Applied Mathematics
Vol.06 No.11(2015), Article ID:60845,10 pages
10.4236/am.2015.611172
The CARSO (Computer Aided Response Surface Optimization) Procedure in Optimization Studies
Mauro Fernandi1, Massimo Baroni2, Matteo Bazzurri2, Paolo Benedetti2, Danilo Chiocchini3, Diego Decastri1, Cynthia Ebert4, Lucia Gardossi4, Giuseppe Marco Randazzo2, Sergio Clementi3*
1Flint Group, Cinisello Balsamo, Milano, Italy
2Biology, Biotechnology and Chemistry Department, Section Chemistry, University of Perugia, Perugia, Italy
3M.I.A. Multivariate Infometric Analysis Srl, Perugia, Italy
4Department of Chemistry and Pharmaceutical Sciences, University of Trieste, Trieste, Italy
Email: *mia@miasrl.com
Copyright © 2015 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/



Received 15 September 2015; accepted 27 October 2015; published 30 October 2015
ABSTRACT
The paper illustrates innovative ways of using the CARSO (Computer Aided Response Surface Optimization) procedure for response surfaces analyses derived by DCM4 experimental designs in multivariate spaces. Within this method, we show a new feature for optimization studies: the results of comparing their quadratic and linear models for discussing the best way to compute the most reliable predictions of future compounds.
Keywords:
CARSO Procedure, Design Strategies, Double Circulating Matrices (DCMs), Optimization

1. Introduction
Following our recent papers [1] - [3] , where we showed a new strategy for collecting the data needed for defining a response surface on the basis of an innovative method that requires only a very low number of experimental data, based on Double Circulant Matrices (DCMs). They are similar to Central Composite Designs (CCDs), which represent the best way to generate response surfaces. The final surface model is obtained by the formerly developed CARSO method [4] , where the surface equation is derived by a PLS model on the expanded matrix, containing linear, squared and bifactorial terms.
This paper is meant to discuss if this statistical form of the X block is the most appropriate within chemical studies of experimental properties either in terms of mixtures, or in terms of literature constants, mainly electronic and steric, reported for each substituent. For a long time, organic chemists collected data for a class of similar compounds (the field was called correlation analysis) and computed the slope of the resulting straight line, thus comparing the behaviour of different chemicals in the same reaction.
After a full immersion of one of us (SC, 1981) in the group of Svante Wold at Umea (Sweden) to learn chemometrics and to use their software SIMCA [5] , a few of us decided to enter into this new fascinating world. In order to drive the interest of our colleagues towards a more rewarding result, e.g. so that the study should find the best conditions for optimizing the property or the process, we published CARSO in 1989 [4] , and GOLPE in 1993 [6] . Since then we recognized that our procedures were children of SIMCA.
Now we discuss the best way to compute reliable predictions. It is clear that, for an optimization study, the first must is a plan of the experiments, but this is outside the objectives of this paper. See, for example, reference [1] .
2. Optimization Studies: The Models
This paper is meant to make clear how an optimization study should be carried out. The first choice should be an experimental plan as round as possible: we selected the double circulant matrices (DCMs) that represent the maximal roundness [1] . The next thing to be done is to find how the y response(s) can be described by the best possible relationship in terms of the x values. In principle it is possible to use linear or non linear models. The linear models are simpler to be interpreted, but it is unusual that the response depends only by a single descriptor. Usually the response is affected by a complex mixture of different effects that contribute simultaneously: therefore a bell form should appear more appropriate to give a better model. Therefore each model will be evaluated by different approaches, namely the variance explained, the standard deviation and the intervals covered by the recalculations or predictions. Indeed we wish to show numerically the relative performances of linear and quadratic models.
2.1. Collection of Results by DCM4
Our experimental data set contains 13 different mixtures (objects) generated by different relative amounts of 4 variables according to a strategy in keeping to the experimental design illustrated by a DCM4 [2] . At the time we wanted to show which could be the minimum number of measurements needed to build a response surface (and we suggested to use only two of the six possible quartets of mixture, namely sm1 and sm3, plus two replicates at the centre point, i.e. 10 points). Indeed, in this case, with the objective to compare the soundness of our models, we prefer using a model with all three first quartets of the DCM4 and one single central point only [2] .
