American Journal of Plant Sciences
Vol.5 No.1(2014), Article ID:41945,7 pages DOI:10.4236/ajps.2014.51014
Family and/or Friends? Gene Mapping at Crossroads
1Department of Plant Breeding and Genetics, Pir Mehr Ali Shah-Arid Agriculture University, Rawalpindi, Pakistan; 2Center of Agricultural Biochemistry and Biotechnology (CABB), University of Agriculture, Faisalabad, Pakistan.
Email: *msajjadpbg@gmail.com, msajjad@uaar.edu.pk
Copyright © 2014 Muhammad Sajjad et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Muhammad Sajjad et al. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received October 25th, 2013; revised December 15th, 2013; accepted January 1st, 2014
KEYWORDS
Family Based Mapping; LD Mapping; NAM
ABSTRACT
Mapping gene(s) underlying a specific trait offers an opportunity to plant breeders to apply marker assisted selection. All gene mapping approaches except LD mapping use family based segregation populations developed by crossing two or more parents. These family based gene mapping approaches include simple interval mapping, composite interval mapping, multiple interval mapping and Bayesian mapping etc. Each approach has its own advantages and disadvantages based on type of population and underlying statistical model. Unlike family based approaches, LD mapping uses population of unrelated individuals which are like friends belonging to different family backgrounds. Relative pros and cons of family and friends based approaches make them complementary to each other. Family based approaches identify wide chromosomal region underlying the trait of interest with relatively lower markers density, and therefore, have low mapping resolution. Conversely, friends based LD mapping identifies chromosomal region of interest with higher resolution using higher marker density. The integration of family and friends based approaches addresses their respective pros and cons successfully to enhance mapping resolution for more valid application of marker assisted selection.
1. Introduction
The germplasm is often exploited to develop improved crop varieties for changing needs and environments [1]. The vast amount of genetic variation present in the form of germplasm can be exploited to the best once the traits of economic importance have been mapped with molecular markers [2]. Gene mapping is the estimation of the sequence of genes and their relative positions on a particular chromosome. The objective of gene mapping is to find molecular markers which are impartially inherited and closely linked due to location within or in close proximity of the genes governing the quantitative traits. Mapping a nucleotide sequence underlying a specific trait offers an opportunity for plant breeders to apply marker assisted (MAS) selection. Most of yield contributing traits are controlled by many loci and their molecular characterization and genetic mapping are called quantitative trait loci mapping (QTL-mapping).
Quantitative variation may be explained as the combined action of many discrete genes, each having a small effect on the overall phenotype and being influenced by the environment. The contribution of each quantitative locus at a phenotypic level is expressed as an increase or decrease in trait value and it is not possible to distinguish the effect of various loci acting in this manner from one another based on phenotypic variation alone. Furthermore, the effect of particular environmental variables is also expressed as a quantitative increase or decrease in the final trait value. The same amount of total genetic variation can be produced by allelic variation at many loci, each having a small effect on the trait or at a few loci having a larger effect. As both genetic and environmental factors contribute in the same positive or negative manner to trait value, it is generally not possible, from the phenotypic distribution of the trait alone, to distinguish the effect of genetic factors from those of environmental factors as sources of variation in traits. Therefore, breeding for quantitative traits tends to be a less efficient and time-consuming process. Tools for directed genetic manipulation of quantitative traits have undergone a crucial revolution since the late 1980s with the development of molecular markers. As a result, interchange between molecular biology and quantitative genetics, which has developed independently for many years, has become apparent since the 1990s [3]. Since then, high-density molecular maps have been constructed in many crops and genome-wide mapping and marker-based manipulation of genes affecting quantitative traits have become possible. Traits which have been improved largely by conventional breeding and biometrical methods in the past can be manipulated now using molecular markers. Location and effect of the genes controlling a quantitative trait can be determined by marker-based genetic analysis. A chromosomal region linked to or associated with a marker which affects a quantitative trait was defined as a quantitative trait locus (QTL) [4]. A QTL that has a large effect and can explain a major part of total variation can be analyzed genetically as a major gene in most cases.
The efficiency of a gene mapping also called QTL mapping approach is judged on the basis accuracy in QTL identification, while playing down the occurrence of false negatives and positives. False negative (Type II error) is a state of lack of marker-trait association when in fact it exists and false positive (Type I error) is a state when there is a marker-trait association when in fact it does not exist.
A brief overview of statistics of all QTL mapping approaches is well described by Xu, [5]. For detailed statistical description following references are highly recommended: [6-9]. Furthermore, many freely accessed websites offer courses on statistical genomics and QTL mapping (e.g. http://www.stat.wisc.edu/yandell/statgen/course/).
