Journal of Cancer Therapy
Vol.5 No.1(2014), Article ID:42123,14 pages DOI:10.4236/jct.2014.51013
Haploidization of Human Diploid Metaphase Cells: Is This Genome Reductive Mechanism Opperational in Near-Haploid Leukemia?
![]()
CROMOS, Richmond, USA.
Email: kwalencromos@gmail.org
Copyright © 2014 Kirsten H. Walen. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. In accordance of the Creative Commons Attribution License all Copyrights © 2014 are reserved for SCIRP and the owner of the intellectual property Kirsten H. Walen. All Copyright © 2014 are guarded by law and by SCIRP as a guardian.
Received December 2nd, 2013; revised December 29th, 2013; accepted January 5th, 2014
KEYWORDS
Nutritional Replicative Stress; Amino Acid Glutamine; Chromosome/Genomic Aberrations; Small Cells; Proliferative Advantage; Loss of Heterozygosity; Genomic Doubling; Uniparentaldisomy; Endoreplication; Genomic Territories; Nuclear Roundness; Nutritional Insufficiency Autophagy
ABSTRACT
The present study presents cytogenetics/cytology of haploidization in the origin of a new, fast growing diploid, small cell-type (F-dPCs). The sequence of events was haploid groupings of the chromosomes in normal, human metaphase cells, followed by genomic doubling to homozygousdiploidy. These events were responses to DNA replication stress fromamino acid glutamine deprivation. Importantly, these homozygous cells outgrew normal fibroblasts in 2 - 3 passages—they had gained proliferative advantage (GPA), presumably from loss (LOH) of tumor suppressor genes. They were morphologically changed cells with rounded nuclei that grew in a “streaming” growth pattern and with changed form and size of mitosis, similar to some hyperplasias. The grouping of the chromosomes in metaphase cells was asymmetric with a narrow range around the median (23) (no micro-nuclei), suggesting genetic control. The root-origin of haploidization was evidenced by maternal and paternal genomes occupying separate territories in metaphase cells, which assumedly permitted independent segregations of bichromatid chromosomes. In near-haploid ALL-L1 leukemia the loss of virtually, whole chromosomal complements was judged by SNP array analyses, as a primary event before genomic doubling to hyperdiploidy with LOH. From the present data such specific, non-random loss of chromosomes strongly suggested, a haploidization process capable of genomic doubling, as observed for the “birth” of the small, F-dPCs. This suggestion was supported by this type of leukemia being the L1-type, where L1 signifies small cells. The possibility now exists that a tumorigenic process can be initiated directly from diploid cells through haploid (near-haploid) distributed chromosomes in normal metaphase cells. This event followed by monosomic doublings to UPDs would lead to massive LOH and a return to para-diploidy, a frequent occurrence in many types of tumors. The present simple, cultural derivations of the extraordinary F-dPCs allow GPA-identification and experimental manipulations, perhaps relevant in a vaccine program.
1. Introduction
Cancer originates from normal diploid cells, stem or progenitor cells, and the initiation process is known neither from molecular nor from gross cellular changes (e.g., aneuploi-dization). But based on evaluation/assessment of data from tumorigenesis, the widely accepted theory is: accumulation of growth favorable mutations over time gives a growth advantage over normal cells of origin (the mutation hypothesis), which is the most fundamental requirement for tumorigenesis [1-3]. Initiation may be of many different types, but widely believed to give rise to growth advantage somehow linked to simple aneuploidy
(gain/loss of single chromosomes), which with increasing time (progression) becomes complex with associated aberrations in a cancerous process [4,5]. The in vivo visualization of progression is first evident from hyperplasia and/or mild dysplasia which most often it presented with normal G-banded karyotypes, occasionally together with a small clone with simple chromosomal aneuploidy (+/−1 - 2 chromosomes) [6,7]. But today, a diploid karyotype has the probability of being aneuploid from loss of whole or segmental chromosomal regions with compensated doubling to homozygous, uniparentaldisomy (UPDs) [8,9]. The resulting loss of heterozygosity (LOH) is believed to remove tumor suppressor genes (e.g., p53), which would give pro-growth-genetics/epigenetics opportunity for expression (i.e., gain of a proliferative advantage-GPA) [10]. This scenario is in agreement with suggestions of UPD as a factor in the aneuploid paradox: the fact that aneuploidy is associated with GPA in cancer cells, whereas in normal cells it is growth-retarding [11, 12].
Recently, several review articles addressed the issue of genomic changes that could have tumorigenic initiating potentials associated with aneuploidy and/or have relevance to new therapy treatments [5,10,13-15]. As reviews in general, old problems thus, become updated from inclusions of new discoveries, but clearly defined, initiating occurrences appear still to be caught in the perception of non-observable (non-visual) “mutational” events. However, one article mentioned the presence of diplochromosomes (4-chromatid chromosomes) in pre-senescence of normal diploid cells [14], which has “mutational” consequences (see below). This is the first verification of a significant difference between simple tetraploidy (genomic doubling, 2n) and tetraploidy from endoreplication to 46 chromosomes with four sister centromere regions [16-19]. The question is the behavior of these latter chromosomes on a mitotic spindle constructed for segregation of bichromatid (2 sister centromeres) chromosome into single chromatids?
Facts have shown that change to endo-replication cycles is associated with a survival process in stressful environments where G2/M chromosomal structure replicates to diplo-chromosomes [20,21]. Recently, in human ovarian cancer giant cells from endo-cycles were shown to divide to near-diploid cells, some containing tumor stem cell markers, and also showing endo-cycles back to giant cells [22-24]. The base for this endo-replication-change is most likely an “awakening” of a primitive division system involving endopolyploid reductive division back to diploidy during the reproductive cycle of some unicellular eukaryotes (e.g., radiolaren Aulachantaschoolymantha) [25-27]. For reproductive purpose this primitive division (mitotic-meiosis) had to be orderly with high fidelity, which for human cells was expressed as a twostep meiotic-like division of endotetraploid cells into four 2n/2C products [17,18]. And interestingly, as the unicellular offspring cells inherits the primitive reproductive machinery, the human progeny cells also showed inheritance of endopolyploid, special, reductive division traits (e.g., co-segregation) [28,29]). Such inheritance however, would have to cope with the endo-derived progeny cells’ innate machinery for normalmitosis. Thus it was observed, that in endo-progeny-cell divisions, the endo-inherited traits had become blended into the mitotic process (e.g., co-segregation of prophase genomes). Mitosis had become a “functional construct” from two division systems which no longer operated with normal mitoticfidelity. One important characteristic was “mild”, loss/ gain of individual chromosomes (aneuploidy) apparently from structural abnormality in the kinetochore regions. Since the karyology was normal for such cells, the theory is that the losses were compensated by UPDs, which led to LOH for tumor suppressor genes, leading to the observed incremental GPA [29].
Most of these latter experiments (except: [29]) tide endopolyploidization to dysfunctional telomeres in presenescence: a phase when division of diploid cells diminished and a sudden peak-increase in endotetraploid cells occurred naturally [14,16-19]. A following of these endohappenings into the senescent phase revealed an unprecedented cellular event: growth of three dimensional (3-D) tumor-like spheres associated with cell polarity loss in weeks old cultures [30,31]. These results sparked the question of whether such growth-changes would occur from young, normal diploid cells for which propagation of endo-offspring cells would not be limited by dysfunctional telomeres.
Earlier works had shown endopolyploidization associated with amino acid deprivation (AAD) [32,33], and glutamine was chosen, because of its importance in cell metabolism and proliferation of normal cells [34,35]. Several experiments with short-term (2 - 4 days) AAD treatments of two normal primary cell strains (L645 & WI-38) revealed not only endo-derived progeny cells, but also a never-before observed or reported small, cell-type [29]. This new cell-type and the larger endo-derived progeny cells (special cells) became present in the same normal cell populations, but could be populated as individual, pure populations. As such, growth kinetic experiments compared to the normal fibroblasts showed increases in the millions for the special cells (1 for endocells and 4 - 5 for small-cells). It was concluded that both cell-types had acquired GPA over the cell of origin. The small cell-type appeared to have gained proliferative advantage connected with the “birth”-process and was called fibroblast derived prolific cells—F-dPCs. Early studies and preliminary observations suggested birth via a haploidization process in normal metaphase cells [29, 36,37]. The objective of the present study is an in debt characterization of these fast growing cells, with emphasis on the haploidization process and its further development to diploid homozygous cells by genomic doubling. The goal was to link the origin of F-dPCs to these extraordinary happenings, and to the unprecedented occurrence of proliferative near-haploid leukemic cells, which also underwent genomic doubling, resulting in multiple UPDs.