Table 1 reports the results of our experimental data set. For each of the 13 different mixtures (objects) are listed the real value of the y (the inverse of the contact angle) and their coded values between 0 and 100, that helps to understand immediately if the following predictions are within or outside the range of the collected data.
Table 2 lists the recalculation by the three statistical models Q1, L1 and L2, indicating the variance explained. The ranges of each of the models are somewhat different. They span the ranges reported in the last rows of the Table, even showing the maximum and minimum values and their mean averages. On comparing the three models we can see that the Q1 model have a range of 103.8 (between −8 and 95.8) with an average of 43.9, while the L1 model have a range of 92.3 (between −16.9 and 98.2) with an average of 29.3, and the L2 model have a range of 114.7 (between −16.1 and 98.2) with an average of 41.3.
The data of the statistical model Q1 is a quadratic model with only one latent variable, which contains all linear, quadratic and bifactorial terms (and the PLS uses all the terms, as if all of these were “new” pseudolinear data), whereas L1 has only linear terms and one latent variable, and L2 has also only linear terms but computed by two latent variables, even if this type of modelling is well known only by researchers with confidence in chemometrics.
Table 2 reports the predictions, for the three models, on the 12 objects of this collection and excluding the central point, because it gives three different predictions and there is no way to choose the best of them.

Table 1. Selected experimental plan (training set).
Table 2. Modelling of the selected experimental plan (training set).
On comparing the three models by their explained variance and STD (standard deviation), it is clear that Q1 is by far the best, showing a 93% of explained variance and a STD of 7.8, followed by L2, showing a 80% of explained variance and a STD of 14.1, and L1 showing a 67% of explained variance and a STD of 20.0.
2.2. Predictions on Hypervertices
In order to evaluate the relative sizes of variations of the three models shown in Table 2, we report the predictions, for each of the three models, on the 16 points located on the hypervertices of the 4D space, to study comparatively the span generated by each model. Therefore the values reported in Table 3 could allow to find out which of the models span a greater interval between the minimum and maximum. The list of the vertices is ordered according to the Yates proposal.
2.3. Comparison of External Predictions (Hypervertices)
The comparison of the predictions listed in Table 3 allows to evaluate the span for each of the three models used on the same set of experimental data. We remind that examining the real data we already reported the standard deviations: the Q1 model gives the best result (7.8), followed by L2 (14.1) and L1 (20).
On inspecting the external predictions we observe that Q1 shows an interval of 130.0 (from −12.7 to 117.3), while L1 shows an interval of 110.3 (from −25.9 to 84.4) and L2 shows an interval of 245.2 (from −93.4 to 151.9). This result clearly shows that this model (linear model with two latent variables), independently of other good parameters, can gives very risky external predictions outside the explored range. Because of that we stopped to continue to explore the characteristics of the L2 model.
2.4. Revision of Recalculations/Predictions
The data discussed after Table 2 for the training set and after Table 3 for the testing set clearly indicate that the quadratic models might be defined to be the most reliable because the intervals of predictions are closer to 100 (similar to the L1 one, but different from the L2 models).
Table 3. Predictions on the 16 hypervertices (testing set).
These results show two significant characteristics to be interpreted: the ranges and the averages of recalculations/predictions of each model. The Q1 models cover roughly the range between 0 and 100. The L1 models have the smaller intervals of variation (92.3 for recalculations and 110.3 for predictions), but they are all shifted towards the lower part of the collected data. The L2 models have by far the larger intervals covering predictions data much larger than the highest ones and lower than the smallest figure, and therefore we did not investigate it any more.
However all these arguments (besides the standard deviations and the ranges of recalculations/predictions) might appear to be somewhat too poor to claim that the quadratic model is better than the linear ones.