The scope of this review article confines to describe comparative advantages and disadvantages of all QTL mapping approaches with special focus on newly developed nested association mapping (NAM) approach.
2. Family Based Approaches
Majority of gene mapping approaches use family based segregation populations developed by crossing two or more parents such as F2, doubled haploids (DHs), recombinant inbred lines (RILs), recombinant inbred chromosomal lines (RICLs) and near isogenic lines (NILs) etc. These family based gene mapping approaches include simple interval mapping, composite interval mapping, multiple interval mapping and Bayesian mapping etc. The comparative advantages and disadvantages of these approaches are briefly described here.
2.1. Single Marker-Based Approaches
The single marker approach (also referred as single point analysis or single factor analysis of variance) has been extensively used with isozyme markers [10]. Single factor ANOVA is made for each marker independently. F-test is used to test the significance between marker genotype classes. Though statistical computations for this approach are simple but it has some major drawbacks: 1) the probability of QTL detection is significantly affected with the distance between marker and QTL; 2) the approach cannot discriminate marker association with one or more QTLs; 3) the QTLs effects are likely to be miscalculated because of their confounding with recombination frequencies.
2.2. Simple Interval Mapping
Simple interval mapping approach was developed by Lander and Botstein [11] exploiting full benefits of linkage maps. The approach detects marker-trait associations at multiple points of targeted interval between two adjacent marker loci. The log of odds ratio (LOD) is used to test the presence of a QTL. If the LOD value for a QTL exceeds critical threshold value, the QTL is considered to be significantly associated with the trait under study. The formula for setting significance levels suitable for simple interval mapping for given number of number of marker interval, number of chromosomes, genome size and false positive rates was devised by Lander and Botstein [11]. Simple interval mapping has been the most widely used approach because of its calculations through statistical software MAPMARKER/QTL (ftp://ftp-genome.wi.mit.edu/distribution/software/newqtl/). In this approach recombination between QTL and marker can be compensated using tightly linked markers. Thus, the probability of detecting QTL and providing accurate estimate of QTL effect is increased. However, simple interval mapping fails to take into account genetic variance of all QTLs when multiple QTLs are segregating in segregating populations. In such cases, simple interval mapping suffers from same limitations of single marker analysis.
2.3. Composite Interval mapping
Composite interval mapping combines interval mapping approach for a single QTL in an interval with multiple regressions on marker associated with few other QTLs [12]. This approach has been used to develop precise models for two or three linked QTL [13,14]. It takes into account a marker interval and some other chosen single markers in each analysis. Consequently, on a chromosome with n markers, n-1 tests of interval-QTL associations are carried out. The advantages of composite interval mapping over single marker analysis and simple interval mapping are: 1) multiple QTLs can be mapped simultaneously; 2) the QTL association tests are not affected by the QTLs outside the specified interval because linked markers are used only as cofactors (this characteristic of composite interval mapping increase the accuracy of QTL mapping); 3) an other factor of increased power of QTL detection is reduced residual variance because of eliminating variance of unlinked QTLs.
2.4. Multiple Interval Mapping
Multiple interval mapping is advancement from interval mapping just as multiple regressions extends analysis of variance. With this approach we can infer the location of QTLs between markers, handle missing data properly and determine interactions between QTLs. Three different statistical approaches are used for multiple interval mapping: 1) maximum likelihood [15] and chronological testing to search model space; 2) multiple imputation which uses pairwise plots, Bayesian log of odds values (LOD) and sequential testing [16]; 3) Markov chain Monte Carlo (MCMC) to search model space [14]. Multiple interval mapping is a multiple-QTL analysis which combines QTL mapping with the analysis of genetic architecture of quantitative traits through an algorithm to identify positions, number, effects and interaction of a QTL.
2.5. Bayesian Mapping
Bayesian paradigm which has been used successfully in different contexts provides a logical approach to statistical modeling [17]. Bayesian analysis treats every factor as an unidentified variable with a prior distribution. It classifies variables into two classes: observable variables and unobservable variables. The observables variables include phenotypic data, pedigrees and marker data etc.
Bayesian approach gained its popularity in QTL mapping because of the availability of Markov chain Monte Carlo (MCMC) algorithms. MCMC approach achieves many analytic goals which are otherwise intricate to achieve [18]. Bayesian mapping approach can also use prior knowledge of QTLs. With MCMC approaches linkage analysis can be performed with any number of marker loci, multiple trait loci and multiple genomic regions. Simultaneously, MCMC allows the use of complex pedigrees of arbitrary size.