2. Materials and Methods
As earlier, the two primary cell strains WI-38 and L645, both from primary lung tissue with fibroblastic growth were used in the present experiments [28,29]. (For simplicity of data-presentation and because, both cell strains responded similarly to AAD, illustrative results are presented mostly from L645 cells, which showed less degenerative debris.) Regular growth medium (M+) consisted of Eagle-essential medium supplemented with 10% fetal calf serum (FBS), 3 mmol glutamine, penicillin/-streptomycin and 2% sodium-carbonate for pH adjustment to 7.4 - 7.5. Slide chamber cultures were seeded to about 1/4 confluence, 1 - 2 days later when they were washed 2× in Hank’ balanced salt solution before addition of glutamine deficient medium (M-) for 48 and 72 hours exposures. Trials with 10% and 2% fetal bovine serum (fbs) in Mshowed presence of mitosis with 10%, and not with 2%, which became the standard M-, AAD medium. Recovery medium (M+) consisted of normal growth mediumwith 3 mmol glutamine and 10% serum. Following preliminary assessments of recovery-cells over eight days with every second day harvests the crucial cytogenetic events were found to be restricted to harvest periods 2 to 3 days in M+. Before fixation the slide cultures were washed 2× with warmed Hank’ balanced salt solution of which a fraction was left (from last wash) on the slides and mixed with an equal fraction of cold Carnoy’s solution (3 methyl alcohol to 1 acidic acid) for slow fixation. After 2 - 3 incremental additions of fixative to this mixture it was discarded and changed to 100% Carnoy’s with 2 - 3 changes. Air dried preparations were stained in 2% Gurr’s Giemsa for 2 - 3 minutes, dipped 2× in water and quickly air dried followed by coverbound application for coverslips. Microphotographs were taken from 10×, 16×, 25×, 40× and 100× objectives by a Zeiss standard microscope with an attached Zeiss camera for 36-mm film. These photographs were enlarged 396×, 619×, 990× and 2475×, respectively.
Weekly passage-increases kept in T-25 flasks were prepared by a standard dilution of 1 to 4 from which 1 part seeded new flask cultures and 2000 to 4000 cells seeded single or double well chamber slides. The phase contrast photographs were taken by a VWR Vista Vision inverted microscope with a Pentax (2x-M) camera attached, at an enlargement of 250× onto a 36-mm film.
3. Results
In pilot experiments for exploration of variable AAD responses expressed in M+, it was observed that 2 - 3 days AAD exposure could give rise topure populations of the small F-dPC cell-type. Thus, chamber slide cultures after such treatments and with 1 to 3 days growth in M+ medium, were harvested for in situ observations of the first appearance (birth) of the small cells. Longer exposures (especially 4 - 5 days) to AAD and with 2 - 5 days recovery-periods in M+ resulted in addition of endopolyploid cells (4n/8n), which underwent meiotic-like division to E-dTMCs (endo-derived transmuted cells) [29]. This later appearance of endopolyploidy is in agreement with time requirement for re-replication of bichromatid-G2 chromosomes into 46diplochromosomes (4C to 8C). In such mixed cell populations, inclusive of normal diploid cells (in G1/G0 during AAD) it was difficult to follow the haploidization process, such that present data are only from short AAD treatments (2 - 3 days) and recovery periods also at mostly 2 to 3 days (4 day harvests were available when needed for assessments.)
AAD treatment of both L645 and WI-38 cell strains produced pronounced slimming of the normal fibroblast cells (Figures 1(A) and (B)), which reverted to the normal phenotype in complete M+ medium. Beginning mitotic cycling in M+ was a sudden burst in day 2 harvests (very few in day 1), which rose to an overall 4% - 6% mitotic index in day 3-(4) M+ harvests, consisting of both F-dPCs and recovered fibroblasts. The main nuclear morphological difference between these two types of cells is that fibroblast nuclei are consistently oblong and almost twice as large as the rounded, smaller nuclei of FdPCs (Figures 1(F) versus 1(C)-(E)). E-dTMCs in 4 day harvests were differentiated from fibroblast nuclei also by a rounded nuclear appearance (Figures 1(F) and 1(G)). To assess the proliferative ability of F-dPCs at “birth” shake-off mitoses from the first wave of mitoses in1-2 day recovery growth were seeded onto chamber and flasks. It was reasoned that this wave would contain cells in S and G2 while under AAD exposure, which would give rise to small-celland fibroblast-growths. The initial ratio (chamber-slide 1-day harvests) was about 1 to 50, which in 5-day growths had changed to about 10 to 0.5 for small-cells versus fibroblasts. In contact inhibited areas the F-dPCs grew much tighter together than the fibroblasts. Thus, the F-dPCs relative to the normal fibroblasts showed shorter cell cycling time and birthlinked proliferative ability. In the flask cultures with
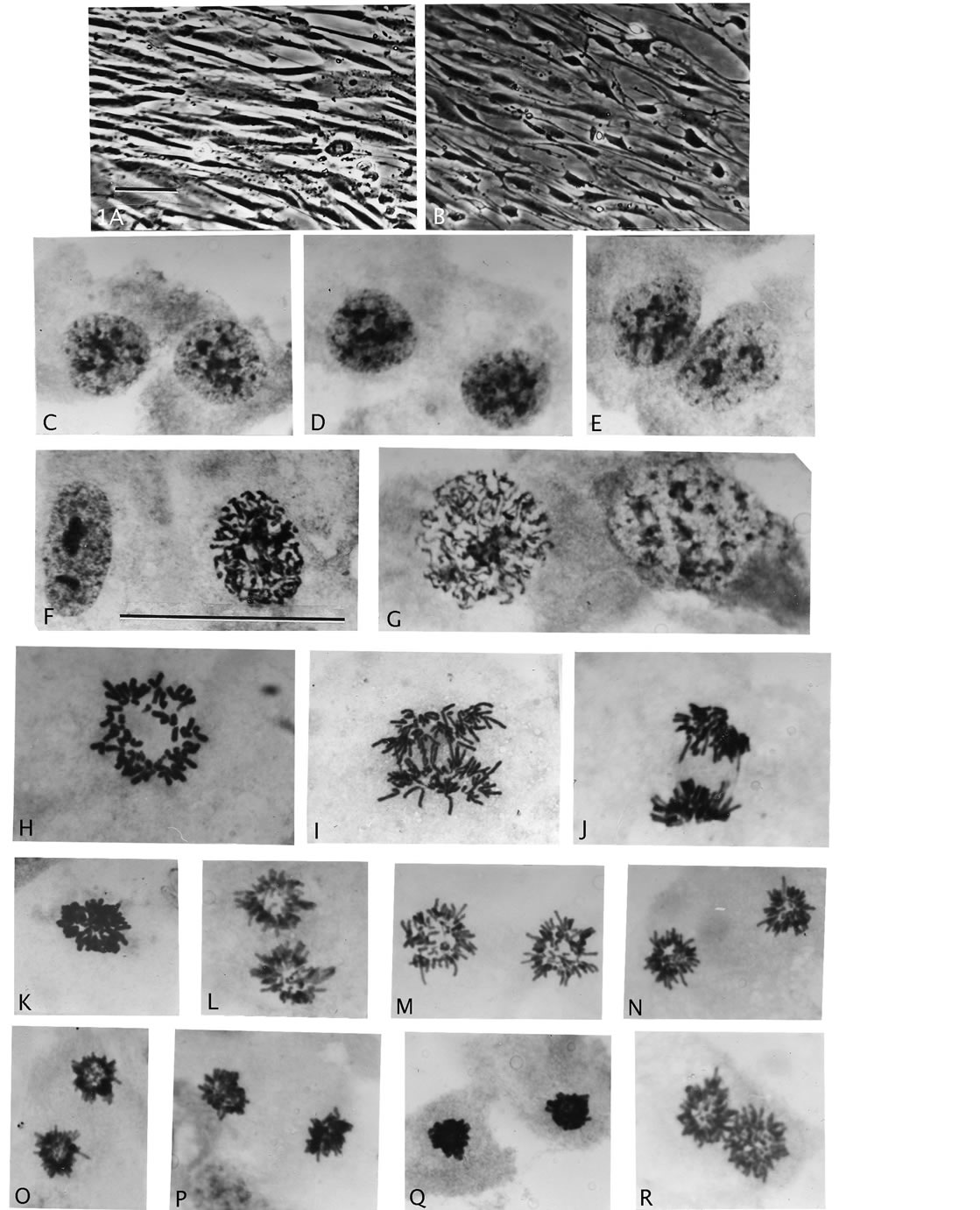
Figure 1. Nuclear size and mitosis of F-dPCs compared to normal fibroblasts. (A), (B), phase-contrast of normal and AAD treated fibroblasts. (C)-(E), pairs of small rounded nuclei different from fibroblast control (F), and from endo-derived larger rounded nuclei (G). (H)-(J), normal mitosis: rosette figure, early anaand telophase stages. (K)-(R), different phases of star-like mitosis of F-dPCs, -note consistent small size compared to control ((H)-(J)). Magnifications: AB = 396×, C-R = 2475×, scale bar 1A = 30 μm.
passage extended proliferations, the fibroblastic presence rapidly disappeared (p3).