Therefore we decided to approach our problem by constructing a unique list of predicted y values obtained by “inner” models, i.e. using the values predicted for each object by modelling using the two submatrices not including the object, and computing a sort of self-predictions of the left out objects for each model based on a couple of the three submatrices, as we always applied for validating each model.
3. Inner Predictions by Each Couple of Submatrices
In order to evaluate the predictions by partial models, so that the objects left out are really “predicted” and not “recalculated”, we computed the two models using only the two submatrices that does not contain the object to be evaluated for correctly obtaining “external” or “inner” predictions.
The quadratic predictions listed in Table 4 are computed for the objects of the left out submatrix, reported in italics, and allow to compute the standard deviations of the predictions. Table 5 lists the linear predictions with one latent variable The results shown in Table 4 and Table 5 allow to compare the models by their standard deviation: 11.8 for the Q1 model, 21.3 for the L1 model.
Moreover we can compare the intervals found for each model. In order to evaluate the predictions by partial models, so that the objects left out are really “predicted” and not “recalculated”, we computed the two models using only two submatrices for correctly obtaining “inner” predictions for each object of the third one. The interval found for the Q1 model is 100.7, while for the L1 model is 91.2. Therefore the intervals of both models Q1 and L1 with inner predictions are very similar.
Table 4. Quadratic inner predictions (1LV) compared with coded experimental data.
Table 5. Linear inner predictions (1LV) compared with coded experimental data.
4. Summary of the Data Collection
The data reported in the Tables give a clear answer to our question: which of the models can be defined the most reliable for new predictions, to be computed by the data of Table 1. The discussion of these results clearly shows that the Q1 model is by far the best, with respect to L1, while L2 is the worst, because its predictions cover intervals from very high to very low, outside from those of the experimental data.
Because of this the best way of describing the trends of a series of compounds appear to be a quadratic model, that finds out reliable results, usually within the explored space. On the contrary the linear model with one latent variable gives predictions within a much smaller interval, which is also shifted download, towards lower numbers, while the linear model with two latent variables (which is less used by researchers) spans a much larger space, with quite higher and lower results.
However the data listed in Table 1 can be considered self-referenced, since they are all experimental values, not evaluated by predictions. On the contrary, the results reported in Table 4 and Table 5 are real predictions obtained by the differences between the coded experimental data and their inner values.
5. Dissection of the Information
This approach can be used as an alternative way to compare the relative reliability of the quadratic and linear models based on the dissection of information according to the Pythagoras’ theorem. Given a non expanded DCM4 it is not possible to execute a Multiple Linear Regression. Indeed the expanded DCM4 cannot be treated by MLR because of two reasons:
a) The number of objects (13) is smaller than the number of variables (14);
b) In the linear blocks each column of the DCM4 is a linear combination of the other three.
The only possibility of running MLR on a DCM4 is eliminating one column (say x4), and adding on the left a column of numbers “ 1” for determining the intercept. The coefficients of MLR are listed in Table 6, and it is possible to recalculate each object listed in Table 7.
Table 6. Coefficients of multiple linear regression.
Table 7. Dissection of information.
Table 7 shows the dissection of information according to the Pythagoras’ theorem: TSS = MSS + RSS, where TSS = sum[yexp − (y ave exp)]2 (total information = 17,971), MSS = sum[rec − (y ave exp)]2 (information explained by the model = 14,927), RSS = sum[yexp-rec (y by MLR)]2 (information not explained by the model = 3044). Formally the word “information” should be substituted by the stastistical term “deviance”, but in che- mometrics we prefer the term information which will be understood by a larger number of readers.
On applying the same computations with any other possible triplet of variables (x2, x3, x4 shown; x1, x3, x4; x1, x2, x4; x1, x2, x3) we obtained always different results for the MLR coefficients (excluding the intercept): see Table 8.
Although we found that the MLR coefficients are different on using diverse triplets of variables it is noteworthy that the vectors of the y values computed (not shown) by the data of one of them (x1, x2, x3) is identical to the other one (x2, x3, x4).