2.6. In Silico Mapping
In silico mapping was developed to identify genes by concurrently exploiting genotypic, phenotypic and pedigree data available in genomic databases and breeding programs without designed mapping experiments. This approach was first used to explore whether chromosomal segments underlying quantitative traits could be predicted with the SNP database and existing phenotypic data from mouse inbred strains [19]. The genotypic and phenotypic data was analyzed in silico to discover candidate QTL intervals. The potential of the computational method to accurately detect QTL intervals was tested. The results of 19 out of 26 experiments verified QTL intervals detected by in silico mapping. Hence, in silico mapping can reduce many months to years of field and laboratory work required to phenotype and genotype experimental progenies, to milliseconds once a large number of relevant data is publically available.
Currently, the most frequently used approaches for genetic mapping are: 1) Linkage analysis (LA) 2) LDbased Association mapping [20].
2.7. Family Based Linkage Analysis Mapping
This is classical approach in which LD is created by developing a population by crossing few founders. For family mapping, the first step is to establish mapping populations like F2, double haploids, back crosses, recombinant inbred lines and near isogenic lines which are then phenotyped to find out segregation of the trait in different environments. In the next step, DNA markers showing polymorphism between the parents and among segregants are identified. For this, a set of markers is screened for polymorphism and the polymorphic markers are used to generate genotypic data to construct linkage map (relative genetic distance) and order (position) of the molecular markers used for genotyping. The genetic map is accomplished by the assessment of recombination frequencies between the markers. The markers located on the linkage map are associated with the phenotypic data of trait(s) being studied and significantly correlated markers with a phenotypic trait are considered to be closely linked with the QTL region affecting the trait being mapped.
In family mapping, the accuracy of mapping a gene relies on the size of mapping population, genetic variation covered by the population, and number of molecular markers applied. Once, the QTLs underlying a specific trait are exactly tagged with molecular markers using linkage analysis mapping approach, the markers can be used to transfer the gene of interest from a donor line to the target genotype (marker assisted selection). Even though, linkage mapping is being used for gene mapping in crop plants, it is very costly, has low resolution and evaluates few alleles simultaneously in a relatively longer time scale [21-24]. Low resolution in linkage analysis mapping is due to lower number of meiotic events happened since experimental crossing in the near past [25]. Although linkage analysis in plants typically localizes QTLs to 10 to 20 cM intervals because of the limited number of recombination events that occur during the construction of mapping populations but it requires relatively less number of markers compared to genomewide association mapping. This advantage of linkage analysis can be used to identify putative genomic regions that can be used as prior information for fine mapping using association mapping. Association mapping is an alternative and/or complementary approach to identify marker-trait associations and has been extensively employed in animal and humangenetics [26,27] in which reasonably large segregating populations are not possible to develop.
3. Friends Based LD Mapping/Association Mapping
Current gene mapping efforts are shifting from conventional linkage analysis based mapping to LD based association mapping [28] which is the most effective approach to utilize natural variation in the form of ex situ conserved crop genetic resources. In association mapping, a natural population of unrelated individuals which can be called as friends is surveyed to determine marker-trait associations using LD [21]. LD refers to historically increased non-equilibrium (reduced level of recombinations) of specific alleles at various loci. The level of LD extent can be measured statistically and therefore has been extensively used in humans to tag and finally clone genes controlling complex quantitative traits [29-32]. This approach was extended to plants in 2001 and a substantially increased mapping resolution over F1-derived mapping populations was reported [33].
Association mapping offers several advantages over familybased mapping [34]. The availability of huge genetic variation in the form of germplasm provides broader allele coverage and saves time and cost to establish tedious and expensive bi-parental mapping populations, and most importantly offers higher resolution due to the exploitation of relatively higher number of meiotic events throughout the history of germplasm development. Association mapping also offers the possibility of using historically measured phenotypic data [35,36]. Furthermore, covering the whole genome with sufficient mapping resolution requires thousands of markers, therefore, the strategy of targeting individual linkage groups is being successfully adopted [37,38].
