophysical Research Communications, 354, 334-339.
http://dx.doi.org/10.1016/j.bbrc.2006.12.216 - 94. Heremans, Y., Van De Casteele, M., Veld, P.I., Gradwohl, G. and Serup, P. (2002) Recapitulation of Embryonic Neuroendocrine Differentiation in Adult Human Pancreatic Duct Cells Expressing Neurogenin 3. Journal of Cell Biology, 159, 303-311.
http://dx.doi.org/10.1083/jcb.200203074 - 95. Yechoor, V., Liu, V., Espiritu, C., Paul, A. and Oka, K. (2009) Neurogenin3 Is Sufficient for Transdetermination of Hepatic Progenitor Cells into Neo-Islets, in Vivo but Not Transdifferentiation of Hepatocytes. Developmental Cell, 16, 358-373.
http://dx.doi.org/10.1016/j.devcel.2009.01.012 - 96. Zhou, Q., Brown, J., Kanarek, A., Rajagopal, J. and Melton, D.A. (2008) In Vivo Reprogramming of Adult Pancreatic Exocrine Cells to Beta-Cells. Nature, 455, U627-U630.
http://dx.doi.org/10.1038/nature07314 - 97. Blyszczuk, P., Czyz, J., Kania, G., Wagner, M. and Roll, U. (2003) Expression of Pax4 in Embryonic Stem Cells Promotes Differentiation of Nestin-Positive Progenitor and Insulin-Producing Cells. Proceedings of the National Academy of Sciences of the United States of America, 100, 998-1003.
http://dx.doi.org/10.1073/pnas.0237371100 - 98. Gefen-Halevi, S., Rachmut, I.H., Molakandov, K., Berneman, D. and Mor, E. (2010) Nkx6.1 Promotes Pdx-1-Induced Liver to Pancreatic Beta-Cells Reprogramming. Cellular Reprogramming, 12, 655-664.
http://dx.doi.org/10.1089/cell.2010.0030 - 99. Roep, B.O. and Peakman, M. (2012) Antigen Targets of Type 1 Diabetes Autoimmunity. Cold Spring Harbor Perspectives in Medicine, 2, a007781.
http://dx.doi.org/10.1101/cshperspect.a007781 - 100. Nicolau, C., Le Pape, A., Soriano, P., Fargette, F. and Juhel, M.F. (1983) In Vivo Expression of Rat Insulin after Intravenous Administration of the Liposome-Entrapped Gene for Rat Insulin I. Proceedings of the National Academy of Sciences of the United States of America, 80, 1068-1072.
http://dx.doi.org/10.1073/pnas.80.4.1068 - 101. Moore, H.P.H., Walker, M.D., Lee, F. and Kelly, R.B. (1983) Expressing a Human Proinsulin Cdna in a Mouse Acth-Secreting Cell—Intracellular Storage, Proteolytic Processing, and Secretion on Stimulation. Cell, 35, 531-538.
http://dx.doi.org/10.1016/0092-8674(83)90187-3 - 102. Stewart, C., Taylor, N.A., Green, I.C., Docherty, K. and Bailey, C.J. (1994) Insulin-Releasing Pituitary Cells as a Model for Somatic Cell Gene Therapy in Diabetes Mellitus. The Journal of Endocrinology, 142, 339-343.
http://dx.doi.org/10.1677/joe.0.1420339 - 103. Stewart, C., Taylor, N.A., Docherty, K. and Bailey, C.J. (1993) Insulin Delivery by Somatic Cell Gene Therapy. Journal of Molecular Endocrinology, 11, 335-341.
http://dx.doi.org/10.1677/jme.0.0110335 - 104. Hughes, S.D., Quaade, C., Johnson, J.H., Ferber, S. and Newgard, C.B. (1993) Transfection of Att-20ins Cells with Glut-2 but Not Glut-1 Confers Glucose-Stimulated Insulin Secretion. Relationship to Glucose Metabolism. The Journal of Biological Chemistry, 268, 15205-15212.
