Open Journal of Pediatrics
Vol. 3 No. 3 (2013) , Article ID: 35962 , 4 pages DOI:10.4236/ojped.2013.33042
Current profile and outcome of 100 esophageal atresia patients in the Kyushu area of Japan
![]()
Department of Pediatric Surgery, Reproductive and Developmental Medicine, Graduate School of Medical Sciences, Kyushu University, Fukuoka, Japan
Email: taguchi@pedsurg.med.kyushu-u.ac.jp
Copyright © 2013 Nagata Kouji et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received 31 May 2013; revised 1 July 2013; accepted 9 July 2013
Keywords: Esophageal Atresia; Outcome; Low-Birth-Weight Infant; Congenital Heart Disease; Operation
ABSTRACT
Objectives: Since Spitz et al. reported the prognostic classification of esophageal atresia (EA) patients in 1994, decades have been past and there have been many advances in surgery and neonatology. Nevertheless, there have been very few reports according to the recent outcome of the neonates with EA, and otherwise, time has come to re-evaluate the credibility of this classification. The aim of this study was to validate the recent prognosis of the EA. Methods: Patient data were collected from 22 cooperative facilities during the 5 year period from 2005 to 2009 in Kyushu area, Japan. Total of 100 EA patients were retrospectively reviewed according to their characteristics and the outcome. Patient who missed the characteristics and outcome was excluded from the respective data. Results: Only 29.8% (28/94) was prenatally diagnosed and 52.0 (52/100) had associated anomalies including major congenital heart disease (CHD), abnormal chromosome, and others. According to the operation, primary anastomosis was performed 57.0% (57/100) and the staged operation was performed 34.0% (34/100). Survival rate in the neonatal period was 89.0% (89/ 100), and overall survival rate was 78.0% (78/100). According to the Spitz classification, if patients with associated anomalies were excluded, survival rate of Group 1 (>1500 g and no CHD) was 93.8% (61/65), Group 2 (<1500 g or CHD) was 68.4% (13/19), and Group 3 (<1500 g and CHD) was 50% (1/2). Conclusion: EA was proved to be rarely diagnosed prenatally. Primary outcome of the Group 1 and Group 3 in Spitz classification were fairly good, but Group 2 was worse as ever. The comprehensive treatment strategy for EA patients with birth weight under 1500 g or CHD should be reconsidered to improve the overall outcome.
1. INTRODUCTION
Esophageal atrasia (EA) is a malformation occurring in 1: 2500 births, and its outcome has been improved since its initial identification [1,2]. Spitz et al. examined the prognostic classification of EA in 1994, both the birth weight under 1500 g and the congenital heart anomaly were recognized as the most valuable prognostic factors [3]. This classification system has proven to be clinically useful for treatment stratification, counseling parents, and comparing outcomes between institutions and the nations [4-8]. With many advances in the fields of neonatal intensive care, nutritional support, anesthesiology and surgical management during the last decade, recent reports have reviewed the patient outcome for EA during the periods between these decades, and documented some improvements in outcome [8,9].
Recent reports have documented that the predictors for adverse outcome changed, with cardiac anomalies becoming the most important risk factor [3,6,9,10]. And other reports have stated that the birth weight <1500 g showed no significant difference when compared to the patients with birth weights >1500 g [6].
However, the operative strategy for the EA patients with the presence of the concomitant congenital heart disease might be different and the multi-disciplinary consideration should be given to improve the morbidity and the mortality [11]. And the initial operative strategy for the high-risk, very low birth weight (VLBW) infant also remains controversial and continues to be a surgical challenge [12,13].
Herein, the aim of this study was to validate the recent profile and outcome of the neonates with EA, treated in the Kyushu area of Japan over the last 5 years, and also to validate the operative strategy and the outcome of the EA patients classified by the Spitz classification.
2. PATIENTS AND METHODS
We sent questionnaires to the Department of Pediatric Surgery at representative institutions throughout the Kyushu area of Japan. The questionnaires were designed to collect data on the current profile and outcome of patients with EA.
Twenty two out of 24 institutions (92%) answered our questionnaires. Among 105 patients for whom data were obtained, 100 (95%) patients were included in this study. Five patients were excluded because there had not written the primary outcome. Primary outcome was defined as the patient’s survival for 6 months after the first operation. Patient demographics, the presence of associated anomalies, initial operative treatment and outcomes were examined. Associated anomalies involved chromosomal abnormalities, congenital heart disease (CHD), anorectal, gastrointestinal, genitourinary, digital, and VACTERL (vertebra, anorectal, cardiac, tracheo-esophageal, radial/renal, limb) and others. CHD was defined as a disease that required medical or surgical treatment. A diagnosis of VACTERL association was made if three or more components of the association were present.