The general approach of association mapping includes six steps as described by Almaskri et al. [39] and adopted by Sajjad et al. [40] 1) a collection of diverse genotypes are selected that may include, land races, elite cultivars, wild relatives and exotic accessions, 2) a comprehensive and precise phenotyping is performed over the traits such as, yield, stress tolerance or quality related traits of the selected genotypes in multiple repeats and years/environments, 3) the genotypes are then scanned with suitable molecular markers (AFLP, SSRs, SNPs), 4) population structure and kinships are determined to avoid false positives followed by 5) quantification of LD extent using different statistics like D, D' or r2. Finally, 6) genotypic and phenotyping data are correlated using appropriate statistical software allowing tagging of molecular marker positioned in close proximity of gene(s) underlying a specific trait. Consequently, the tagged gene can be mobilized between different genotypes and/or cloned and annotated for a precise biological function.
In a set of unrelated individuals, mapping power using association mapping approach is the probability of detecting the true marker-trait associations that depends on 1) the evolution and extent of LD in the genomic region harboring the loci for trait(s) being mapped and mapping population; 2) the type of gene action of the trait; 3) size and composition of population; 4) field design and accuracy of phenotyping, genotyping and data analysis. The power of AM can be increased by better data recording and analysis and increasing population size. In AM there are specific statistical methods to determine the falsepositives (Type 1 error) such as permutation [41] or false recovery rates [42]. For association mapping study in the presence of population structure Pritchard et al. [43] established a useful technique for structured association (SA). Structured association (SA) uses Bayesian approach [44] to search sub-populations using Q matrix to avoid false positives. Population structure (Q-matrix) and kinship coefficient (K-matrix) can be estimated in subpopulations using the program STRUCTURE (Pritchard and Wen 2004). Recently, Yu et al. [45] established another approach called a mixed linear model (MLM) to bloc structure information (Q-matrix) and kinship information (K-matrix) in AM analysis. Later on, the Q+K MLM model performed better even in highly structured population of Arabidopsis as compared to any other model that used Qor K-matrix alone [46].
Some mixed model approaches also combine QTL and LD, where, QTLs or already known genes are used as a priori information in population mapping [47]. This is the effective approach in association mapping that reduces the number of markers and populations size. This approach also increases the precision and power of marker-trait associations [48].
4. Family and Friends Based Nested Association Mapping (NAM)
The most commonly used approaches to genetic mapping are family based linkage analysis and LD based association mapping [49]. Considering their advantages and disadvantages these two approaches are complementary. Linkage analysis identifies wide chromosomal region underlying the trait of interest with relatively lower number of marker coverage, therefore, it has low mapping resolution. On the other hand, association mapping identifies chromosomal region of interest with high resolution using higher marker density [33]. Nested association mapping integrates family based linkage analysismapping and association mapping to combine their respective advantages to enhance mapping resolution without using very dense marker maps. The creation of NAM population is pre-requisite for NAM study. The first NAM population was developed in model crop maize (Zea mays L.) because of immediate availability of highly diverse germplasm and possibility of generating segregating progenies and their selfing to make immortal RIL genotypes [50].
The NAM strategy is to generate an immortal common mapping population that could be exploited efficiently by researches for genomic, genetic and system biology tools to dissect complex traits. First NAM population of Pakistani wheats is being developed at the Department of Plant Breeding and Genetics, PMAS-Arid Agriculture University Rawalpindi. The procedure of developing this mapping population is outlined in figure 1.
The procedure for developing of nested association mapping population in wheat being adopted at Department of Plant Breeding and Genetics PMS-Arid Agriculture University Rawalpindi is given below.
1) Selection of highly diverse wheat genotypes as founders and crossing with a reference genotype (e.g. Inqlab-91-the most successful cultivar), followed by selfpollination of each hybrid for six generations and selecting 200 homozygous recombinant inbred lines (RILs) per family (total 6000 RILs).
2) Genotyping of each founder with large number of molecular markers for which Inqlab-91 will have rare alleles.
3) Genotyping with a smaller number of tagging markers on both the founders and the progenies to identify the inheritance of chromosome segments and to project the high-density marker information from the founders to the progenies.
4) Phenotyping of progenies for various complex traits.
5) Conducting genome-wide association analysis connecting phenotypic traits with high-density markers of the progenies.