Mitosis of these small F-dPCs in p18 and p19 recovery growth wascytologic strikingly different from normal mitosis (Figures 1(K)-(R) compare normal 1(H)-(J)). All these illustrations of division figures have the same magnifications which again demonstrate a significant reduction in the size of the F-dPCs mitoses relative to the fibroblastic mother-cell. The F-dPC-division started with a noticeably tight, hyperchromatic, small metaphase rosette figure (Figure 1(K) compare 1(H)) which appeared to contain normal chromosomal arrangement with kinetochores pointing into and arms pointing out of the circle. Strangely, this arrangement (star-like) was maintained through anaand into progressive chromatin compaction intelophase (Figures 1(N)-(P)), which was followed by cytokinesis (Figure 1(Q)). In next passage growths these peculiar forms of anaphase figures were still present (Figure 1(R)). The principle difference from normal mitosis besides smallness was absence of kinetochore-orientations in a polar direction which gives rise to trailing chromosomal arms (compare stars to Figures 1(I) and (J)). Typical cytology of normal, early telophase with trailing arms (Figure 1(J)) has not been observed for F-dPCsin countless mitotic observations, which raises the question of spindle apparatus involvement or not in these different forms of mitosis. Chromosomal counts in naturally and mildly, hypotonic spread metaphases showed that the number-range was tightly around 46 (43 - 48) (Table 1). Karyotypes showed absence of gross chromosomal abnormalities [29]. Thus, these new, fast growing cells show inherited changes to smallness and to a different form of mitotic cycling (star-like), which can be used as morphological markers for presence of F-dPCs.
The further expanded proliferation of these cells in passage growth clearly showed cell morphology and growth pattern differences from normal fibroblastic growth (Figure 2(A)). In loose growth, the cells and nuclei were uniformly of same size, either polarized with triangular shape or roundish (Figures 2(C) and (D)). They contained several small nucleoli. In more denser growths the cells could grow on top of each other which is shown from a live culture (Figure 2(B)), or they formed hyperplasia-like “streaming” growth patterns from spindle-shaped cells (Figures 2(E) and (F)). Compared to the normal polarized pattern from fibroblasts (Figure 2(A)), the cells in the F-dPC-pattern are more spindle shaped and grow tighter together, as mentioned above. Different streams showed the same pattern, and growth seemed to start with a narrow end (Figure 2(E)) which broadened with added rows of cells (Figure 2(F)). All together, the F-dPCs demonstrate a series of different characteristics from the cell of origin, which point to inheritedgenetic/epigenetic changes.
The question of the mode of origin of F-dPCs in normal, fibroblastic cell populations was suggested to be from a haploidization process in normal diploid metaphase cells [29,36,37]. The present study showed that this event can be observed when the chromosomes are aligned on the equatorial plate in rosette configuration. At this stage two very different microscopic views are possible: sideand polar-views and only polar-view was informative for observations of the grouping process, and for evaluations of chromosomal numbers in each group [29]. From such a vantage ground, rosette figures, without any overt change in their specific morphology were
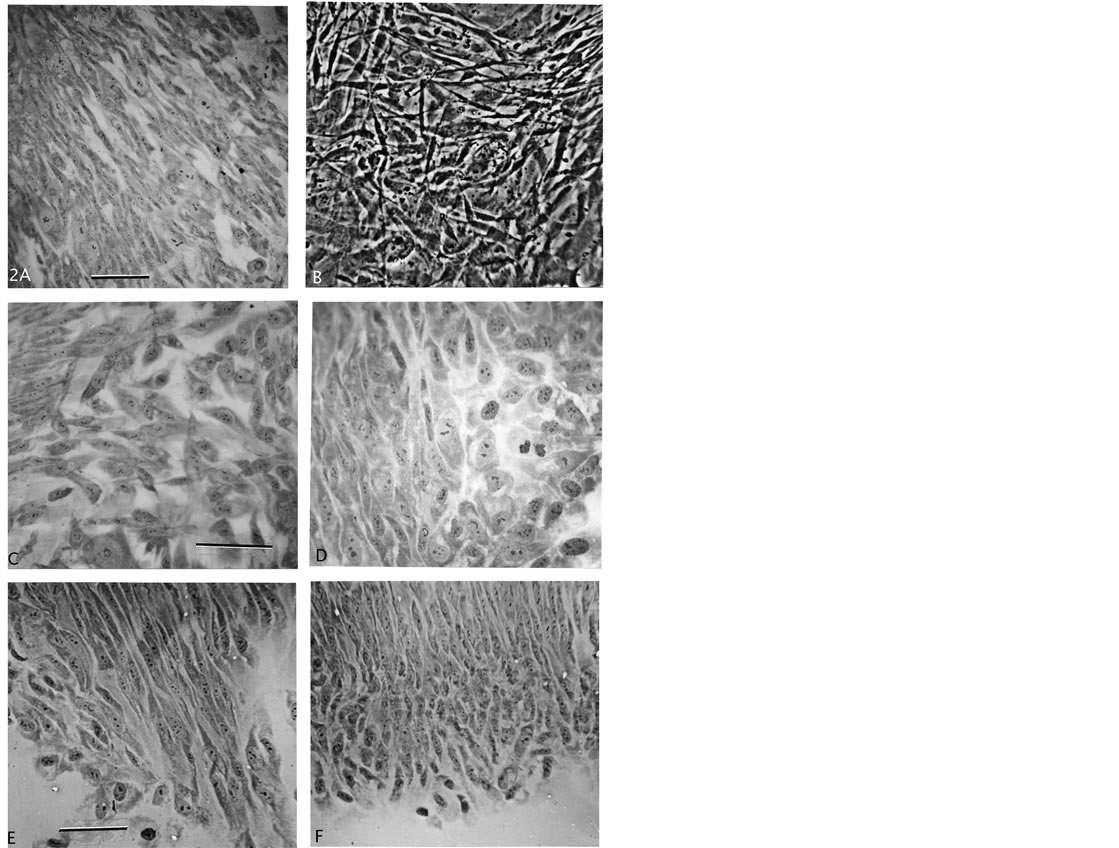
Figure 2. Growth patterns of normal fibroblasts and F-dPCs. (A) Normal fibroblast growth-pattern. (B) Phase-contrast of layered, disoriented focal growth. (C)-(F) Different shapes and forms of F-dPCs in loose ((C), (D)) versus in polarized tight, streaming growth patterns ((E), (F)). Magnification: (A), (E), (F) = 247×, (B) = 396×, (C), (D) = 619× scale bar 2A = 30 μm.

Table 1. Chromosome number distribution.
observed to segregate into two halves (near-halves) (Figures 3(A)-(C), (E)-(G) and (I)). The direction of the movements of the two halves was not in accord with the cytoskeleton axis (judged from disagreement with neighbor
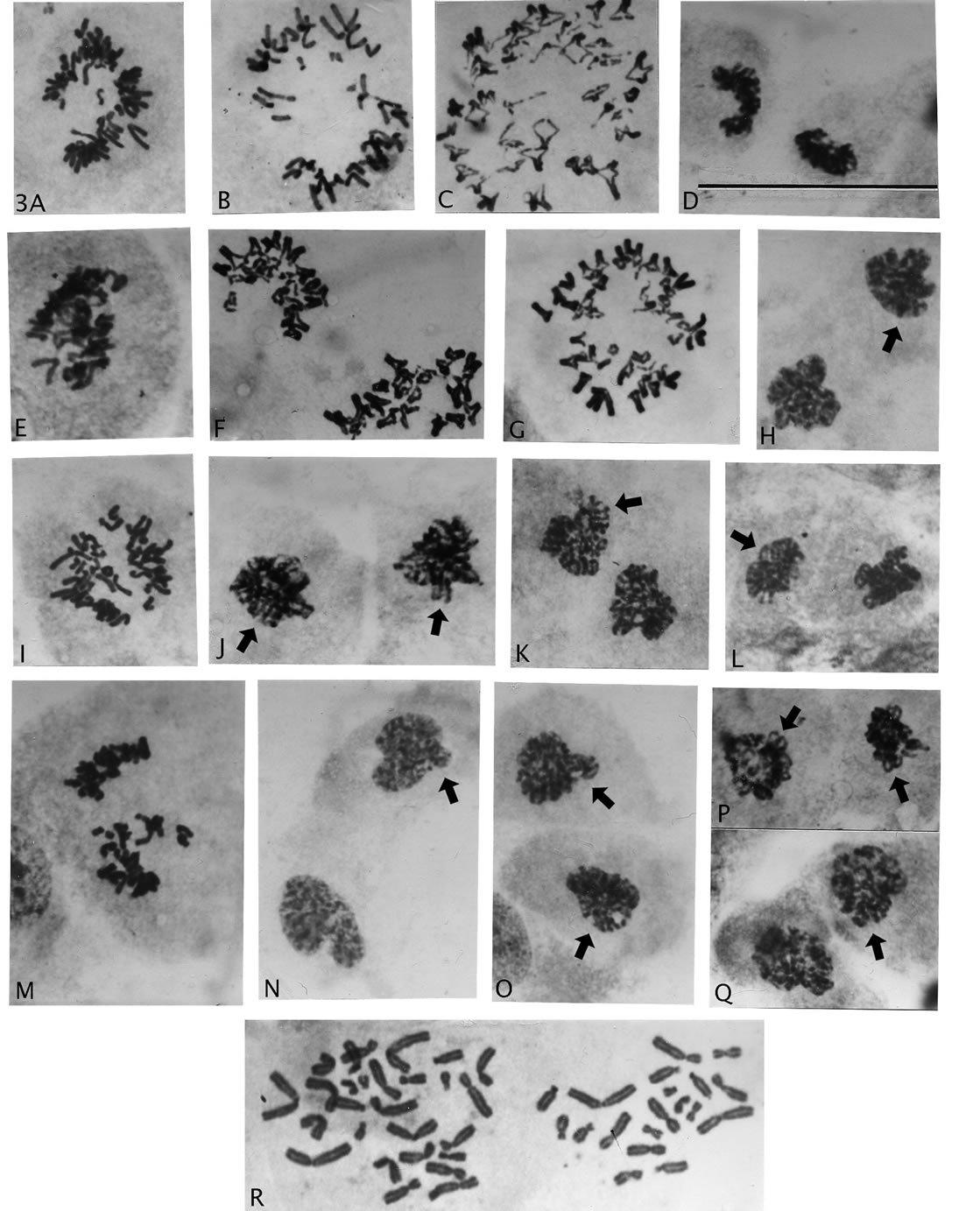
Figure 3. Sequence of cellular events following haploid-groupings ofrosette metaphases. (A), (B), (I), (M), rosette halving, (C), (F), (G) sister chromatid exchange chromosomes and (D), (E), (F) retained rosette chromosomal arrangement in segregated groups. (H), (J)-(L), (O)-(Q), various examples of dysmorphic nuclear pairs with uneven circumference, paired chromatin strands (arrows), nuclear membrane formation ((H) arrow) and normal appearing cyokinetic division furrow ((J), (Q)). (N), nuclei with normal appearing, early interphase chromatin cytology (arrow, uneven circumference). Magnification: all illustrations 2475×, scale bar 3D = 120 μm.
