Paper Menu >>
Journal Menu >>
 J. Biomedical Science and Engineering, 2010, 3, 109-113 JBiSE doi:10.4236/jbise.2010.32016 Published Online February 2010 (http://www.SciRP.org/journal/jbise/). Published Online February 2010 in SciRes. http://www.scirp.org/journal/jbise Novel method for discerning the action of selection during evolution Ming Yang1, Ada Solidar2, Gerald J. Wyckoff1 1Division Molecular Biology and Biochemistry, University of Missouri-Kansas City, Kansas City, Missouri, USA 2VaSSA Informatics, LLC Kansas City, Missouri, USA Email: wyckoffg@umkc.edu, ada@vassainformatics.com Received 10 October 2009; revised 10 December 2009; accepted 15 December 2009. ABSTRACT A common problem in molecular comparative geno- mics is the identification of genes that are under posi- tive, adaptive selection [1]. Such genes are likely to be crucial for speciation, species differentiation, and func- tional specialization. However, discerning the differ- ence between positive selection and relaxation of func- tional constraint can be difficult using current meth- ods. Both processes generally increase the rate of ami- no acid change relative to synonymous changes within coding regions, and unless the amino acid rate is over- whelmingly high across an entire gene, the signature of positive selection can be obscured [2]. Some meth- odologies do not explicitly determine the difference be- tween a relaxation of functional constraint and posi- tive selection, leaving researchers to determine via other means whether the trajectory of a gene has been spe- cialization or creation of a new function, or removal from the genome via a process of degeneration. Keywords: Utilizing Information Theory; Action of Selection during Evolution 1. INTRODUCTION Most current methods evaluate the possibility of positive selection based on the exchangeabilities of amino acids. The rationale is that if an observed amino acid substitu- tion has a low probability in terms of their amino acids’ physio-chemical properties, then it is more probable that the substitution may be driven by selection events. There are several kinds of matrices that can be used to evaluate the probability of substitutions. Function, charge, and amino acid structural properties (via Karlin and Ghandour [3]) and genetic and structural similarity (from Feng et al., [4]) are common methods. However, Dayhoff's PAM-250 matrix is easily the most common. Based on evolutionary distance measures from a 1,572 amino acid change data set in 71 closely related proteins, PAM stands for “percent-accepted matrix” [5]. It set the path for most matrices to come. Henikoff and Henikoff proposed the BLOSUM (BLO- cks of Amino Acid Substitution Matrix) matrices based on a large number of proteins to get a better measure of differences between two proteins specifically for more distantly related proteins [6]. To create the matrices, the BLOCKS database was searched for ungapped, highly conserved protein domains within protein families and amino acid frequency substitutions were determined, scaled by relative amino acid frequency. [7] They then calculated a log- odds score for each of the 210 possible substitutions of the 20 standard amino acids. BLOSUM was designed for search algorithms when relatively close protein relationships are being examined, such as FASTA and BLAST. However, this work set the stage for other research looking at more fine-grained matrices for evolutionary comparison and ultimately led to the work described in this paper. Contrasting both PAM and BLOSUM matrices that are based on amino acids, Tang and co-workers proposed a universal evolutionary