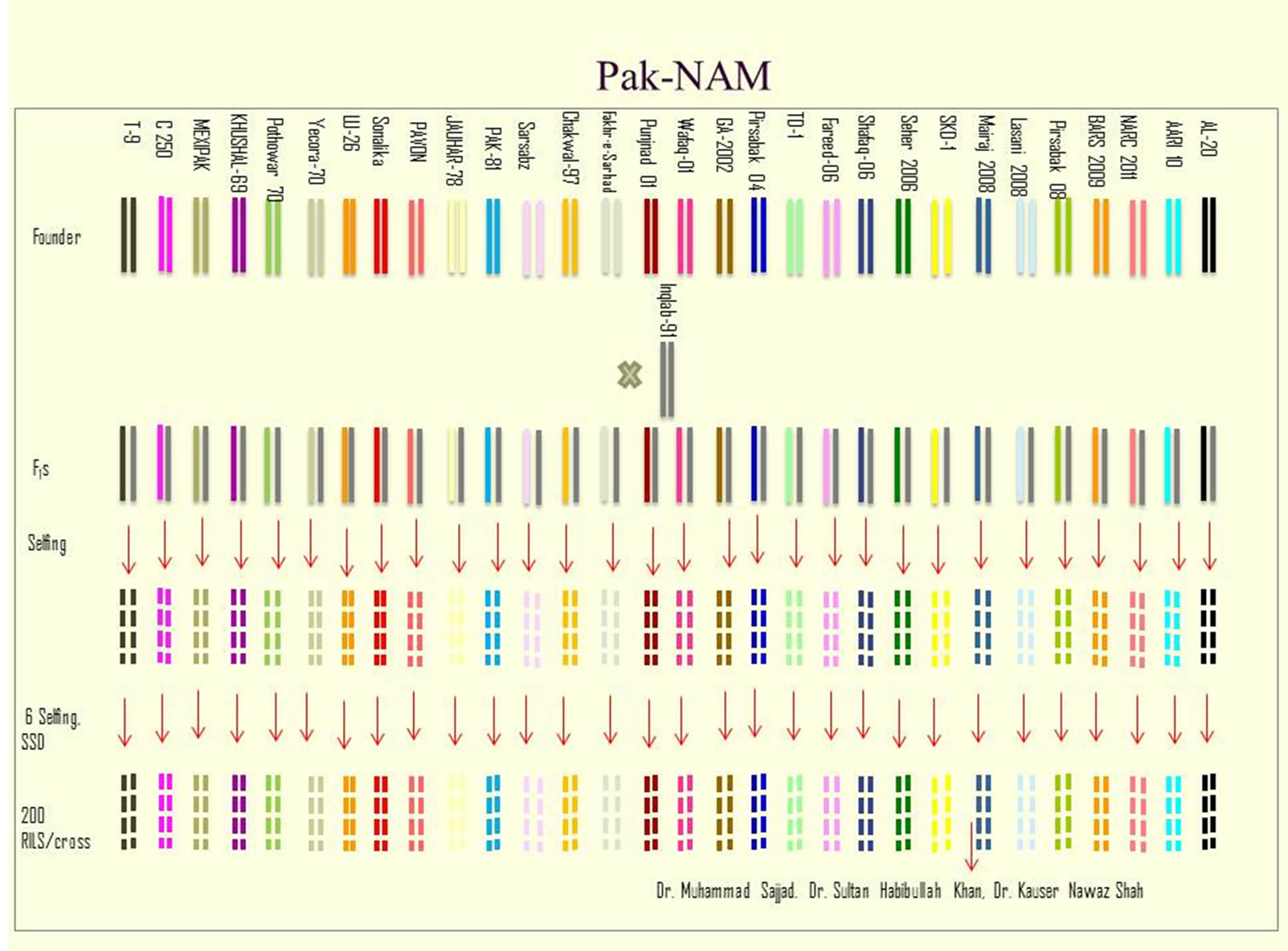
Figure 1. Diagrametic presentation of development of wheat Pak-NAM population.
5. Conclusion
The integration of family based linkage analysis and LD mapping approaches in the form of nested association mapping approach would enhance QTL mapping resolution power resulting in precise marker-trait association. Since a NAM population is stable and immortal, multilocations and multi-years phenotyping would enhance the validity of QTLs leading to more accurate marker assisted selection in future.
REFERENCES
- M. Sajjad, S. H. Khan and A. S. Khan, “Exploitation of Germplasm for Grain Yield Improvement in Spring Wheat (Triticumaestivum),” International Journal of Agriculture and Biology, Vol. 13, No. 5, 2011, pp. 695-700.
- M. Akbari, P. Wenzl, V. Caig, J. Carling, L. Xia, S. Yang, G. Uszynski, V. Mohler, A. Lehmensiek, H. Kuchel, M. J. Hayden, N. Howes, P. Sharp, P. Vaughan and B. Rathmell, “Diversity Arrays Technology (DArT) for HighThroughput Profiling of the Hexaploid Wheat Genome,” Theoretical and Applied Genetics, Vol. 113, No. 8, 2006, pp. 1409-1420. http://dx.doi.org/10.1007/s00122-006-0365-4
- P. A. Peterson, “Quantitative Inheritance in the Era of Molecular Biology,” Maydica, Vol. 37, 1992, pp. 7-18.
