Case Reports in Clinical Medicine
Vol.2 No.1(2013), Article ID:28917,5 pages DOI:10.4236/crcm.2013.21014
De novo duplication 3q in an infant with a vascular ring and features overlapping Cornelia de Lange phenotype
![]()
Department of Pediatrics and Human Development, College of Human Medicine, Michigan State University, East Lansing, USA; *Corresponding Author: Sainan.Wei@hc.msu.edu
Received 26 January 2013; revised 28 February 2013; accepted 7 March 2013
Keywords: Cornelia de Lange; 3q Duplication; Trisomy 3q; Pure 3q Duplication
ABSTRACT
Partial duplication of chromosome 3q is a recognizable syndrome with characteristic facial features, microcephaly, digital anomalies, genitourinary and cardiac defects as well as growth retardation and developmental delays. While there is clinical overlap with the unrelated Cornelia de Lange syndrome (CDLS), there are distinguishing features and molecular etiologies. Most cases of 3q duplication appear to be the result of an unbalanced translocation or inversion and therefore accompanied by additional cytogenetic anomalies. Consequently, pure duplications of 3q are very rare; we are aware of only 12 such cases that have been reported previously. Here, we present a new case of pure, partial 3q duplication in a 3-month-old female who displayed a number of clinical signs consistent with previously reported phenotypes and the additional novel finding of a vascular ring.
1. INTRODUCTION
Individuals with partial duplication of chromosome 3q typically display characteristic facial features including low frontal hairline, bushy eyebrows, synophrys, hypertelorism, long eyelashes, downslanting palpebral fissures, epicanthal folds, wide nasal bridge with bulbous nasal tip, prominent philtrum, down-turned corners of the mouth and low-set, malformed ears. Additional physical findings include microcephaly, digital anomalies such as brachydactyly and clinodactyly, hirsutism, genitourinary and cardiac defects in addition to preand postnatal growth retardation and developmental delay [1]. Less common reported features include ocular anomalies, neuroimaging findings, hip dysplasia, central sleep apnea and conductive hearing loss [2]. Approximately one-third of patients die within the first year of life due to infections and cardiac anomalies [3]. Although 3q duplication syndrome has overlapping features with and clinically resembles the unrelated Cornelia de Lange syndrome (CDLS), there are distinguishing features and molecular etiologies. Most cases of 3q duplication appear to be the result of an unbalanced translocation or inversion; as a result, there is often an accompanying cytogenetic anomaly such as deletion of another chromosomal segment [4]. Therefore, pure duplications of 3q are very rare; we are aware of only 12 such cases that have been reported previously [2, 6]. Here, we present a new case of a pure, partial 3q duplication in a 3-month-old female who displayed a number of clinical signs consistent with previously reported phenotypes and the additional novel finding of a vascular ring.
2. CASE REPORT
A Hispanic female, the third child of non-consanguineous parents, was born after an unremarkable pregnancy at 38 weeks gestation. Birth weight was at the 10th to 25th percentile with length and head circumference at minus 2 to minus 3 standard deviations and 50 - 75th percentile, respectively. The patient was admitted to the Neonatal Intensive Care Unit for meconium aspiration, jaundice and asymmetric growth retardation. She was noted to have a wide and open anterior fontanelle, hypertrichosis with overgrowth of scalp hair onto the forehead, synophrys, bushy eyebrows, anteverted nares, long philtrum, downturned corners of the mouth, low-set and malformed pinnae, maxillary prognathism and retrognathia (Figure 1C). The nipples were laterally displaced and the hands showed bilateral 5th finger clinodactyly and bilateral single palmer creases. A chromosomal microarray was requested because of the aforementioned dysmorphology.
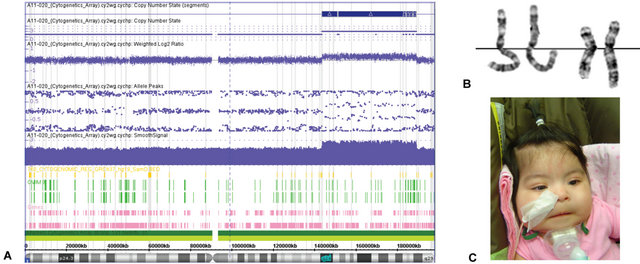
Figure 1. A: SNP microarray results showing copy number gain of probes in the region of 3q24q28; B: Partial karyogram showing two pairs of chromosome 3 from two metaphase cells. The chromosome 3 with dup(3) (q24q28) is to the right of each pair; C: Hypertrichosis with overgrowth of scalp hair onto the forehead, bushy eyebrows, anteverted nares, and downturned corners of the mouth in our patient with 3q duplication.