index (EI) for amino acid chan- ges based on the genetic code [8]. The EIs are defined as the observed/expected amino acid changes based on the transition and transversion rate between related codons. The high correlations between EIs derived from genes with various functions in divergent species suggest that the amino acid properties are strong determinants of their substitution patterns. The EIs can be used to clas- sify proteins based on their exchangeability and detect the positive selection in each of the groups. There is another category of methodologies that are based on the sequence information content at specific sites. In an alignment of DNA or amino acid sequences, the information content for each position is calculated based on the distribution of the variations at that site, and they are measured in bits [9]. The information con- tents are smaller for divergent sites and larger for con- served sites. It can therefore be thought of as giving a measure of the tolerance for substitutions at the position: higher information content indicates that the site can  M. Yang et al. / J. Biomedical Science and Engineering 3 (2010) 109-113 Copyright © 2010 SciRes. JBiSE 110 tolerate less replacements and so is more conserved, and a lower information content in a site means it can tolerate more substitutions and has been subjected to more muta- tions. Sequence LOGOS are graphical representations of sequence alignments [10]. Each LOGO consists of “stacks” of nucleotide or amino acid symbols, with the overall height of the stack representative of the “total” information content at that position. The height of each symbol corresponds to the relative contribution to in- formation content of each symbol at that position within the alignment. Although the above methods are useful, only one evo- lutionary variable is examined. Further sequence logos, though useful, are essentially graphic methods of illus- trating the sequence conservation for the sites in an align- ment, but not for the each individual sequence. Given the above problems, we are aiming at utilizing two indepen- dent parameters to access the nature of the amino acid substitutions more reliably. As Tang’s EI is included, ano- ther parameter should be evolution-independent; protein structure for example, however the structure data are ex- pensive to collect and there is no proven methods to jus- tify the differences. Linear sequence complexity is a pro- mising parameter as the technique is inexpensive and can be quantified for comparison across a wide range of data types. We argue that information theory allows us to deter- mine the gain or loss of entropy within a sequence mar- ried to evolutionary methodologies that look at the like- lihood of amino acid change and rate changes allow us to determine whether a gene is evolving in an essentially neutral fashion, whether it is specializing it’s function, likely gaining a new function, or heading towards non- functionalization. While information theory has been ap- plied to non-coding regions to examine transcription fac- tor binding sites and regulatory elements and to coding regions to examine intron/exon boundaries and alterna- tive protein splicing [11-13], its application to comparative genomics in combination with other proven methodologies yields an interesting analysis tool for further study. 