- H. Geldermann, “Investigations on Inheritance of Quantitative Characters in Animals by Gene Markers. I. Methods,” Theoretical and Applied Genetics, Vol. 46, 1975, pp. 319-330. http://dx.doi.org/10.1007/BF00281673
- Y. Xu, “Molecular Plant Breeding,” CAB International, USA, 2010. http://dx.doi.org/10.1079/9781845933920.0000
- R. Wu, C. Ma and G. Casella, “Statistical Genetics of Quantitative Traits: Linkage, Maps and QTL,” Statistics for Biology and Health, Springer, Berlin, 2007.
- D. Sorensen and D. Gianola, “Likelihood, Bayesian and MCMC Methods in Quantitative Genetics,” Springer-Verlag Inc., New York, 2002.
- B. H. Liu, “Statistical Genomics: Linkage, Mapping and QTL Analysis,” CRC Press, Boca Baton, 1998, p. 611.
- M. Lynch and B. Walsh, “Genetics and Analysis of Quantitative Traits,” Sinauer Associates, Sunderland, 1998, p. 980.
- M. D. Edwards, C. W. Stuber and J. F. C. Wendel, “Molecular Marker Facilitated Investigations of Quantitative trait Loci in Maize,” Genetics, Vol. 116, 1987, pp. 113- 125.
- E. S. Lander and D. Botstein, “Mapping Mendelian Factors Underlying Quantitative Traits Using RFLPlinkage Maps,” Genetics, Vol. 121, 1989, pp. 185-199.
- Z. B. Zeng, “Precision Mapping of Quantitative Trait Loci,” Genetics, Vol. 136, 1994, pp. 1457-1468.
- O. Martinez and R. N. Curnow, “Estimating the Locations and the Sizes of the Effects of Quantitative Trait Loci Using Flanking Markers,” Theoretical and Applied Genetics, Vol. 85, 1992, pp. 480-488. http://dx.doi.org/10.1007/BF00222330
- J. M. Satagopan, B. S. Yandell, M. A. Newton and T. G. Osborn, “A Bayesian Approach to Detect Quantitative Trait Loci Using Markov Chain Monte Carlo,” Genetics, Vol. 144, 1996, pp. 805-816.
- C. H. Kao and Z. B. Zeng, “General Formulas for Obtaining the MLEs and the Asymptotic Variance-Covariance Matrix in Mapping Quantitative Trait Loci When Using the EM Algorithm,” Biometrics, Vol. 53, 1997, pp. 653-665. http://dx.doi.org/10.2307/2533965
- S. Sen and G. A. Churchill, “A Statistical Framework for Quantitative Trait Mapping,” Genetics, Vol. 159, 2001, pp. 371-387.
- D. Malakoff, “Bayes Offers a ‘New’ Way to Make Sense of Numbers,” Science, Vol. 286, 1999, pp. 1460-1464. http://dx.doi.org/10.1126/science.286.5444.1460
- S. Xu, “QTL Analysis in Plants,” In: N. J. Camp and A. Cox, Eds., Methods in Molecular Biology, Vol. 195, Quantitative Trait Loci: Methods and Protocols, Humana Press, Totowa, 2002, pp. 283-310.
- A. Grupe, S. Germer, J. Usuka, D. Aud, J. K. Belknap, R. F. Klein, M. K. Ahluwalia, R. Higuchi and G. Peltz, “In Silico Mapping of Complex Disease-Related Traits in Mice,” Science, Vol. 292, 2001, pp. 1915-1918. http://dx.doi.org/10.1126/science.1058889
- I. Mackay and W. Powell, “Methods for Linkage Disequilibrium Mapping in Crops,” Trends in Plant Science, Vol. 12, No. 2, 2007, pp. 57-63. http://dx.doi.org/10.1016/j.tplants.2006.12.001
- S. A. Flint-Garcia, J. M. Thornsberry and E. S. Buckler, “Structure of Linkage Disequilibrium in Plants,” Annual Review of Plant Biology, Vol. 54, 2003, pp. 357-374. http://dx.doi.org/10.1146/annurev.arplant.54.031902.134907
- P. K. Gupta, S. Rustgi and P. L. Kulwal, “Linkage Disequilibrium and Association Studies in Higher Plants: Present Status and Future Prospects,” Plant Molecular Biology, Vol. 57, No. 4, 2005, pp. 461-485. http://dx.doi.org/10.1007/s11103-005-0257-z
- B. Stich, H. P. Maurer, A. E. Melchinger, M. Frisch, M. Heckenberger, J. R. van der Voort, J. Peleman, A. P. Sø- rensen and J. C. Reif, “Comparison of Linkage Disequilibrium in Elite European Maize Inbred Lines Using AFLP and SSR Markers,” Molecular Breeding, Vol. 17, No. 3, 2006, pp. 217-226. http://dx.doi.org/10.1007/s11032-005-5296-2
- J. Ross-Ibarra, P. L. Morrell and B. S. Gaut, “Plant Domestication, a Unique Opportunity to Identify the Genetic Basis of Adaptation,” Proceedings of the National Academy of Sciences, Vol. 104, No. 1, 2007, pp. 8641-8648. http://dx.doi.org/10.1073/pnas.0700643104
- J. L. Jannink and B. Walsh, “Association Mapping in Plant Populations,” In: M. S. Kang, Ed., Quantitative Genetics, Genomics and Plant Breeding, CAB International, Oxford, 2002, pp. 59-68.