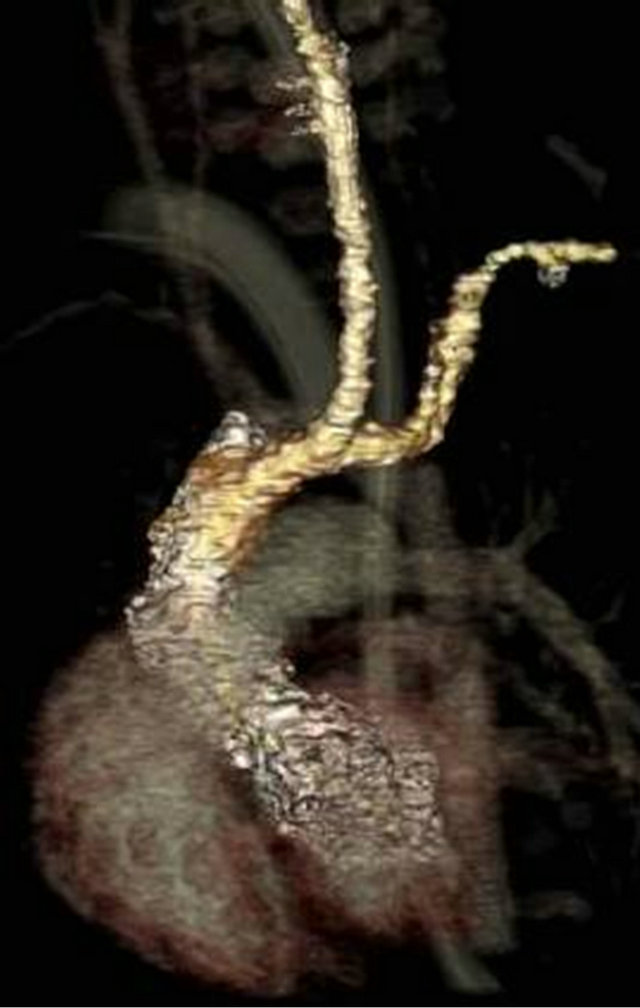
At one month of age, the patient was readmitted with respiratory distress that progressed to respiratory failure and was found to be positive for metapneumovirus. She failed extubation, prompting a bronchoscopy which identified tracheomalacia. A brain MRI and EEG were normal. Further workup included a multislice computed tomography (MSCT) showing double aortic arch with fourvessel sign and narrowing of the distal trachea above the carina consistent with a vascular ring (Figures 2(a) and (b)). An echocardiogram additionally noted a very small patent ductus arteriosus with small left-to-right shunt. The patient underwent a PDA ligation and division of the vascular ring. At the time of this report, the patient was 3 months old and at 14 days post-operative; she remained with a tracheostomy.