2. METHODS 2.1. Universal Evolutionary Index For each pair-wise protein alignment, we adopted the universal evolutionary index to quantify the likelihood of the amino acid substitution [8]. This index is a uni- versal ranking of the likelihood of amino acid change and was proposed based upon the high correlation of EIs from different sets of genes of different taxa. Comparing with other indexes, the universal evolutionary index is scaled such that its weighted average is 1, and it is easy for comparison and can be adjusted to specific species by multiplying the average Ka/Ks ratios of the given dataset. 2.2. Information Content Analysis We adopted the program VaSSA program from VaSSA Informatics, LLC to examine linear information content. The change of information content in aligned sequences is checked and their functional meaning is accessed. The sequence subsections with fixed size are scanned across, and their linear information content is measured. The con- tribution of each single position in the sequence to the total information content of the sequence is evaluated. Information content, in this specific example, is es- sentially a measure of the entropy rate of a particular sequence (vis a vis Shannon [14]); that is the measure of the ability to compress the sequence via some encoding. In our usage, this measure is then normalized by the channel carrier capacity of the sequence; that is, given the lexicon and it’s representation, how complex could the sequence be if the symbols were arranged such that they were minimally subject to compression. Formally, the channel carrier capacity is the limiting rate for infor- mation transmittal in the medium. While over an entire genome, this rate can be calculated and would be rela- tively fixed, it varies within the genome based on codon usage, representation of the lexicon, and other factors (such as rate of duplication). In this case, then, we’re examining what the local channel (gene or locus) could have carried across evolution vs. what the observed en- tropy rate within that channel is at a given point in time. This is rather different than standard definitions of bit- wise information content used in LOGOS and BLOCKS (and other usage), as in those cases information is said to be transmitted across species and the measure of the data transmittal rate is measured as a function of the frequency of inter-species change for a particular point within a sequence. By combining the information content and universal evolutionary index, we can examine each amino acid change between sequences and plots them on a two-axis chart (Figure 1); the chart is broken into quadrants, and where the majority of amino acid changes sit within the chart determines the likely evolutionary pressures acting upon a gene. This quadrants division is based on an em- pirical study that shows the sequences without functions (e.g. introns, intergenic sequences) are less complicated than the functional ones. Thus an unlikely amino acid change (low EI) that increases the complexity of the se- quence (positive information content change) is more probable to be driven under positive selection; similarly an unlikely amino acid change (low EI) that decreases the complexity of the sequence (negative information content change) is more probable to be driven under non-functionalization of the protein. A likely (high EI) amino acid change is within the constraint. Positive in- formation change may indicate it is within functional con-  M. Yang et al. / J. Biomedical Science and Engineering 3 (2010) 109-113 Copyright © 2010 SciRes. JBiSE 111 Figure 1. Functional sorting of the amino acid substitution using the information content and evolutionary index. Figure 2. The information content of the 28 zinc finger pro- teins. straint, while a negative one may hint the protein is los- ing some of the unrelated functions. 