- 105. Simpson, A.M., Tuch, B.E., Swan, M.A., Tu, J. and Marshall, G.M. (1995) Functional Expression of the Human Insulin Gene in a Human Hepatoma Cell Line (Hep G2). Gene Therapy, 2, 223-231.
- 106. Simpson, A.M., Marshall, G.M., Tuch, B.E., Maxwell, L. and Szymanska, B. (1997) Gene Therapy of Diabetes: Glucose-Stimulated Insulin Secretion in a Human Hepatoma Cell Line (Hep G2ins/G). Gene Therapy, 4, 1202-1215.
http://dx.doi.org/10.1038/sj.gt.3300527 - 107. Tuch, B.E., Szymanska, B., Yao, M., Tabiin, M.T. and Gross, D.J. (2003) Function of a Genetically Modified Human Liver Cell Line That Stores, Processes and Secretes Insulin. Gene Therapy, 10, 490-503.
http://dx.doi.org/10.1038/sj.gt.3301911 - 108. Ganesan, L.P., Mohanty, S., Kim, J., Clark, K.R. and Robinson, J.M. (2011) Rapid and Efficient Clearance of Blood-Borne Virus by Liver Sinusoidal Endothelium. PLoS Pathogens, 7.
http://dx.doi.org/10.1371/journal.ppat.1002281 - 109. Chen, R.H., Meseck, M.L. and Woo, S.L.C. (2001) Auto-Regulated Hepatic Insulin Gene Expression in Type 1 Diabetic Rats. Molecular Therapy, 3, 584-590.
http://dx.doi.org/10.1006/mthe.2001.0299 - 110. Chen, R., Meseck, M., McEvoy, R.C. and Woo, S.L. (2000) Glucose-Stimulated and Self-Limiting Insulin Production by Glucose 6-Phosphatase Promoter Driven Insulin Expression in Hepatoma Cells. Gene Therapy, 7, 1802-1809.
http://dx.doi.org/10.1038/sj.gt.3301306 - 111. Burkhardt, B.R., Parker, M.J., Zhang, Y.C., Song, S.H. and Wasserfall, C.H. (2005) Glucose Transporter-2 (Glut2) Promoter Mediated Transgenic Insulin Production Reduces Hyperglycemia in Diabetic Mice. FEBS Letters, 579, 5759-5764.
http://dx.doi.org/10.1016/j.febslet.2005.09.060 - 112. Hsu, P.Y.J., Kotin, R.M. and Yang, Y.W. (2008) Glucose- and Metabolically Regulated Hepatic Insulin Gene Therapy for Diabetes. Pharmaceutical Research, 25, 1460-1468.
http://dx.doi.org/10.1007/s11095-008-9539-x - 113. Nakayama, M. (2011) Insulin as a Key Autoantigen in the Development of Type 1 Diabetes. Diabetes/Metabolism Research and Reviews, 27, 773-777.
http://dx.doi.org/10.1002/dmrr.1250 - 114. Ramshur, E.B., Rull, T.R. and Wice, B.M. (2002) Novel Insulin/Gip Co-Producing Cell Lines Provide Unexpected Insights into Gut K-Cell Function in Vivo. Journal of Cellular Physiology, 192, 339-350.
http://dx.doi.org/10.1002/jcp.10139 - 115. Park, F., Ohashi, K., Chiu, W., Naldini, L. and Kay, M.A. (2000) Efficient Lentiviral Transduction of Liver Requires Cell Cycling in Vivo. Nature Genetics, 24, 49-52.
http://dx.doi.org/10.1038/71673 - 116. Duckworth, W.C., Bennett, R.G. and Hamel, F.G. (1998) Insulin Degradation: Progress and Potential. Endocrine Reviews, 19, 608-624.
http://dx.doi.org/10.1210/er.19.5.608 - 117. Hausl, M.A., Zhang, W., Muther, N., Rauschhuber, C. and Franck, H.G. (2010) Hyperactive Sleeping Beauty Transposase Enables Persistent Phenotypic Correction in Mice and a Canine Model for Hemophilia B. Molecular Therapy: The Journal of the American Society of Gene Therapy, 18, 1896-1906.
http://dx.doi.org/10.1038/mt.2010.169
NOTES

*Corresponding author.