3. METHODS
EDTA-anticoagulated blood of the patient, and subsequently her parents were sent to the MSU Clinical Genetics Laboratory for both genome-wide SNP microarray and cytogenetic analysis. Genome-wide microarray-CGH analysis was performed using Affymetrix cytogenetics whole genome 2.7 M array with 2.7 million markers including 2,361,876 non-polymorphic probes/markers and 400,103 SNP probes/markers. Genomic DNA was extracted from whole blood, then amplified and purified. Following denaturation of probe DNA, hybridization was carried out with the Affymetrix cytogenetic 2.7 M array assay kit following the manufacturers’ standard protocol. The data was analyzed by ChAS (Chromosome Analysis Suite) and compared against a reference model file provided by Affymetrix to detect gains and losses. Chromosome slides were made by conventional methods from
 (a)
(a)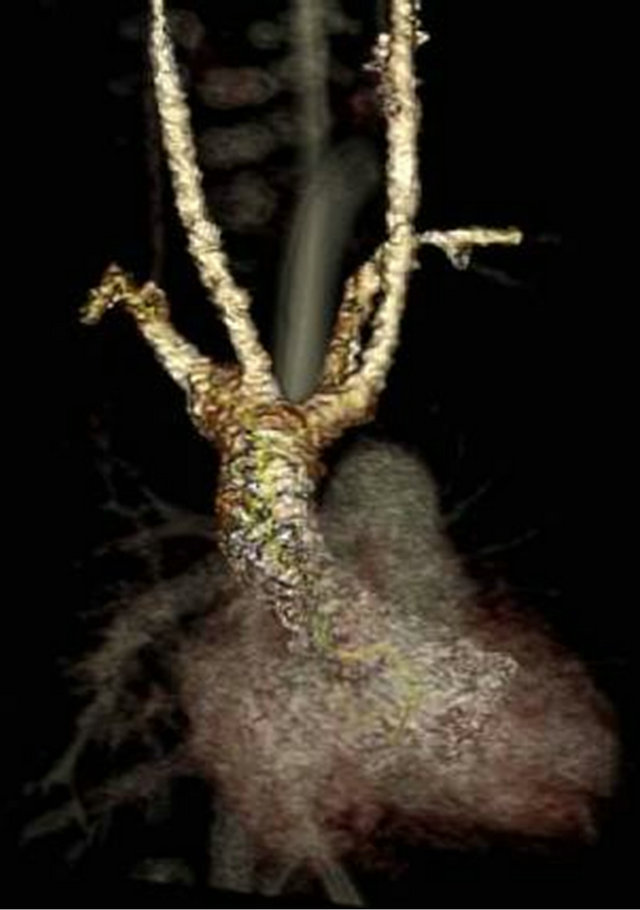 (b)
(b)
Figure 2. (a) and (b), Thorax multislice computed tomography illustrating features of vascular ring.
phytohaemagglutinin-stimulated peripheral blood cultures. Subsequently, routine chromosome analysis was performed on standard GTG-banded metaphase spreads. High resolution genome wide microarray-CGH analysis revealed a copy number gain of 29,436 probes in the region of 3q24q28 (Figure 1A) indicating a duplication estimated to be at least 45.6 Mb in size. Chromosome analysis confirmed this to be a segmental duplication (Figure 1B). Parental karyotypes were normal (data not shown), confirming the patient’s 3q duplication was a de novo occurrence.
4. DISCUSSION
Pure duplications of chromosome 3q are rare as the majority appear to occur in conjunction with other structural anomalies such as unbalanced translocations and insertions [7,10]. This report represents a new case of pure 3q duplication due to a de novo interstitial duplication event detected in a 3-month-old female with multiple congenital anomalies, tracheomalacia and vascular compression from a double aortic arch. Until now, only 12 cases of pure 3q duplication have been reported. Table 1 summarizes the clinical features of these cases, including our patient.
Table 1. Clinical summary of thirteen pure 3q duplication patients.
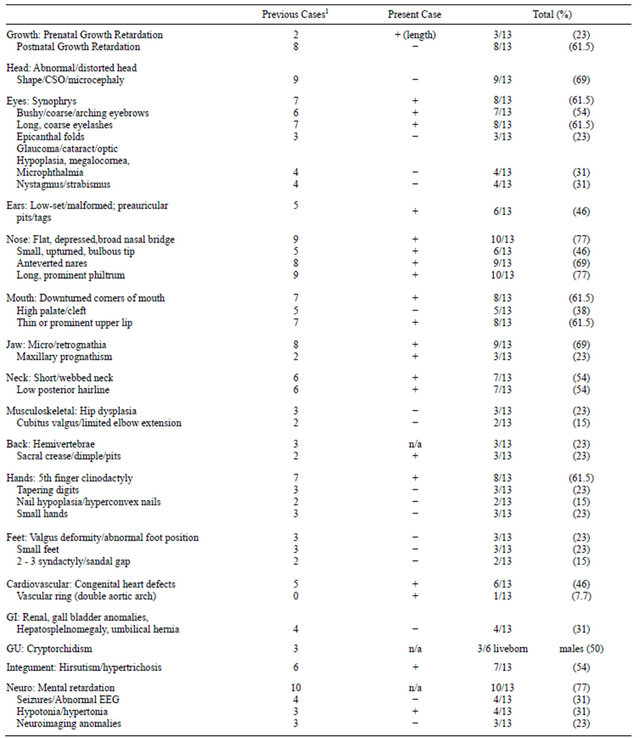
1Incidence among 12 previously published cases, n/a not applicable; + present, − absent/not reported [2,3,5,7-9,11,12,21-23].