3. RESULTS Zinc finger proteins are a group of protein families clas- sified based upon their conserved sequence motif, and they are capable of binding DNA, RNA, protein and/or lipid substrates following their coordination with one or more zinc atoms [15-17]. The primary amino acid se- quences, the folding, the number of fingers and their spatial arrangement jointly determine the protein’s bind- ing properties. Among the many zinc finger families with various binding modes and unique functions, the Cys2/His2(C2H2) zinc fingers were the first group to be characterized [18,19]. This subset of zinc finger proteins plays pivotal roles in DNA transcription and develop- ment in organisms. About 400 C2H2 zinc finger proteins known exist in humans, which makes them one of the largest protein families in animals. The C2H2 zinc fin- gers are identified by their conserved sequence motif (CX2–4FX8HX3–5H). A zinc atom can be coordinated with the two cysteines and two histidines within the mo- tif to form a compact structure that can bind sequence specifically to DNA in its major groove. More recently, Laity and co-workers [20] found a sub-set of C2H2 zinc fingers that contains two interacting fingers, and they are evolutionarily distinct. We performed an evolutionary analysis of these data using our information theory based methods. The data set contains 28 interacting two-finger C2H2 zinc fingers, and there is a conserved tryptophan be- tween the first two Cysteine residues of the proteins. The information content of each sequence is calculated using VaSSA (Figure 2). To illustrate the site-specific change of information content, we align the DNA sequence 26 and 27 based on their peptide alignment (Figure 3), and calculate their information content (Figure 4). The site-specific comparison of the proteins is plotted 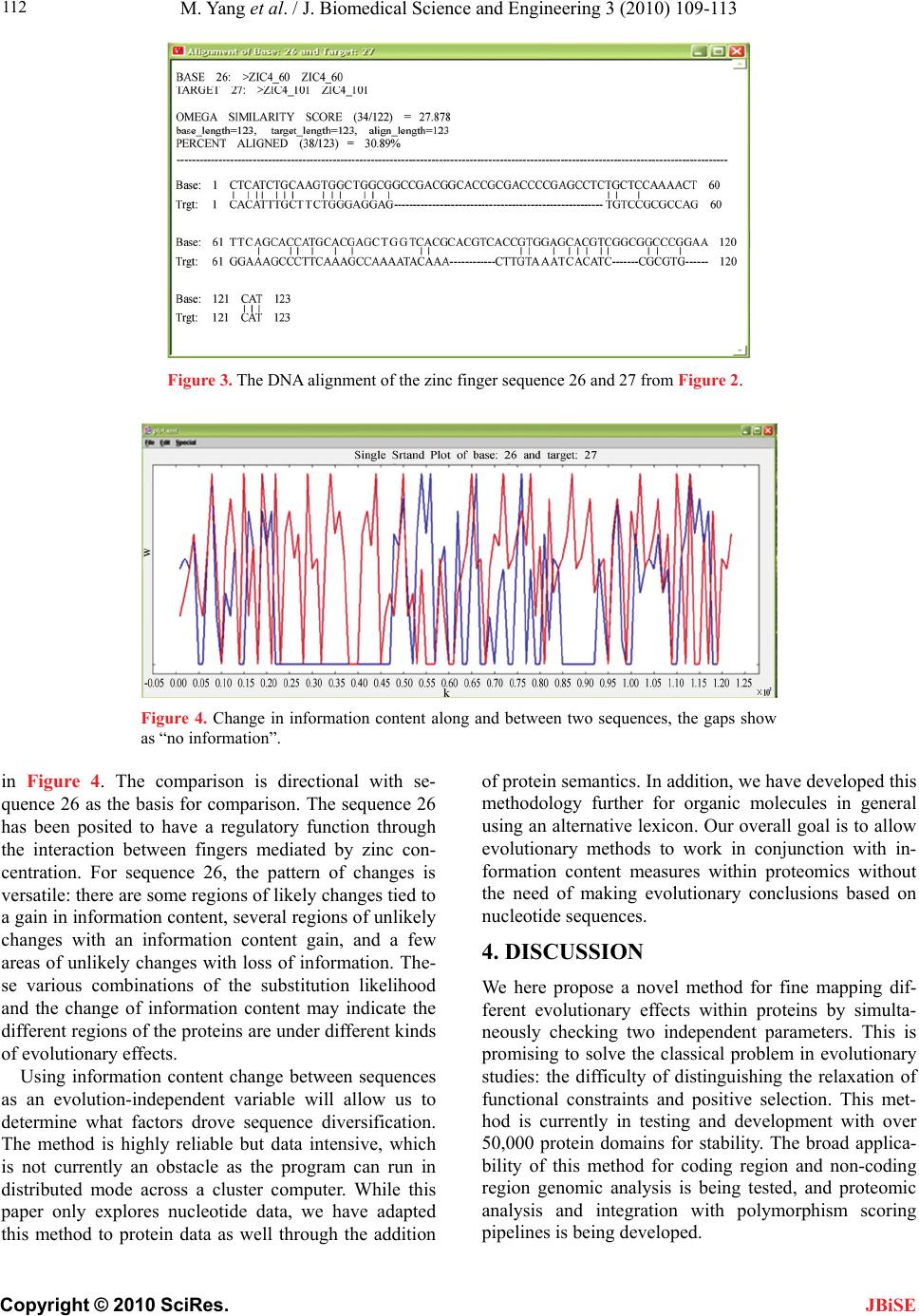 M. Yang et al. / J. Biomedical Science and Engineering 3 (2010) 109-113 Copyright © 2010 SciRes. JBiSE 112 Figure 3. The DNA alignment of the zinc finger sequence 26 and 27 from Figure 2. Figure 4. Change in information content along and between two sequences, the gaps show as “no information”. in Figure 4. The comparison is directional with se- quence 26 as the basis for comparison. The sequence 26 has been posited to have a regulatory function through the interaction between fingers mediated by zinc con- centration. For sequence 26, the pattern of changes is versatile: there are some regions of likely changes tied to a gain in information content, several regions of unlikely changes with an information content gain, and a few areas of unlikely changes with loss of information. The- se various combinations of the substitution likelihood and the change of information content may indicate the different regions of the proteins are under different kinds of evolutionary effects. Using information content change between sequences as an evolution-independent variable will allow us to determine what factors drove sequence diversification. The method is highly reliable but data intensive, which is not currently an obstacle as the program can run in distributed mode across a cluster computer. While this paper only explores nucleotide data, we have adapted this method to protein data as well through the addition of protein semantics. In addition, we have developed this methodology further for organic molecules in general using an alternative lexicon. Our overall goal is to allow evolutionary methods to work in conjunction with in- formation content measures within proteomics without the need of making evolutionary conclusions based on nucleotide sequences. 