cells’ polarity), and was not led by kinetochores with trailingchromosomal arms as in normal mitosis (Figures 1(I) and (J)). The separated groups often showed the original bichromatid, chromosomal arrangements of the rosette circle in either one or both division products (Figures 3(A), (B), (D), (F), (G), (I) and (M)), before a gathering of chromosomes to rounded pairs of condensed nuclear bodies (Figures 3(H), (J)-(L), (N)-(Q)). The uneven circumference and the clear bichromatid content (Figures 3(J)-(L), (P) and (Q)) in some of these bodies show that these nuclear pairs are not from normal mitosis. They can only have originated from the haplodized grouped chromosomes as also is indicated by new membrane formation (Figure 3(H), arrow), and the further cycling to involving cytokinesis (Figure 3(J) and (Q)). (Time-laps photography is not available.) In other words, the mentioned illustrations demonstrate a cycling continuum from the grouping process to interphase cells (Figure 3(N)). Interestingly, the grouping process continued in presence of repairing chromosomes, shown by sister chromatid exchange figures (Figures 3(C), (F) and (G)).
The asymmetric divisions (Figures 4(A)-(H)) show a
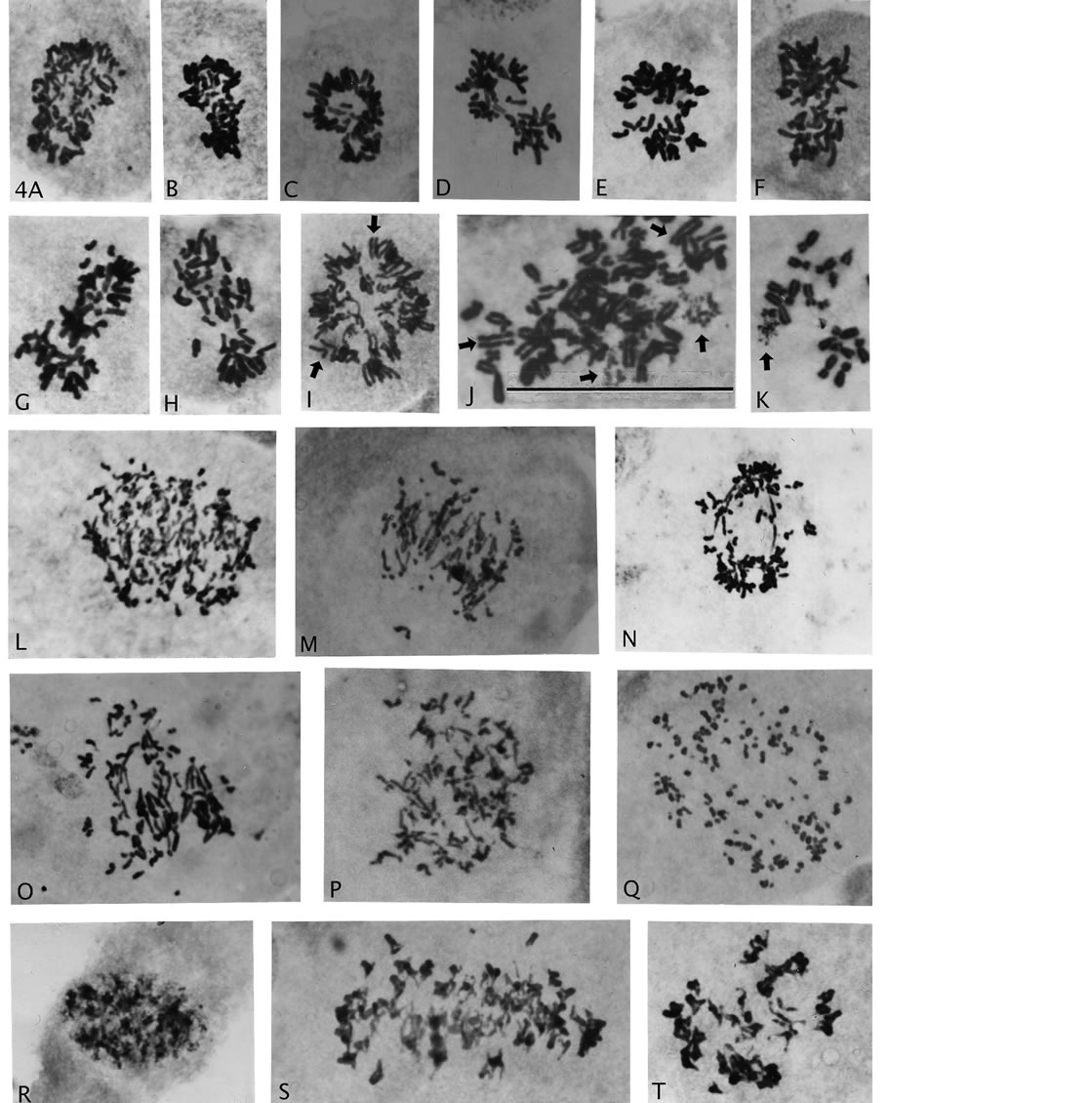
Figure 4. Asymmetric haploidization and various examples of chromosomal and mitotic abnormalities associated with AAD treatment. (A)-(H), asymmetric chromosome distribution. (I)-(K) premature sister chromatid separation ((I), (J) arrow heads) and single chromosome breakage ((J), (K) arrows). (L)-(Q), severe breakage with/without associations to anaphase bridges. (R)-(T), sister chromatid exchange chromosomes: in prophase nucleus (R), in side viewed metaphase (S) and with condensed expression in polar viewed metaphase (T). Magnification: all illustrations 2475×, scale bar 4J = 120 μm.
“pinching-off” of a comparatively small number of chromosomes from rosette figures. Importantly, micro-nuclei from single or a few chromosomes were not observed, which would have indicated random loss. A previous count/estimate of numbers of chromosomes in such groupings showed a narrow asymmetric distribution around the median (23), which suggested a genetic controlled haploidization process [29]. One such asymmetric division with rarer, good spreading of the chromosomes (Figure 3(R)) shows 27 and 19 chromosomes (total 46) in the two groups. Chromosome identification in the smaller groupindicates that the larger group is nullisomic for 2 - 3 (?) homologous pairs, and therefore, most likely would not be viable. But asymmetric divisions with no nullisomy may well have chance of proliferative survival, because of thebichromatid chromosomal constitution (see Discussion).
Figure 4 also show “primary” genomic destructive behavior following AAD treatment (Figures 4(L)-(Q)) with anaphase bridges associated with extensive breakage to small, pieces of chromosomes (Q). The pieces likely include short arms of chromosomes and probably also from breakage in fragile site. Also shown is an interphase nucleus (early prophase?) and a cell with sister chromatid exchanges demonstrating heterochromatization with stickiness causing chromosomal clumping (Figure 4(R) and (T)). Figure 4(S) is a side-view of chromosomes with sister chromatid exchanges, which demonstrate the inferiority of this type of view as compared to rosette figures in analyses of metaphase cells (Figure 4(S) compare Figures 3(C), (F) and (G)). Furthermore, the less common abnormality of premature chromatid segregation (Figures 4(I) and (J), arrow heads) was also present in these recovery growths, and recent interest in so-called chromotrypsis appears also to be a product (Figures 4(J) and (K) arrows). Three chromosomes in Figure 4(J) and one and in Figure 4(K) showed breakage while the rest of the chromosomes show normal metaphase structure.