A specific critical region responsible for the phenotype has yet to be clearly defined, but it has been suggested to be between 3q26.3 to 3q27 [11] or 3q26.3 to 3q29 [3,7]. By comparing the phenotype of our patient with those of other pure 3q duplications, evidence for previously proposed critical regions is supported and suggests that the minimal region of overlap may be at band 3q26.3 [3,10]. This region therefore may contain the gene(s) responseble for the dup(3q) phenotype.
Duplication 3q syndrome has attracted particular interest because of its recurrent features and the clinical similarity to CDLS. Similarities in the facial features of children with duplication 3q and CDLS have been repeatedly noted [12-14].
5. CONCLUSION
Our patient represents the thirteenth case of pure partial trisomy 3q, with features consistent with those previously reported. We additionally report the novel finding of a vascular ring. The availability of increasingly dense microarray technology, whole exome and full genome sequencing will undoubtedly result in further clarification of the phenotype and natural history of this rare and poorly defined syndrome.
REFERENCES
- Schinzel, A. (2001) A catalog of unbalanced chromosome aberration in man. 2nd Edition, De Gruyter Verlag, Berlin, New York.
- Shanske, A.L., Leonard, J., Nahum, O., Hu, T. and Desai, J.P. (2004) Soft-tissue material properties under large deformation: Strain rate effect. Proceedings of the 26th Annual International Conference of the IEEE EMBS, San Francisco, 1-5 September 2004, 2758-2761.
- Faas, D.H.W., de Vries, B.B.A., van Es-van Gaal, J., Merk, G., Draaisma, J.M.T. and Smeets, D.F.C.M. (2002) A new case of dup(3q) syndrome due to a pure duplication of 3qter. Clinical Genetics, 62, 315-320. doi:10.1034/j.1399-0004.2002.620411.x
- Salazar, D., Rosenfeld, W., Verma, R.S., Jhaveri, R.C. and Dosik, H. (1979) Partial trisomy of chromosome 3 (3q12 leads to qter) owing to 3q/18p translocation. A trisomy 3q syndrome. American Journal of Diseases of Children, 133, 1006-1008.
- Holder, S., Grimsley, L., Palmer, R., Bulter, L. and Baraitser, M. (1994) Partial trisomy 3q causing mild Cornelia de Lange phenotype. American Journal of Medical Genetics, 31, 150-152.
- Dundar, M., Uzak, A., Erdogan, M., Saatci, C., Akdeniz, S., Luleci, G., Keser, I. and Karauzum, S. (2011) Partial trisomy 3q in a child with sacrococcygeal teratoma and Cornelia de Lange syndrome phenotype. Genetic Counseling, 22, 199-205.
- Grossmann, V., Muller, D., Muller, W., Fresser, F., Erdel, M., Janecke, A.R., Zchocke, J., Utermann, G. and Kotzot, D. (2009) “Essential” pure trisomy 3q27->qter: Further delineation of the partial trisomy 3q phenotype. American Journal of Medical Genetics, 149A, 2522-2526. doi:10.1002/ajmg.a.33058
- Aqua, M.S., Rizzu. P., Lindsay, E.A., Shaffer, L.G., Zackai, E.H., Overhauser, J. and Baldini, A. (2005) Duplication 3q syndrome: Molecular delineation of the critical region. American Journal of Medical Genetics, 55, 33-37. doi:10.1002/ajmg.1320550111
- Meins, M., Hagh, J.K., Gerresheim, F., Einhoff, E., Olschewski, H., Strehl, H. and Epplen, J.T. (2005) Novel case of dup(3q) syndrome due to a de novo interstitial duplication 3q24-q26.31 with minimal overlap to the dup (3q) critical region. American Journal of Medical Genetics, 132A, 84-89. doi:10.1002/ajmg.a.30384
- Falek, A., Schmidt, R. and Jervis, G.A. (1966) Familial de Lange syndrome with chromosome abnormalities. Pediatrics, 37, 92-101.