4. DISCUSSION We here propose a novel method for fine mapping dif- ferent evolutionary effects within proteins by simulta- neously checking two independent parameters. This is promising to solve the classical problem in evolutionary studies: the difficulty of distinguishing the relaxation of functional constraints and positive selection. This met- hod is currently in testing and development with over 50,000 protein domains for stability. The broad applica- bility of this method for coding region and non-coding region genomic analysis is being tested, and proteomic analysis and integration with polymorphism scoring pipelines is being developed.  M. Yang et al. / J. Biomedical Science and Engineering 3 (2010) 109-113 Copyright © 2010 SciRes. JBiSE 113 REFERENCES [1] Graur, D. and Li, W.H. (2000) Fundamentals of molecu- lar evolution. Sinauer Associates. [2] Fay, J.C., Wyckoff, G.J. and Wu, C.I. (2001) Positive and negative selection on the human genome. Genetic s, 158, 1227-34. [3] Karlin, S. and Ghandour, G. (1985) Multiple-alphabet amino acid sequence comparisons of the immunoglobu- lin kappa-chain constant domain. Proc Natl Acad Sci U S A, 82, 8597-601. [4] Feng, D.F., Johnson, M.S. and Doolittle, R.F. (1985) Aligning amino acid sequences: Comparison of com- monly used methods. J. Mol. Evol., 21, 112-125. [5] Dayhoff, M.O., Schwartz, R.M. and Orcutt, B.C. (1978) A model of evolutionary change in proteins, in Dayhoff, M.O. Edition, Atlas of Protein Sequence and Structure. Natl. Biomed. Res. Found., Washington DC, 5(3), 345- 352. [6] Henikoff, S. and Henikoff, J.G. (1992) Amino acid sub- stitution matrices from protein blocks. Proc Natl Acad Sci U S A, 89, 10915-9. [7] Henikoff, J.G. and Henikoff, S. (1996) Blocks database and its applications. Methods Enzymol, 266, 88-105. [8] Tang, H., Wyckoff, G.J., Lu, J. and Wu, C.I. (2004) A universal evolutionary index for amino acid changes. Mol Biol Evol, 21, 1548-56. [9] Minsky, A. (2004) Information content and complexity in the high-order organization of DNA. Annu Rev Biophys Biomol Struct, 33, 317-42. [10] Schneider, T.D. and Stephens, R.M. (1990) Sequence logos: A new way to display consensus sequences. Nu- cleic Acids Res, 18, 6097-100. [11] Smith, A.D., Sumazin, P. and Zhang, M.Q. (2005) Iden- tifying tissue-selective transcription factor binding sites in vertebrate promoters. Proc Natl Acad Sci U S A, 102, 1560-5. [12] Nalla, V.K. and Rogan, P.K. (2008) Automated splicing mutation analysis by information theory. Hum Mutat, 29, 1168. [13] Nalla, V.K. and Rogan, P.K. (2005) Automated splicing mutation analysis by information theory. Hum Mutat, 25, 334-42. [14] Shannon, C.E. (1948) A mathematical theory of commu- nication. Bell System Technical Journal, 27, 379-423, 623-656. [15] Hall, T.M. (2005) Multiple modes of RNA recognition by zinc finger proteins. Curr Opin Struct Biol, 15, 367-73. [16] Brown, R.S. (2005) Zinc finger proteins: Getting a grip on RNA. Curr Opin Struct Biol, 15, 94-8. [17] Klug, A. (1999) Zinc finger peptides for the regulation of gene expression. J Mol Biol, 293, 215–8. [18] Schuh, R. et al. (1986) A conserved family of nuclear proteins containing structural elements of the finger pro- tein encoded by Kruppel—a drosophila segmentation gene. Cell, 47, 1025-32. [19] Miller, J., McLachlan, A.D. and Klug, A. (1985) Repeti- tive zinc-binding domains in the protein transcription factor IIIA from Xenopus oocytes. Embo J, 4, 1609-14. [20] Wang, Z. et al. (2006) Solution structure of a Zap1 zinc- responsive domain provides insights into metalloregula- tory transcriptional repression in Saccharomyces cere- visiae. J Mol Biol, 357, 1167-83. |