Finally, an unanswered question is whether the haploid-groupings in normal metaphase cells rely on any preconditions in such cells. This appears to be true, because one study claimed a two territorial separation of maternal and paternal (mat-pat) genomes in lymphocyte cultures of several mammalian species, which appeared to segregate from each other in a haploidization process [38]. Therefore, since human cells were not included in these studies, G-banding of mildly hypotonic spread L645- cells (present primary cell-strain) were analyzed for cellpositioning of homologous chromosomes relative to each other (Table 2). Of a total of 30 metaphases only 5 were not overspread for this purpose. The demarcation “line” for two hemispheres were sometimes indicated, naturally by open space, which for pen-marking was followed, but with the adjusted inclusion of group A-homologs (#1, #2, #3) into different hemispheres. This approach put a significant number of B and C group homologs into separate hemispheres. Although, the number of analyzable cells was small, Table 2 shows a definite pattern of homologous chromosomes separated into different territories. This conclusion is supported by the single analysis of a tetraploid cell, and the consistency of #4 chromosomes in one territory, all pointing to a non-random distribution of homologous chromosomes relative to each other in these normal diploid metaphase cells. Additionally, mouse embryonic cells maintained a mat and pat genomic territorial separation for several divisions [39], and the present human cell-strain is of similar origin.
4. Discussion
The original objective: characterization of the new small, very likely homozygous (see below) cell-type, F-dPCs, has been fulfilled to the degree that a simple methodology (AAD) can be of futureuse in procurement of these unprecedented cells. The main conclusions regarding geno-phenotypic change and origin of the F-dPCs are as follows: 1) The cells are inherently different from fibroblasts, their cell of origin, by small size, cell-shape, growth-pattern and form of mitosis, which expressed the reduced cell-size in all phases, but left unanswered the question of chromosomal-spindle attachmentin star-like segregations [40,41]. 2) The origin from a haploidization
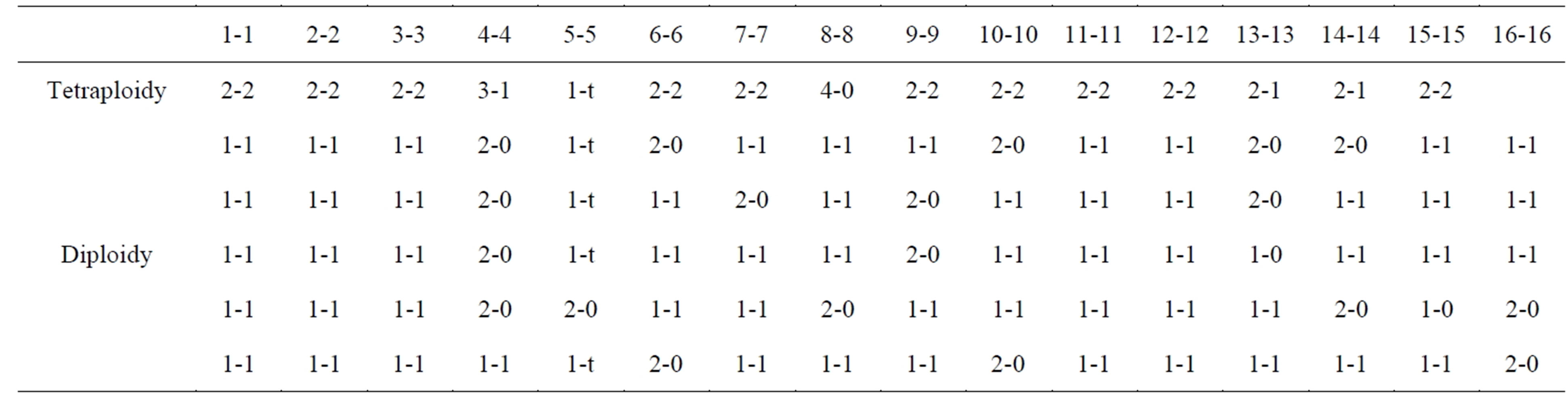
Table 2. Two territorial positions for individual chromosomes with placements from 6 metaphases.
process is strongly supported by cycling of haploid, grouped chromosomes to paired interphase cells through normal appearing cytokinesis furrows. The cytological un-even circumference and the distinct presence of paired chromatin threads (i.e., bichromatids, Figure 3(J)) in these division products, are unique features, only known for larger products from endo-meiotic-like first divisions [17]. 3) The genomic constitution of these interphase cells is likely near-1n/2C frombichromatid chromosomes, which by dissolution of sister chromatid cohesion can change to near-2n/2C. This constitution is a normal G1 condition for licensing of an S-period that would lead to diploid homozygous cells. This sequence of events is supported by a tight correlated occurrence of the haploidization process with the appearance of the FdPCsin a narrow time-window, which was interpreted as cause and effect [29]. 4) The expressed GPA of the FdPCs is probably not surprising considering, the extensive homozygous determined LOH creating loss of tumor suppressor genes (Coschi and Dick, 2012) [10]. This present GPA-type has never before been attained from normal diploid cells in vitro, and should not be compared to immortalization of cells. This latter process involves a period of chromosomal breakage-union-crisis with gain of telomerase activity for very, rarer survivors [42]. (The multi-birth process of proliferating F-dPCs does not support a gain of telomerase activity.)
As previously suggested [32] DNA replication stress from AAD treatment may have led to under-supply of proteins involved in this process with result of stalled replication forks, which resumed activity in M+ conditions [43]. The observed extensive, chromosomal breakage, anaphase bridges, chromosomal compaction (Figures 4(L)-(T)), and premature sister strand separation (Figures 4(I) and (J)), plus genomic repair by sister chromatid exchanges, are all abnormalities suggested to occur in the cancerous process [43]. The small F-dPCs were “born” in this environment of genomic damage, which indicates that genetic diversity among these cells would be expected. The single chromosomes with breakage (Figures 4(J) and (K)) and degenerate morphology is reminiscent of DNA asynchronous replication in heterokaryons, where fusion between metaphase and cells in S/G2 resulted in fragmentation of the chromatin in the latter cell [44].
Indirectly, the suggested diploid homozygosity of the F-dPCs is strongly supported by two in vivo happenings: Firstly, human placental teratoma cells are also complete, complement homozygous diploid from the fact that an X-bearing sperm fertilized an empty egg cell, which with whole genome duplication led to proliferating homozygous cells [45,46]. Secondly, the following karyotypic example show the extent of haploidy in one case of early childhood ALL-L1 leukemia: 28, XX, -1, -2, -3, -4, -5, -6, -7, -8, -9, -11, -12, -13, -15, -16, -17, -19, -20, -22 with heterozygous disomy (mat/pat genomes) for only X andnumbers 10, 14, 18 and 21 [47]. The proliferative ability of thesenear-haploid leukemic cells, and not for the present haploid cells, was ascribed to nutritional-help from the in vivo environment (neighboring cell-secretion), which was not an option for the in vitro cells [29].
Recently, SNP array studies of these very, rarer leukemic cases (above) showed that “loss of chromosomes”, was a primary happening (before genome duplication, see below) [48], suggested to originate from multipolar mitosis or non-disjunction with clonal selection [9,49]. From present results, a random loss of 18 different chromosomes, almost a complete genome appears very unlikely compared to an asymmetric haploidization process (Figures 4(A)-(H)). Moreover, the perception is that cancers are mostly in the triploid range, when in fact there are a very large numbers of different types of cancers with chromosome numbers around 46 (noteworthy, from high 30th to 48) [47]. For example, asymmetric haploidization to 31 total chromosomes would be missing 15 different chromosomes, which leaves 16 as disomics. Depending on whether the endoreplication process is for all or a fraction of the monosomics to UPDs the total number of chromosomes can restore to a limit of 46 or below, but above 31. This suggestion, which results in extensive homozygosity, leading to loss of tumor suppressor genes, can unfortunately only be decided from future, cost-effective SNP array haplotyping [48].
However, chromosome doubling from endo-replication (i.e., replication of a G2/M chromosomal structure) of monosomics to UPDs is well known and, was an occurrence for the leukemic example above. Ahyperdiploid clone co-existed with the near-haploid condition: 56, XX, +X, +X, +10, +10, +14, +14, +18, +18, +21, +21 [47,49]. The additions of X, 10, 14, 18, and 21 in tetrasomic conditions and all other “missing” homologs as disomics (UPDs) shows that one cell underwent a complete genomic doubling from an endoreduplication process. Another example from breast tumor, with similar, seeming non-random loss of chromosomes: 35, XX, -1, -3, -4, -5, -7, -8, -10, -11, -12, -13, -14, -15, -16, -17, -18, -19, -20, -21, -22, +8 mar [47], would also be in the hyperdiploid range with 54 + 8 mar chromosomes if the monosomics only, reduplicated to UPDs. This idea of a primary loss of chromosomes from diploid cells followed by polyploidization (disomy) and clonal evolution, has very recently been proposed to be a route to types of cancerous karyotypes [50,51]. Hyperdiploidy and higher levels of plody has traditionally been ascribed to multipolar mitosis or to chromosomal loss from the 2n-type of tetraploidy [13,52], which is more likely to be the endo-type with diplochromosomes [28,53,54]. All in all, the important message to be drawn from all these considerations is that diploid cells directly, (not via a polyploidization process) can perform chromosomal mechanistic events that can lead to potentials for tumorigenesis. From the present study the key diploid-event brings it all back to where it all started over a century ago i.e., asymmetric chromosomal distribution [55,56], but noteworthy, in these cases from an initiating haploidization process.