This surprising result, observed also for the triplet (x1, x2, x4) even if the variables are not listed in sequence, may be attributed to the roundness of the DCM. As a consequence this happens even if the variables are not in an ordered sequence.
6. Using PLS Instead of MLR
In this section we show what happens on using PLS instead of MLR.
The first appearance of the PLS algorithm in the chemical literature was a merit by Herman Wold in 1966, and followed by many others, among which his son Svante. Half century later PLS showed to be much more reliable for finding quantitative relationships between chemical structure and properties.
Obviously the old version of MLR cannot work both on the expanded and the non expanded matrices, because their rank is not the same of the number (k) of independent variables. In other words the squared matrix (XtX), of order k, cannot be inverted.
Applying PLS on the expanded matrix (model called Q1) and on the non expanded matrix (model called L1, because it keeps only the first latent variable) we obtained the results reported in Table 9.
Table 8. MLR coefficients for two triplets of variables.
Table 9. Dissection of the information for models Q1 and L1.
The results reported in Table 9 show that only Q1 mimics the relationship
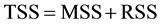
which has an interesting geometrical interpretation in the 2D space. In other words this relationship simulates the geometrics of the Pythagoras’ theorem for a right triangle having a hypotenuse of lenghth2 equal to TSS, the longer side having a lenghth2 equal to MSS, and the shorter one having a lenghth2 equal to RSS.
Furthermore the data of Table 9 also allows to demonstrate the superiority of the Q1 model over L1 because:
a) Q1 shows that the weight of the MSS component is greater than that of RSS;
b) Q1 is the closest to the ideal null value of Abs(TSS-MSS-RSS), the indicator of the geometric idealistic, while L1 is much more distant;
c) In conditions of almost idealistic geometrics (typical of MLR) Q1 shows a RSS value of 1195, whereas L1 shows a value of 5461: this means that the Q1 model preserves only the 22% of the data involved in the L1 model, but gives a better picture of the situation.
d) This means that, under these conditions, we can eliminate 78% of information that appears to be non systematic.
7. Revaluation of the CARSO Software
Besides the comments given so far, we wish to remember that the core of this paper is the CARSO procedure illustrated in ref. 4, published in spring 1989. A few months later a similar paper was published in ref. 8 by our Swedish friends. At the time we suggested to apply this new approach to any data set to be used in optimization studies. Therefore we considered CARSO a possible new module to be inserted into the SIMCA software.
The CARSO module makes a simple, but significant, change of the matrix, that is expanded, on adding the squares and the cross products terms to the linear ones. This approach, some 25 years ago, allowed to model by PLS the expanded matrix, that is still used, in the mode now called Q1, as we showed in this paper.
Indeed, at the time, the main interest of the quadratic model (the linear one cannot give this information) was focused on the search of the operative intervals of each independent variable for the optimization of the y variable(s). Because of this we could use the canonical analysis, searching the coordinates for a maximum, if it exists, or for the stationary points, within the explored space, or even the extreme points on the frontier of the experimental domain. In other words the CARSO method is a full software tool for optimization studies.
Today this practice is not widely used [7] [8] , because a complex system cannot be described by linear terms only, but it requires to involve deviations from linearity (squared terms) and the synergic and antagonist effects (cross product terms). In general, on increasing the polynomial degree used for interpolating the data, grows the fitting and decreases the predictability. The second degree function needed for describing the response surface is the best compromise between these two properties, without involving higher terms.
The equality [TSS = RSS + MSS], that can be represented graphically by the relationships of the areas of the squares built on the sides of a rectangular triangle can be applied (high degree of approximation) only to the Q1 model. Furthermore the Q1 model (CARSO) lowers the unexplained information (RSS) with respect to the linear model. To sum up we can state that the CARSO power in this dataset is the result of both the Q1 power multiplied by the DCM power, so that also objects sum up to zero.
Four years later we published a further paper [6] on almost the same problems (GOLPE: Generating Optimal Linear PLS Estimations) where the declared main field of application was PCA and PLS models. The software, that includes the CARSO modules, adds partial information not shown in SIMCA, but the most important data are limited to the results of the module COEFFER that transforms the PLS loadings (called also gamma) into the coefficients of the second degree polynomial.