Chi square tests (c2) were used to compare the categorical variables between the alive group and the died group, and the threshold of significance was considered to be P < 0.05.
3. RESULT
The data from a total of 100 EA patients was collected from the cooperating institutions. Twenty-eight out of 94 (30%) patients were diagnosed prenatally. There were 59 males and 41 females. With regard to birth weight, twenty one (21%) infants weighed less than 1500 g, thirtyseven (37%) more than 1500 g but less than 2500 g, and forty-one (41%) weighed more than 2500 g. A total of fifty-two patients (52%) had associated anomalies, including twenty-three (23%) with CHD, nine (9%) with anorectal, seven (7%) gastrointestinal, six (6%) genitourinary, and three (3%) digital abnormalities, two cases (2%) had a VACTERL association, one (1%) had an omphalocele, and ten (10%) other anomlies were not written in detail. Thirteen patients (13%) had chromosmal abnormalities, including two trisomy 21 patients, six trisomy 18 patients, one partial trisomy 3 patient, one mosaic pattern of chromosome 14. Three chromosomal abnormalities were not written in detail.
According to the anatomical variations with EA, ninety (90%) patients had EA with a distal traceo-esophageal fistula (TEF), six (6%) patients had isolated EAthree (3%) patients had EA with proximal and distal TEF, and one (1%) patient had TEF without EA. Figure 1 shows the treatments used for the EA. One patient did not receive any surgery due to the presence of a lethal chromosomal abnormality, trisomy 13. Fifty-seven out of 99 (58%) patients received a primary anastomosis and thirty-four (34%) out of 99 patients received a staged operation. A total of 8 patients received a palliative operation, including a gastrostomy, tracheostomy, or gastrostomy with esophageal banding.
The overall survival rate was 78%, where one patient died without surgery (trisomy 13), 10 patients died within 30 days after the operation, and 11 patients died more than 30 days after the operation. The comparisons between the alive group and the died group were shown in Table 1. Birth at the gestational age <33 weeks, birth weight under 1500 g, trisomy 18 and primary repair were significant prognostic factors. Among them, only primary repair was the favorable prognostic factor.
The comparisons of outcome for EA according to the Spitz classification were shown in Table 2. In this study, the survival rate in Group 1 (birth weight > 1500 g, no CHD was 92.5% (62/67), Group 2 (birth weight < 1500 g or with CHD) was 58.6% (14/26) and Group 3 (birth weight < 1500 g and with CHD) was 12.5% (1/7), respectively. In this study, thirteen patients with chromosomal abnormalities were included and when these patients were excluded from the study, survival rate in Groups 1, 2 and 3 became up to 93.8% (61/65), 68.4% (13/19) and 50.0% (1/2), respectively.
4. DISCUSSION
The preoperative prognostic classification is important for giving clinicians and parents a realistic prognosis for children, and to compare results among institutions. In 1994, Spitz et al. proposed a new system of risk classification, using both major cardiac anomalies and low birth weight as preoperative risk factors [4]. Almost two decades have passed since this proposed classification, and there have been many advances in perinatal and neonatal intensive care, including the centralization of perinatal support, transport systems, nutritional support and the
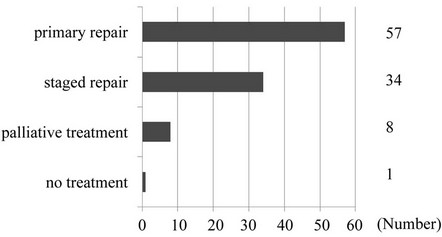
Figure 1. Treatments for EA.

Table 1. Comparosons between the alive group and the died group.
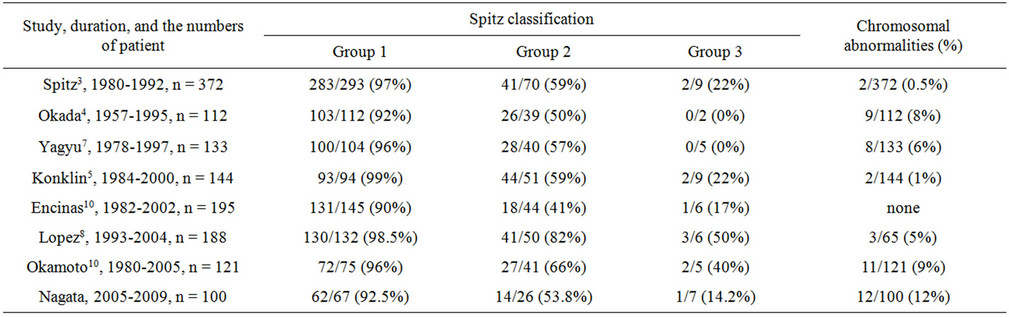
Table 2. Comparosons of outcome for EA according to the Spitz classifiation and the rates of chromosomal abnormalities.
management of respiratory distress syndrome, which have contributed to the further improvement of outcome in patients. In fact, Lopez et al. reported data analyzed using this Spitz classification, and documented a significant improvement in Group 2 infants, thus speculating that the improved survival was the result of advances in the management of the low-birth-weight infants [8].