Interestingly, this proposal has support from cell morphology. In ALL-L1 leukemia subtype, L1 denotes characteristic small cells as compared to the large cell-type of the L2 classification. Small-cells are a consequence of the haploidization process, clearly demonstrated for the different phases of mitosis (Figures 1(K)-(R) compare 1(H)-(J)) and for interphase nuclei. How this morphological change happens from the larger diploid cell-size, appears today to rest upon the growing evidential material for mat and pat genomes being separated in two different territories (Table 2) [38]. Chromosomal painting of rosette figures support this view by the finding of homologous chromosomes being in a diametrically opposite position to each other in the metaphase circle [57,58]. Halving (near-halving) were observed for rosette figures (Figures 3(A), (B), (I) and (M)) during the haploidization process, meaning: the reduced numbers of chromosomes in the metaphase groups were derived from a single territory, which upon genomic doubling to homozygous cells did not (as expected?) result in diploid-sized nuclei or mitosis (Figures 1(C)-(E) and (K)-(R)). Thus, single territorial origin of proliferative cells as the FdPCs, stay permanently small. Support for this speculation comes from “beginning” tumorigenesis in atypical squamous cells in gynecological specimens [59,60]. The nuclear areas measured 1/2 of diploid sized, squamous nuclei (75 - 125 μm versus 150 - 225 μm), and were even referred to as “small cells”. Furthermore, small and large cell, lung carcinomas (and others), do not show cell-size difference due to chromosomal numerical difference [47]. An addendum to this discussion is that evolution from mitosis to primitive one-step meiosis has been argued to involve a type of pairing between homologs in diploid eukaryotes, undergoing haploid, reproductive divisions [61]. The present haploidization process argues that whole complements as units can segregate independent of each other. The fallibility of this system, expressed as asymmetric divisions within narrow limits around a mean, which is indicative of genetic control [29] thus, becomes a reasonable expectation.
The GPA of F-dPCs expressed in a “streaming” growth-pattern has similarity to some types of hyperplasia, but not to others that show nuclear-size (abnormal DNA content) and -shape variability (atypia) [62-64]. Interestingly, the new-born F-dPCs displayed rounded cells and nuclei (Figures 1(C)-(E)), as in some hyperplasias, which became extremely polarized to spindle-shaped cells in the cell-tight streams (Figures 2(E) and (F)). These growths displayed foci of multilayered, crisscrossed cells with change in cell polarity (Figure 2(B)), which was reminiscent of earlier observed 3D-pattern as tumor like spheres in senescence [30]. Loss of cell polarity, disrupting cellular architecture with associated change in cell morphology, has been discussed as a result from skewed division relative to the cytoskeleton axis [28-30, 65]. Skewed divisions were events in aggressive oral cancers [66]. Moreover, cancer pathology regards tissue architectural changes from loss/change of cell polarity as “onset” of tumorigenesis [67].
The pressing question now is how short term AAD treatment can have such genomic/-chromosomal different, abnormal effects on normal diploid cells? The answer lie in the choice of amino acid, glutamine, which is special among all amino acids in that it is a “fuel” in normal cell proliferation, adding to the maintenance of cellular homeostasis [34,35]. Cells normally proliferate under aerobic conditions, in which glucose through glycolysis produce pyruvate, carbondioxide and energy source, ATP in mitochondria. ATP is the energy engine for all macromolecular protein syntheses. Glutamine is an added “nutrient” to glucose by providing nitrogen and carbon to for example, biosynthesis of nucleotides and enzymes for DNA replication (e.g., topoisomerase I & II). In serum, glutamine is the most plentiful, free amino acid, which explains the present reduction from 10% to 2% in M-, and two-times wash of cells for a total stop of mitosis during glutamine-deficiency treatments. It was also noted, that maintenance of the high proliferative capacity of F-dPCs was not kept up when feeding was done with 4 - 5 day old medium (M+), unless supplemented by a new dose of fresh glutamine.
From a human health perspective the present results raises the question about diet compositions, which has a tendency to be time-lasting in individual cases, and for many elderly, a potential problem. Cancer incidence is highest in this group of people, but total deficiency is likely not one of the problems. Malnutrition from inadequate amounts of any amino acid is more likely, when it comes to “feeding” of millions of cells that daily “turnover” in replacement proliferation (e.g., intestine/skin). Nutritional stress as in the present experiments has been found to cause defective autophagy for proper elimination of cell-waste-products [68]. The autophagic process(s) is rather complex in tumorigenesis by being both tumor repressive and promoting [69]. Avoidance of under-nourished metabolism from deregulated mTOR-kinase (Back and Kim [70] is avoidance of a waste-product, toxic tissue environment. Under such conditions cells are killed and others are injured, and it is these “sick” cells with genomic damage that can seek survival mechanisms different from normal mitosis as presently expressed by haploidization to F-dPCs with high proliferative ability. GPA, being a fundamental characteristic for a beginning of a potential cancerous process is therefore, to be avoided, and one factor may be adequate amounts of amino acids in all different diets.
5. Conclusion
Present and earlier studies of normal, diploid, human cells have demonstrated visual, beginning/initiating cellevents that led to genetic changes, positive, for gain of proliferative advantage (GPA) in cell cultures. GPA is the ultimate requirement for a tumorigenic process. Therefore, it is suggested that in addition to the “mutation-theory”, a new cell division mechanistic theory can be formulated for potential tumorigenesis. Basically this theory involves change to a mildly, unstable mitotic machinery with propensity for the progeny cells having gained homozygous determined LOH. The basic proposition is a one-step gain of multiple cell-behavioral changes from inheritance of primitive, unicellular, reproductive division traits. Normal cells under DNA replication stress can revert to primitive cell division-systems, which are bestowed by inheritance new cellular characteristics on consequent progeny cells as compared to the cell of origin. Such progeny cells have by inheritance, “foreign” division traits embedded into their mitotic machinery. A simile for such happenings is primitive unicellular mitotic-meiosis division to genome reduced offspring cells that inherit the mother-cell’s reproductive machinery for yet another generation to come. When such divisions happen in human cells the inherited “reproductive-traits” by the progeny cells must become blended into the innate, mitotic process for survival (nonapototic) and for orderly, continuing proliferation. For example, the haploidization process induced by amino acid glutamine deficiency, demonstrated co-segregation of half-rosette, bichromatid groups of chromosomes, which are in total disagreement with normal mitotic single chromatid segregation, without complement co-segregation. Furthermore, this genome reductive division did not demonstrate mitotic pole-oriented, leading kinetochores with trailing chromosomal arms, which is indicative of normal spindle operation. A question is also why this abnormal division (haploidization) in metaphase cells was not arrested from mitotic checkpoint control, but allowed to cycle from haploidy through genomic doubling to small homozygous diploid cells with GPA (F-dPCs) (see Discussion for smallness). Mitosis for these new, small cells showed changed form both morphologically and operationally. The tumorigenic “genetic” danger of this division system was especially, evidenced by near-haploid cell proliferation in a type of childhood leukemia that also existed in hyperdiploid form from genomic doubling. These abnormal genomic happenings can occur from for example, inadequate, dietary amino acids, leading to toxic tissue environments from improper autophagic waste disposal, and are certainly to be avoided.
Conflict of Interest Statement
The author of this manuscript has no conflict of interest to declare.
REFERENCES
- B. Vogelstein and K. W. Kinzler, “The Multistep Nature of Cancer,” Trends in Genetics, Vol. 9, No. 4, 1993, pp. 138-141. http://dx.doi.org/10.1016/0168-9525(93)90209-Z
- I. Bozic, T. Antal, H. Ohtsuki, H. Carter, D. Kim, S. Chen, R. Karchin, et al., “Accumulation of Driver and Passenger Mutations during Tumor Progression,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 107, No. 43, 2010, pp. 18545-18550. http://dx.doi.org/10.1073/pnas.1010978107
- D. Hanahan and R. A. Weinberg, “Hallmarks of Cancer: The Next Generation,” Cell, Vol. 144, No. 5, 2011, pp. 646-674. http://dx.doi.org/10.1016/j.cell.2011.02.013
- S. E. Shackney and T. V. Shanky, “Genetic and Phenotypic Heterogeneity of Human Malignancies: Finding Order in Chaos,” Cytometry, Vol. 21, No. 1, 1995, pp. 2- 5. http://dx.doi.org/10.1002/cyto.990210103
- D. J. Gordon, B. Resio and D. Pellman, “Causes and Consequences of Aneuploidy in Cancer,” Nature Reviews. Genetics, Vol. 13, No. 3, 2012, pp. 189-203.