- E. K. Karlsson, I. Baranowska, C. M. Wade, N. H. C. S. Hillbertz, M. C. Zody, N. Anderson, T. M. Biagi, N. Patterson, G. R. Pielberg, E. J. I. Kulbokas, K. E. Comstock, E. T. Keller, J. P. Mesirov, H. Euler, O. Kämpe, A. Hedhammar, E. S. Lander, G. Andersson, L. Andersson and K. Lindblad-Toh, “Efficient Mapping of Mendelian Traits in Dogs through Genome-Wide Association,” Nature Genetics, Vol. 39, 2007, pp. 1321-1328. http://dx.doi.org/10.1038/ng.2007.10
- A. DeWan, M. Liu, S. Hartman, S. S. Zhang, D. T. L. Liu, C. Zhao, P. O. S. Tam, W. M. Chan, D. S. C. Lam, M. Snyder, C. Barnstable, C. P. Pang and J. Hoh, “HTRA1 Promoter Polymorphism in Wet Age-Related Macular Degeneration,” Science, Vol. 314, 2006, pp. 989-992. http://dx.doi.org/10.1126/science.1133807
- D. B. Goldstein and M. E. Weale, “Population Genomics: Linkage Disequilibrium Holds the Key,” Current Biology, Vol. 11, No. 14, 2001, pp. 576-579. http://dx.doi.org/10.1016/S0960-9822(01)00348-7
- N. Risch and K. Merikangas, “The Future of Genetic Studies of Complex Human Diseases,” Science, Vol. 273, No. 5281, 1996, pp. 1516-1517. http://dx.doi.org/10.1126/science.273.5281.1516
- K. M. Weiss and A. G. Clark, “Linkage Disequilibrium and Mapping of Human Traits,” Trends in Genetics, Vol. 18, No. 1, 2002, pp. 19-24. http://dx.doi.org/10.1016/S0168-9525(01)02550-1
- J. M. Chapman, J. D. Cooper, J. A. Todd and D. G. Clayton, “Detecting Disease Associations Due to Linkage Disequilibrium Using Haplotype Tags: A Class of Tests and the Determinants of Statistical Power,” Human Heredity, Vol. 56, No. 1-3, 2003, pp. 18-31. http://dx.doi.org/10.1159/000073729
- H. Taniguchi, C. E. Lowe, J. D. Cooper, D. J. Smyth, R. Bailey, S. Nutland, B. C. Healy, A. C. Lam, O. Burren, N. M. Walke, L. J. Smink, L. S. Wicker and J. A. Todd, “Discovery, Linkage Disequilibrium and Association Analyses of Polymorphisms of the Immune Complement Inhibitor, Decay Accelerating Factor Gene (DAF/CD55) in Type 1 Diabetes,” B. M. C. Genetics, Vol. 7, No. 22, 2006. http://dx.doi.org/10.1186/1471-2156-7-22
- J. M. Thornsberry, M. M. Goodman, J. Doebley, S. Kresovich, D. Nielsen and E. S. Buckler, “Dwarf8 Polymorphisms Associate with Variation in Flowering Time,” Nature Genetics, Vol. 28, 2001, pp. 286-289. http://dx.doi.org/10.1038/90135
- M. Sajjad, S. H. Khan and A. M. Kazi, “The Low Down on Association Mapping in Hexaploid Wheat (Triticumaestivum L.),” Journal of Crop Science and Biotechnology, Vol. 15, No. 3, 2012, pp. 147-158. http://dx.doi.org/10.1007/s12892-012-0021-2
- A. T. W. Kraakman, R. E. Niks, P. M. M. M. Van den Berg, P. Stam and F. A. Van Eeuwijk, “Linkage Disequilibrium Mapping of Yield and Yield Stability in Modern Spring Barley Cultivars,” Genetics, Vol. 168, No. 1, 2004, pp. 435-446. http://dx.doi.org/10.1534/genetics.104.026831