However, the operative strategy for the EA with CHD or with VLBW infants still remains controversial and affects the outcome. Indeed, birth at the gestational age < 33 weeks, birth weight under 1500 g and trisomy 18 were significantly bad prognostic factors in our study. And the survival rates in Groups 2 and 3 were lower, compared to the recent study reported in the literature. However, the numbers of patients who had chromosomal abnormalities were relatively higher in this study, it might be difficult to compare. According to treatment strategy for the EA with CHD, Encinas et al. documented the low rate of diagnose for the cardiac anomalies before EA operation and showed relatively higher mortality in Spitz classification [10]. And Hayashi et al. described the treatment strategy for their 6 EA patients with CHD. In their study, they stated that surgical repair of EA usually performed ahead of palliative cardiac surgery for CHD, although the necessity of cardiac surgery in the early neonatal period should be anticipated for patients with duct-dependent circulations [11]. Because CHD associated with EA contains a wide variety of types and levels of severity, it is important to realize that the treatment strategy should be tailored to each patient and the multidisciplinary consideration should be given to improve the morbidity and the mortality.
According to the operative strategy for the VLBW infant, there also remains controversial. There is no definition or criteria about the appropriate body weight for primary anastomosis. And those VLBW infants with associated congenital anomalies as well as infants with severe respiratory distress syndrome are less likely to tolerate single lung ventilation and the operative time required to perform the primary repair [12,13]. Although Choudly et al. stated that low birth weight does not affect the outcome [6], Petrosyan et al. showed the significantly lower complication rate of anastomotic complications and overall morbidity should be considered to be the preferable surgical approach to the VLBW infants [13]. Prospective study will be needed to conclude the efficacy of the treatment strategy for EA with VLBW infant.
This study included 13 EA patients with chromosomal abnormalities. And when the chromosomal abnormalities were excluded from this series, survival rates in the Group 2 and the Group 3 increased. Several reports documented that EA with lethal congenital anomalies for whom surgery would be of no benefit [3,8]. Those include major cerebral anomalies, Grade IV intraventricular hemorrhage, pulmonary atresia, trisomy 18, and complex CHARGE syndrome [3,8]. These data deletion of the patient number should be cautioned because the outcome will be easily changed to better and therefore, the selection bias will be occurred.
One of the limitations of this study was that this study was the query formation, so that there exist as the limitation of the detailed analysis. And the other limitation was that the treatment strategy for EA was definitely different in each institution. Nevertheless, this study described the recent issue around EA patients which will continue to be discussed for future. To elucidate the proper treatment for the EA, centralization and the tailored treatment strategy might be needed in Kyushu area.
In summary, we have herein described the characteristics and outcomes of EA patients treated in the Kyushu area of Japan. Primary outcome of the Group 1 in Spitz classification was fairly good, but Group 2 was worse as ever. The comprehensive treatment strategy for EA patients with birth weight under 1500 g or CHD should be reconsidered to improve the overall outcome.
5. ACKNOWLEDGEMENTS
Author would like to thank you for the departments participated in this survey; Fukuoka Children’s Hospital and Medical Center for infectious disease, Department of Pediatric surgery (Fukuoka); Fukuoka University Hospital, Department of Thoracic, Endocrine and Pediatric surgery (Fukuoka); Iizuka Hospital, Department of Pediatric surgery (Fukuoka); Japanese Red Cross Hospital, Department of Pediatric surgery (Kumamoto); Kagoshima University School of Medicine, Department of Pediatric surgery (Kagoshima); Kitakyushu Municipal Medical Center, Department of Pediatric surgery (Fukuoka); Kumamoto City Hospital, Department of Pediatric surgery (Kumamoto); Kumamoto University, postgraduate school of medical science, Department of Transplantation and Pediatric surgery (Kumamoto); Kurume University School of Medicine, Department of Pediatric surgery (Fukuoka); Kyushu KouseiNenkin Hospital, Department of Pediatric surgery (Fukuoka); Kyushu Medical Center, Department of Pediatric surgery (Fukuoka); Miyazaki Prefectual Miyazaki Hospital, Department of Pediatric surgery (Miyazaki); Nakatsu Municipal Hospital, Department of Pediatric surgery (Ooita); Nagasaki University School of Medicine and Dentistry, Department of Surgery (Nagasaki); Okinawa Prefectural Nanbu Medical Center and Children’s Hospital, Division of Pediatric surgery (Okinawa); Ooita Prefectural Hospital, Department of Pediatric surgery (Ooita); Ooita University Hospital, First Department of Surgery (Ooita); Saga Prefectural Hospital Kouseikan, Department of Pediatric surgery (Saga); Shimonoseki City Central Hospital, Department of Pediatric surgery (Yamaguchi); St. Mary’s Hospital, Kurume, Department of Pediatric surgery (Fukuoka); University of Occupational and Environmental Health, First Department of Surgery (Fukuoka); University of the Ryukyus, First Department of Surgery (Okinawa).