8. Conclusions
This paper was done for showing that in optimization studies it is needed to use a quadratic model. In other words, it means that only this model can be used for deriving reliable predictions of further compounds. This has been shown numerically here, but this is also implied into this problem the need of requiring a hyperbell for finding out the operative intervals. Because of this, the best way of describing the trends of a series of compounds is a quadratic model that finds out reliable results, usually within the explored space. On the contrary, the linear model with one latent variable gives much lower data, which seems unreliable.
This choice is in keeping with the position referred by Rosipal [7] , who discussed the nonlinear PLS models on the basis introduced by Svante Wold et al. [8] . Because of this, the best way of describing the trends of a series of compounds is a quadratic model that can be found by using a linear model on the expanded matrix, as we did since the beginning.
The main goal of this paper was finding out which statistical method was more reliable for computing the predictions of new objects outside the training set used: we took into account a quadratic model and a linear model and we could demonstrate that the quadratic model is by far the best.
Acknowledgements
The authors wish to thank Dr. Matthias Henker (Flint Group Germany) for financing a partnership contract with MIA srl and Prof. Svante Wold and his former coworkers in Umeå (Sweden) for introducing SC to chemometrics.
Cite this paper
MauroFernandi,MassimoBaroni,MatteoBazzurri,PaoloBenedetti,DaniloChiocchini,DiegoDecastri,CynthiaEbert,Giuseppe MarcoRandazzo,SergioClementi,LuciaGardossi, (2015) The CARSO (Computer Aided Response Surface Optimization) Procedure in Optimization Studies. Applied Mathematics,06,1947-1956. doi: 10.4236/am.2015.611172
References
- 1. Clementi, S., Fernandi, M., Decastri D. and Bazzurri, M. (2014) Mixture Optimization by the CARSO Procedure and DCM Strategies. Applied Mathematics, 5, 3026-3039.
http://dx.doi.org/10.4236/am.2014.519290 - 2. Clementi, S., Fernandi, M., Baroni, M., Decastri, D., Randazzo, G.M. and Scialpi, F. (2012) MAURO: A Novel Strategy for Optimizing Mixture Properties. Applied Mathematics, 3, 1260-1264.
http://dx.doi.org/10.4236/am.2012.330182 - 3. Fernandi, M., Bazzurri, M., Decastri D. and Clementi S. (2015) Experimental Design for Optimizing a Mixture of Materials plus an Evaporating Solvent. Applied Mathematics, 6, 1740-1746.
http://dx.doi.org/10.4236/am.2015.610154 - 4. Clementi, S., Cruciani, G., Curti, G. and Skagerberg, B. (1989) PLS Response Surface Optimization: The CARSO Procedure. Journal of Chemometrics, 3, 499-509.
http://dx.doi.org/10.1002/cem.1180030307 - 5. SIMCA 4.01. www.umetrics.com
- 6. Baroni, M., Costantino, G., Cruciani, G., Riganelli, D., Valigi, R. and Clementi, S. (1993) Generating Optimal Linear PLS Estimations (GOLPE): An Advanced Chemometric Tool for Handling 3D-QSAR Problems. Quantitative Structure-Activity Relationships, 12, 9-20.
http://dx.doi.org/10.1002/qsar.19930120103 - 7. Rosipal, R. (2011) Non Linear Least Squares: An Overview. In: Lodhi, H. and Yamanishi, Y., Eds., Chemoinformatics and Advanced Machine Learning Perspectives: Complex Computational Methods and Collaborative Techniques, ACCM, IGI Global, 169-189.
- 8. Wold, S., Ketteneh-Wold, N. and Skagerberg, B. (1989) Nonlinear PLS Modeling. Chemometrics and Intelligent Laboratory Systems, 7, 53-65.
http://dx.doi.org/10.1016/0169-7439(89)80111-X
NOTES

*Corresponding author.