- Sciorra, L.J., Bahng, K. and Lee, M. (1979) Trisomy in the distal end of the long arm of chromosome 3: A condition clinically similar to the Cornelia de Lange syndrome. American Journal of Diseases of Children, 133, 727-730.
- Wilson, G.N., Hieber, V.C. and Schmickel, R.D. (1978) The association of chromosome 3 duplication and the Cornelia de Lange syndrome. Journal of Pediatrics, 93, 783-788. doi:10.1016/S0022-3476(78)81077-4
- Francke, U. (1978) Clinical syndromes associated with partial duplications of chromosomes 2 and 3: dup(2p), dup(2q), dup(3p), dup(3q). Birth Defects Original Article Series, 14, 191-217.
- Francke, U. and Opitz, J.M. (1979) Chromosome 3q duplication and the Brachmann-De Lange syndrome (BDLS). Journal of Pediatrics, 95, 161-163. doi:10.1016/S0022-3476(79)80124-9
- Krantz, I.D., Tonkin, E., Smith, M., Devoto, M., Bottani, A., Simpson, C., Hofreiter, M., Abraham, V., Jukofsky, L., Conti, B.P., Strachan, T. and Jackson, L. (2001) Exclusion of linkage to the CDL1 gene region on chromosome 3q26.3 in some familial cases of Cornelia de Lange syndrome. American Journal of Medical Genetics, 101, 120- 129. doi:10.1002/1096-8628(20010615)101:2<120::AID-AJMG1319>3.0.CO;2-G
- Krantz, I.D., McCallum, J., DeScipio, C., Kaur, M., Gillis, L.A., Yaeger, D., Jukofsky, L., Wasserman, N., Bottani, A., Morris, C.A., Nowaczyk, M.J., Toriello, H., Bamshad, M.J., Carey, J.C., Rappaport, E., Kawauchi, S., Lander, A.D., Calof, A.L., Li, H.H., Devoto, M. and Jackson, L.G. (2004) Cornelia de Lange syndrome is caused by mutations in NIPBL, the human homolog of Drosophila melanogaster Nipped-B. Nature Genetics, 36, 631-635. doi:10.1038/ng1364
- Tonkin, E.T., Wang, T.J., Lisgo, S., Bamshad, M.J. and Strachan, T. (2004) NIPBL, encoding a homolog of fungal Scc2-type sister chromatid cohesion proteins and fly Nipped-B, is mutated in Cornelia de Lange syndrome. Nature Genetics, 115, 139-148.
- Liu, J. and Baynam, G. (2010) Cornelia de Lange syndrome. Advances in Experimental Medicine and Biology, 685, 111-123. doi:10.1007/978-1-4419-6448-9_11
- Fineman, R.M., Hecht, F., Ablow, R.C., Howard, R.O. and Breg, W.R. (1978) Chromosome 3 duplication q/deletion p syndrome. Pediatrics, 61, 611-618.
- Mulcahy, M.T., Pemberton, P.J. and Sprague, P. (1979) Trisomy 3q: Two clinically similar but cytogenetically different cases. Annales De Genetique, 22, 217-220.
- Lim, A.S., Lim, T.H., Chia, P., Raman, S., Pickering, D.L., Zaleski, D.H., Sanger, W.G. and Tien, S.L. (2004) A case of pure partial duplication 3q in a fetus due to a maternally inherited der(5)ins(5;3)(q33.1;q26.2q27) delineated by FISH. Prenatal Diagnosis, 24, 923-936. doi:10.1002/pd.1041
- Stengel-Rutkowski, S., Murken, J.D., Pilar, V., Dutrillaux, B., Rodewald, A., Goebel, R. and Bassermann, R. (1979) New chromosomal dysmorphic syndromes: 3. partial trisomy 3q. European Journal of Pediatrics, 130, 111-125. doi:10.1007/BF00442348
- Van Essesn, A.J., Kok, K., van den Berg, A., de Jong, B., Stellink, F., Bos, A.F., Scheffer, H. and Buys, C.H.C.M. (1991) Partial 3q duplication syndrome and assignment of D3S5 to 3q25-3q28. Human Genetics, 87, 151-154. doi:10.1007/BF00204171