REFERENCES
- Spitz, L. (2006) Esophageal atresia. Lessons I have learned in a 40-year experience. Journal of Pediatric Surgery, 41, 1635-1640. doi:10.1016/j.jpedsurg.2006.07.004
- Spitz, L. (1996) Esophageal atresia: Past, present, future, and future. Journal of Pediatric Surgery, 31, 19-25. doi:10.1016/S0022-3468(96)90313-9
- Spitz, L., Kiely, E.M., Morecroft, J.A. and Drake, D.P. (1994) Oesophageal atresia: At-risk groups for the 1990s. Journal of Pediatric Surgery, 29, 723-725. doi:10.1016/0022-3468(94)90354-9
- Okada, A., Usui, N., Inoue, M., Kawahara, H., Kubota, A., Imura, K. and Kamata, S. (1997) Esophageal atresia in Osaka: A review of 39 years’ experience. Journal of Pediatric Surgery, 32, 1570-1574. doi:10.1016/S0022-3468(97)90455-3
- Konklin, D.E., O’Hali, W.A., Webber, E.M. and Blair, G.K. (2003) Outcomes in esophageal atresia and tracheoesophageal fistula. Journal of Pediatric Surgery, 38, 1726- 1729. doi:10.1016/j.jpedsurg.2003.08.039
- Chroudhury, S., Aschcraft, K.W., Sharp, R.J., Murphy, J.P., Snyder, C.L. and Sigalet, D.L. (1999) Survival of patients with esophageal esophageal atresia: Influence of birth weight, cardiac anomaly, and late respiratory complications. Journal of Pediatric Surgery, 34, 70-74. doi:10.1016/S0022-3468(99)90231-2
- Yagyu, M., Gitter, H., Richter, B. and Boose, D. (2000) Esophageal atresia in Bremen, Germany—Evaluation of preoperative risk classification in esophageal atresia. Journal of Pediatric Surgery, 35, 584-587. doi:10.1053/jpsu.2000.0350584
- Lopez, P.J., Keys, C., Pierro, A., Drake, D.P., Kiely, E.M., Curry, J.I. and Spitz, L. (2006) Oesophageal atresia: Improved outcome in high-risk group? Journal of Pediatric Surgery, 41, 331-334. doi:10.1016/j.jpedsurg.2005.11.009
- Okamoto, T., Takamizawa, S., Arai, H., Bitoh, Y., Nakao, M., Yokoi, A. and Nishijima, E. (2009) Esophageal atresia: Prognostic classification revised. Surgery, 145, 675- 681. doi:10.1016/j.surg.2009.01.017
- Encinas, J.L., Luis, A.L., Avila, L.F., Martinez, L., Guereta, L., Lassaletta, L. and Tovar, J.A. (2006) Impact of preoperative diagnosis of congenital heart disease on the treatment of esophageal atresia. Journal of Pediatric Surgery, 22, 150-153. doi:10.1007/s00383-005-1595-2
- Hayashi, T., Inuzuka, R., Shiozawa, Y., Shindo, T., Shimizu, N. and Katori, T. (2013) Treatment strategy and long-term prognosis for patients with esophageal atresia and congenital heart diseases. Pediatric Cardiology, 34, 64-69. doi:10.1007/s00246-012-0386-5
- Alexander, F., Johanningman, J. and Martin, L.W. (1993) Staged repair improves outcome of high risk premature infants with esophageal atresia and tracheoesophageal fistula. Journal of Pediatric Surgery, 28, 151-154. doi:10.1016/S0022-3468(05)80261-1
- Petrosyan, M., Estrada, J., Hunter, C., Woo, R., Stein, J., Ford, H.R. and Anselmo, D.M. (2009) Esophageal atresia/tracheoesophageal fistula in very low-birthweight neonates: Improved outcomes with staged repair. Journal of Pediatric Surgery, 44, 2278-2281. doi:10.1016/j.jpedsurg.2009.07.047