- A. T. W. Kraakman, F. Martinez, B. Mussiraliev, F. A. van Eeuwijk and R. E. Niks, “Linkage Disequilibrium Mapping of Morphological, Resistance, and Other Agronomically Relevant Traits in Modern Spring Barley Cultivars,” Molecular Breeding, Vol. 17, No. 1, 2006, pp. 41- 58. http://dx.doi.org/10.1007/s11032-005-1119-8
- J. Yao, L. Wang, L. Liu, C. Zhao and Y. Zheng, “Association Mapping of Agronomic Traits on Chromosome 2A of Wheat,” Genetica, Vol. 137, 2009, pp. 67-75. http://dx.doi.org/10.1007/s10709-009-9351-5
- L. Liu, L. Wang, J. Yao, Y. Zheng and C. Zhao, “Association Mapping of Six Agronomic Traits on Chromosome 4A of Wheat (Triticumaestivum L.)” Molecular Plant Breeding, Vol. 1, 2010, pp. 1-10.
- A. H. Al-Maskri, M. Sajjad and S. H. Khan, “Association Mapping: A Step Forward to Discovering New Alleles for Crop Improvement,” International Journal of Agriculture and Biology, Vol. 14, No. 1, 2012, pp. 153-160.
- M. Sajjad, S. H. Khan, M. Qadir, A. Rasheesd, A. M. Kazi and I. A. Khan, “Association Mapping Identifies QTLs on Wheat Chromosome 3A for Yield Related Traits,” Cereal Research Communication.
- G. A. Churchill and R. W. Doerge, “Empirical Threshold Values for Quantitative Trait Mapping,” Genetics, Vol. 138, No. 3,1994, pp. 963-971.
- Y. Benjamini and Y. Hochberg, “Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing,” Journal of the Royal Statistical Society: Series B, Vol. 57, No. 1, 1995, pp. 289-300.
- J. K. Pritchard, M. Stephens and P. Donnelly, “Inference of Population Structure Using Multilocus Genotype Data,” Genetics, Vol. 155, 2000, pp. 945-959.
- P. Marttinen and J. Corander, “Efficient Bayesian Approach for Multilocus Association Mapping Including Gene-Gene Interactions,” BMC Bioinformatics, Vol. 11, 2010, p. 443. http://dx.doi.org/10.1186/1471-2105-11-443
- J. Yu, G. Pressoir, W. H. Briggs, I. V. Bi, M. Yamasaki, J. F. Doebley, M. D. McMullen, B. S. Gaut, D. M. Nielsen, J. B. Holland, S. Kresovich and E. S. Buckler, “A Unified Mixed-Model Method for Association Mapping that Accounts for Multiple Levels of Relatedness,” Nature Genetics, Vol. 38, 2006, pp. 203-208. http://dx.doi.org/10.1038/ng1702
- K. Zhao, M. J. Aranzana, S. Kim, C. Lister, C. Shindo, C. Tang, C. Toomajian, H. Zheng, C. Dean, P. Marjoram and M. Nordborg, “An Arabidopsis Example of Associationmapping in Structured Samples,” PLoS Genetics, Vol. 3, No. 1, 2007, p. e4. http://dx.doi.org/10.1371/journal.pgen.0030004
- B. R. Thumma, M. F. Nolan, R. Evans and G. F. Moran, “Polymorphisms in Cinnamoyl CoA Reductase (CCR) Are Associated with Variation in Microfibril Angle in Eucalyptus spp.” Genetics, Vol. 71, No. 3, 2005, pp. 1257-1265. http://dx.doi.org/10.1534/genetics.105.042028
- R. D. Ball, “Experimental Designs for Reliable Detection of Linkage Disequilibrium in Unstructured Random Population Association Studies,” Genetics, Vol. 170, No. 2, 2005, pp. 859-873. http://dx.doi.org/10.1534/genetics.103.024752
- T. F. Mackay, “Quantitative Trait Loci in Drosophila,” Nature Reviews Genetics, Vol. 1, 2001, pp. 11-20. http://dx.doi.org/10.1038/35047544
NOTES
*Corresponding author.