- A. A. Sandberg, “Chromosome Abnormalities in Human Cancer and Leukaemia,” Mutation Research, Vol. 247, No. 2, 1991, pp. 231-240. http://dx.doi.org/10.1016/0027-5107(91)90019-K
- R. R. Burbano, A. Medeiros, M. I. Medeiros de Amorim, E. M. Lima, A. Mell, J. B. Neto and C. Casartelli, “Cytogenetics of Epiyhelil Hyperplasias of the Human Breast,” Cancer Genetics and Cytogenetics, Vol. 119, No. 1, 2000, pp. 62-66. http://dx.doi.org/10.1016/S0165-4608(99)00175-2
- J. Fitzgibbon, L.-L. Smith, M. Raghavan, M. L. Smith, S. Debernardi, S. Skoulakis, D. Lillington, T. A. Lister and B. D. Young, “Association between Uniparental Disomy and Homozygous Gene Mutation in Acute Myeloid Leukemias,” Cancer Research, Vol. 65, 2005, pp. 9252-9154. http://dx.doi.org/10.1158/0008-5472.CAN-05-2017
- L. P. Gondek, R. Tiu, L. O’Keefe, M. A. Sekeres, K. S. Theil and J. P. Maciejewski, “Chromosomal Lesions and Uniparental Disomy Detected by SNP Arrays in MDS, MDS/MPD and MDS-Derived AML,” Blood, Vol. 111, No. 3, 2008, pp. 1534-1542. http://dx.doi.org/10.1182/blood-2007-05-092304
- C. H. Coschi and F. A. Dick, “Chromosome Instability and Deregulated Proliferation: An Unavoidable Duo,” Cellular and Molecular Life Sciences, Vol. 69, No. 12, 2012, pp. 2009-2024. http://dx.doi.org/10.1007/s00018-011-0910-4
- A. J. Holland and D. W. Cleveland, “Losing Balance: The Origin and Impact of Aneuploidy in Cancer,” EMBO, Vol. 13, 2012, pp. 501-514. http://dx.doi.org/10.1038/embor.2012.55
- S. J. Pfau and A. Amon, “Chromosomal Instability and Aneuploidy in Cancer: From Yeast to Man,” EMBO, Vol. 13, 2012, pp. 515-527. http://dx.doi.org/10.1038/embor.2012.65
- I. Vitale, L. Galluzzi, L. Senovilla, A. Criollo, M. Jemaa, M. Castedo and G. Kroemer, “Illicit Survival of Cancer Cells during Polyploidization and Depolyploidization,” Cell Death and Differentiation, Vol. 18, 2011, pp. 1403- 1413. http://dx.doi.org/10.1038/cdd.2010.145
- T. Davoli and T. de Lange, “The Causes and Consequences of Polyploidy in Normal Development and Cancer,” Annual Review of Cell and Developmental Biology, Vol. 27, 2011, pp. 585-610. http://dx.doi.org/10.1146/annurev-cellbio-092910-154234
- L. M. Zasadil, E. M. C. Britigan and B. A. Weaver, “2n or Not 2n: Aneuploidy, Polyploidy and Chromosomal Instability in Primary and Tumor Cells,” Seminars in Cell & Developmental Biology, Vol. 24, No. 4, 2013, pp. 370- 379. http://dx.doi.org/10.1290/0603019.1
- K. H. Walen, “Human Diploid Fibroblast Cells in Senescence: Cycling Through Polyploidy to Mitotic Cells,” In Vitro Cellular & Developmental Biology—Animal, Vol. 42, No. 7, 2006, pp. 216-224. http://dx.doi.org/10.1290/0603019.1
- K. H. Walen, “Bipolar Genome Reductional Division of Human Near-Senescent, Polyploid Fibroblast Cells,” Cancer Genetics and Cytogenetics, Vol. 173, No. 1, 2007, pp. 43-50. http://dx.doi.org/10.1016/j.cancergencyto.2006.09.013
- K. H. Walen, “Origin of Diplochromosomal Polyploidy in Near-Senescent Fibroblast Cultures: Heterochromatin, Telomeres and Chromosomal Instability (CIN),” Cell Biology International, Vol. 31, No. 12, 2007, pp. 1447- 1455. http://dx.doi.org/10.1016/j.cellbi.2007.06.015
- K. H. Walen, “Genetic Stability of Senescence Reverted Cells: Genome Reduction Division of Polyploid Cells, Aneuploidy and Neoplasia,” Cell Cycle, Vol. 7, No. 11, 2008, pp. 1623-1629. http://dx.doi.org/10.4161/cc.7.11.5964
- H. O. Lee, J. M. Davidson and R. J. Duronio, “Endoreplication: Polyploidy with a Purpose,” Genes & Development, Vol. 23, 2009, pp. 2461-2477. http://dx.doi.org/10.1101/gad.1829209
- S. K. Pandit, B. Westendorp and A. de Bruin, “Physiological Significance of Polyploidization in Mammalian Cells,” Trends in Cell Biology, Vol. 23, No. 11, 2013, pp. 556-566.
- S. Zhang, I. Mercado-Uribe, B. Sun, J. Kuang and L. Liu, “Generation of Cancer-Stem-Like Cells through the Formation of Polyploid Giant Cancer Cells,” Oncogene, Vol. 33, No. 1, 2014, pp. 116-128.
- D. N. Wheatley, “Growing Evidence of the Repopulation of Regressed Tumors by the Division of Giant Cells,” Cell Biology International, Vol. 32, No. 9, 2008, pp. 1029-1030. http://dx.doi.org/10.1016/j.cellbi.2008.06.001
- J. Erenpreisa, K. Salmina, A. Huna, E. A. Kosmacek, M. S. Cragg, F. Ianzini and A. Anisimov, “Polyploid Tumor Cells Elicit Paradiploid Progeny through Depolyploidizing Divisions and Regulated Autophagic Degradation,” Cell Biology International, Vol. 35, No. 7, 2011, pp. 687- 695. http://dx.doi.org/10.1042/CBI20100762
- I. B. Raikov, “The Protozoan Nucleus, Morphology and Evolution,” In: M. Alfert, W. Beermann, L. Goldstein, K. R. Porter and P. Sitte, Eds., Cell Biology Monographs, Vol. 9, Springer-Verlag, Wien-New-York, 1982, pp. 209- 214.
- A. Hunding, “Possible Prepatterns Coverning Mitosis: The Mechnism of Spindle-Free Chromosome Movement in Aulacanthascolymantha,” Journal of Theoretical Biology, Vol. 89, 1981, pp. 353-385.
- K. G. Grell and A. Ruthmann, “Uber die Karyologie des Radiolars Aulachantascolymantha und Feinstruktur Seiner Chromosomen,” Chromosoma, Vol. 15, No. 2, 1964, pp. 185-211. http://dx.doi.org/10.1007/BF00285729
- K. H. Walen, “Genome Reversion Process of Endopolyploidy Confers Chromosome Instability on the Descendent Diploid Cells,” Cell Biology International, Vol. 36, No. 2, 2012, pp. 1-9. http://dx.doi.org/10.1042/CBI20110052
- K. H. Walen, “Normal Human Cells Acquiring Proliferative Advantage to Hyperplasia-Like Growth-Morphology: Aberrant Progeny Cells Associated with Endopolyploid and Haploid Divisions,” Cancer and Clinical Oncology, Vol. 2, No. 2, 2013, pp. 1-15. http://dx.doi.org/10.5539/cco.v2n2p19
- K. H. Walen, “Normal Human Cell Conversion to 3-D Cancer-Like Growth: Genome Damage, Endopolyploidy, Senescence Escape, and Cell Polarity Change/Loss,” Journal of Cancer Therapy, Vol. 2, No. 2, 2011, pp. 181- 189. http://dx.doi.org/10.4236/jct.2011.22023
- K. H. Walen, “Senescence Arrest of Endopolyploid Cells Renders Senescence into a Mechanism for Positive Tumorigenesis,” In: Tumor Dormancy and Cellular Quiescence and Senescence, Vol. 1: Aging, Cancer, and Noncancer Pathologies, Springer, Berlin, 2013.
- J. J. Freed and S. A. Schatz, “Chromosome Aberrations in Cultured Cells Deprived of Single Essential Amino Acid,” Experimental Cell Research, Vol. 55, No. 3, 1969, pp. 393- 409. http://dx.doi.org/10.1016/0014-4827(69)90574-6
- R. Phillip, E. Campbell and D. N. Wheatley, “Arginine Aeprivation, Growth Inhibition and Tumor Cell Death: 2. Enzymatic Degradation of Arginine in Normal and Malignant Cell Cultures,” British Journal of Cancer, Vol. 88, No. 4, 2003, pp. 613-623. http://dx.doi.org/10.1038/sj.bjc.6600681
- R. J. Deberardinis and T. Cheng, “Q’s Next: The Diverse Functions of Glutamine in Metabolism, Cell Biology and Cancer,” Oncogene, Vol. 29, No. 3, 2010, pp. 313-324. http://dx.doi.org/10.1038/onc.2009.358
- J. R. Cantor and D. M. Sabatini, “Cancer Cell Metabolism: One Hallmark, Many Faces,” Cancer Discovery, Vol. 2, No. 10, 2012, pp. 899-905.
- C. L. Huskins, “Segregation and Reduction in Somatic Tissues. I. Initial Observations on Allium cepa,” Journal of Heredity, Vol. 39, 1948, pp. 310-325.
- E. Glass, “Das Problem der Gesonderung in den Mitosen Unbehandelter Rattenlebern,” Chromosoma, Vol. 8, No. 1, 1957, pp. 468-492. http://dx.doi.org/10.1007/BF01259515
- T. T. Glazko, “Chromosomes Subdividing to Haploid Sets in Diploid Metaphase Plates of Some Mammalian Species,” Proceedings of the 15th International Chromosome Conference, Brunel University, 5-10 September 2004, p. 63.
- W. Mayer, A. Smith, R. Fundele and T. Haaf, “Spatial Separation of Parental Genomes in Preimplantation Mouse Embryos,” Journal of Cell Biology, Vol. 148, No. 4, 2000, pp. 629-634. http://dx.doi.org/10.1083/jcb.148.4.629
- J. Travis, “Return of the Matrix,” Science, Vol. 318, No. 5855, 2007, pp. 1400-1401. http://dx.doi.org/10.1126/science.318.5855.1400
- J. Picket-Heaps and A. Forer, “Mitosis: Spindle Evolution and Matrix Model,” Protoplasma, Vol. 235, No. 1-4, 2009, pp. 91-99. http://dx.doi.org/10.1007/s00709-009-0030-2
- M. V. Blagosklonny, “Cell Immortality and Hallmarks of Cancer,” Cell Cycle, Vol. 2, No. 4, 2003, pp. 296-299. http://dx.doi.org/10.4161/cc.2.4.470
- K. L. Chan, T. Palmai-Pallal, S. Ying and I. D. Hickson, “Replication Stress Induces Sister Chromatid Bridging at Fragile Site in Mitosis,” Nature Cell Biology, Vol. 11, No. 6, 2009, pp. 753-760. http://dx.doi.org/10.1038/ncb1882
- K. H. Walen, “Spontaneous Cell Transformation: Karyoplasts Derived from Multinucleated Cells Produce New Cell Growth in Senescent Human Epithelial Cell Cultures,” In Vitro Cellular & Developmental Biology, Vol. 40, No. 5-6, 2004, pp. 150-158. http://dx.doi.org/10.1290/1543-706X(2004)40<150:SCTKDF>2.0.CO;2
- G. E. Sarto, P. A. Stubblefield, J. Lurain and E. Therman, “Mechanisms of Growth in Hydatidiform Moles,” American Journal of Obstetrics & Gynecology, Vol. 148, No. 7, 1984, pp. 1014-1023. http://dx.doi.org/10.1016/0002-9378(84)90545-3
- Y. Kukita, K. Miyatake, R. Stokowski, D. Hinds, K. Higasa, N. Wake, et al., “Genome-wide Definitive Haplotypes Determined Using a Collection of Complete Hydatidiform Moles,” Genome Research, Vol. 15, No. 11, 2013, pp. 1511-1518. http://dx.doi.org/10.1101/gr.4371105
- F. Mitelman, “Catalog of Chromosome Aberrations in Cancer,” Alan Liss, Inc., New York, 1988.
- S. Safavi, E. Forestier, I. Golovleva, G. Barbany, K. H. Nord, A. V. Moorman, C. J. Harrison, B. Johansson and K. Paulsson, “Loss of Chromosomes Is the Primary Event in Near-Haploid and Low-Hypodiploid Acute Lymphoblastic Leukaemia,” Leukemia, Vol. 27, No. 1, 2013, pp. 248-250. http://dx.doi.org/10.1038/leu.2012.227
- S. Heim and F. Mitelman, “Cancer Cytogenetics: Chromosomal and Molecular Genetic Aberrations of Tumor Cells,” 2nd Edition, Wiley-Liss, Inc., New York, 1995.
- L. Olsson, K. Paulsson, J. V. Bovee and K. H. Nord, “Clonal Evolution Through Loss of Chromosomes and Subsequent Polyploidization in Chondrosarcoma,” PLoS ONE, Vol. 6, No. 9, 2011, Article ID: e24977.
- N. Mandahl, B. Johansson, F. Mertens and F. Mitelman, “Disease-Associated Patterns of Disomic Chromosomes in Hyperhaploid Neoplasms,” Genes, Chromosomes and Cancer, Vol. 51, No. 6, 2012, pp. 536-544. http://dx.doi.org/10.1002/gcc.21947
- Z. Storchova and D. Pellman, “From Polyploidy to Aneuploidy, Genome Instability and Cancer,” Nature Reviews Molecular Cell Biology, Vol. 5, 2004, pp. 45-54. http://dx.doi.org/10.1038/nrm1276
- A. Levan and T. S. Hauschka, “Endomitotic Reduplication Mechanisms in Ascites Tumors,” Journal of the National Cancer Institute, Vol. 14, No. 1, 1953, pp. 1-43.
- T. Davoli, E. L. Denchi and T. de Lange, “Persistent Telomere Damage Induces Bypass of Mitosis and Tetraploidy,” Cell, Vol. 141, No. 1, 2010, pp. 81-93. http://dx.doi.org/10.1016/j.cell.2010.01.031
- T. Boveri, “ZurFrage der Entstehung Maligner Tumoren,” Fischer, Jena, 1914.
- L. P. Bignold, B. L. D. Coghlan and H. P. A. Jersmann, “David von Hansemann: Contributions to Oncology: Context, Comments and Translations,” Birkhauser Verlag, Basel, 2007.
- R. G. Nagele, M. R. Jones, H. G. Nguyen, D. J. McCrann, C. St. Hilaire, B. Schreiber, et al., “Precise Spatial Positioning of Chromosomes During Prometaphase: Evidence for Chromosomal Order,” Science, Vol. 270, No. 5243, 1995, pp. 1831-1835.
- A. Bolzer, G. Kreth, I. Solovei, D. Koehler, K. Saracoglu, C. Fauth, et al., “Three-Dimensional Maps of all Chromosomes in Human Male Fibroblast Nuclei and Prometaphase Rosettes,” PLoS Biology, Vol. 3, No. 5, 2005, pp. 826-842.
- S. F. Patten, “Diagnostic Cytopathology of the Uterine Cervix,” Monographs in Clinical Cytology, Vol. 3, S. Karger, Basel, 1978, pp. 20-25.
- [61] M. H. Stoler, “Does Every Little Cell Count? Don’t ASCUS,” Cancer Cytopathology, Vol. 87, No. 2, 1999, pp. 45-47.
- [62] A. S. Wilkins and R. Holliday, “The Evolution of Meiosis from Mitosis,” Genetics, Vol. 181, No. 1, 2009, pp. 3-12. http://dx.doi.org/10.1534/genetics.108.099762
- [63] L. M. Shabad, “Precancerous Morphological Lesions,” Journal of the National Cancer Institute, Vol. 50, 1973, pp. 1421-1428.
- [64] P. P. Rosen, “Proliferative Breast ‘Disease’: An Unresolved Diagnostic Dilemma,” Cancer, Vol. 71, No. 12, 1993, pp. 3798-3807. http://dx.doi.org/10.1002/1097-0142(19930615)71:12<3798::AID-CNCR2820711203>3.0.CO;2-S
- [65] G. Belge, L. Roque, J. Soares, S. Bruckmann, B. Thode, E. Fonseca, A. Clode, S. Bartnitzke, S. Castedo and J. Bullerdiek, “Cytogenetic Investigations of 340 Thyroid Hyperplasias and Adenoma Revealing Correlations Between Cytogenetic Findings and Histology,” Cancer Genetics and Cytogenetics, Vol. 101, No. 1, 1998, pp. 42-48. http://dx.doi.org/10.1016/S0165-4608(97)00057-5
- [66] C. Royer and X. Lu, “Epithelial Cell Polarity: A Major Gatekeeper against Cancer?” Cell Death and Differentiation, Vol. 18, No. 9, 2011, pp. 1470-1477. http://dx.doi.org/10.1038/cdd.2011.60
- [67] W. S. Saunders, M. Shuster, X. Huang, B. Gharaibe, A. H. Enyenihi, I. Petersen and S. M. Gollin, “Chromosomal Instability and Cytoskeletal Defects in Oral Cancer Cells,” Proceedings of the National Academy of Sciences of the United States of America, Vol. 97, No. 1, 2000, pp. 303- 308.
- [68] R. G. Steinbeck, “Dysplasia in View of the Cell Cycle,” European Journal of Histochemistry, Vol. 48, No. 3, 2004, pp. 203-211.
- [69] E. White and R. S. DiPaola, “The Double-Edge Sword of Autophagy Modulation in Cancer,” Clinical Cancer Research, Vol. 15, No. 17, 2009, pp. 5308-5316.
- [70] E. Morselli, L. Galluzzi, O. Kepp, J. M. Vicencio, A. Criollo, M. C. Maiuri and G. Kroemer, “Antiand Pro-Tumor Functions of Autophagy,” Biochimica et Biophysica Acta, Vol. 1793, No. 9, 2009, pp. 1524-1532.
- [71] J. H. Back and A. L. Kim, “The Expanding Relevance of Nuclear mTOR in Carcinogenesis,” Cell Cycle, Vol. 10, No. 22, 2011, pp. 3849-3852. http://dx.doi.org/10.4161/cc.10.22.18329
Abbreviations
AAD: amino acid deprivation GPA: gain of proliferative advantage UPD: uniparentaldisomy F-dPCs: fibroblast derived prolific cells LOH: loss of heterozygosity SNP: single nucleotide polymorphism