Advances in Bioscience and Biotechnology
Vol.3 No.1(2012), Article ID:17201,5 pages DOI:10.4236/abb.2012.31013
A microliter incubator array for understanding culture condition selectivity*
![]()
1Department of Mechanical and Materials Engineering, University of Denver, Denver, USA
2Department of Biological Sciences, University of Denver, Denver, USA
3Department of Biochemistry & Molecular Biology, University of Texas Medical Branch, Galveston, USA
Email: Corinne.Lengsfeld@du.edu
Received 6 October 2011; revised 17 November 2011; accepted 21 December 2011
Keywords: Microfluidics; Infectious Diseases; Incubation
ABSTRACT
The emergence of antibiotic-resistant bacterial strains and the weaponization of rare bacterial strains pose a potential threat of pandemic disease. Those involved in the treatment and control of infectious diseases have called for the development of a device capable of rapidly, simultaneously, and safely investigate a myriad of culture conditions. In response to this need, a microliter incubator array system is described and results from a proof-of-concept study using yeast cells to determine optimal growth conditions is presented.
1. INTRODUCTION
With the development of effective sanitation techniques and the widespread use of antibiotics, infectious bacterial diseases have had a diminishing influence on the health and lifespan of the developed world’s population [1]. In contrast, infectious diseases still have a major impact on the developing world’s population. With the increasing emergence of new antibiotic-resistant bacterial strains of previously controlled diseases and the weaponization of normally rare bacterial strains, the threat of a pandemic disease and its fall-out on the developed world has reemerged; a threat society is currently unprepared to meet [2,3]. Recognition of this threat has led to efforts to address the prevention (quarantine, inoculation) and cure (antibiotics) of these diseases, as well as the study of the complex interaction between the host, pathogen, and environmental factors that impact the progression and severity of the disease [3]. The genomic revolution and the advent of reverse vaccinology promise a rapid response to infectious diseases [4]; however, many of these infectious agents are difficult or impossible to grow in vitro (e.g. Hepatitis C or bacillus causing Whipple disease) [4-6], limiting the analysis of many of the most severe threats (e.g. respiratory syncytial virus).
An understanding of host-pathogen interactions and pathogen virulence factors should lead to new therapies, and even cures [3,7], for many infectious diseases. Complete epidemiology and pathogenesis studies require the pathogen to be cultivated in the laboratory [4,6]. Unfortunately, the conditions for growth are often very specific and difficult to determine. At times this means that an organism simply will not grow in culture, as in the cases of Hepatitis B and C [8]. In other instances, the culture conditions actually create selection pressures that result in mutated strains of the pathogen [9,10]. Mutations can be either harmful or helpful in the development of effecttive vaccines, but characterizing the events that lead to the mutation must be investigated first. Conventional incubators are large and service only one condition at a time, making the process to find the necessary conditions a time-consuming practice of trial and error [6]. To address these limitations, a novel, microliter incubator array system to rapidly, simultaneously, and safely investtigate a myriad of culture conditions is described. Additionally, results from a proof-of concept study where the device is used to impart and observe the effects of variable nutrient conditions on wild-type yeast are presented.
2. DESIGN
Incubators are characterized by providing three main functions-constant temperature, humidified air, and ventilation—in an enclosed space. The incubator introduced in this paper is a multi-layered, polycarbonate device designed for time-efficient laboratory practices where specific liquid nutrients and/or gas compositions can be delivered via channels to yield 25 different well conditions—a matrix of five wells by five wells (Figure 1). The array is scalable to hundreds of wells, limited only by the equal filling of wells along a single fluid channel. The overall outer dimensions of the incubator are 101.6
 (a)
(a)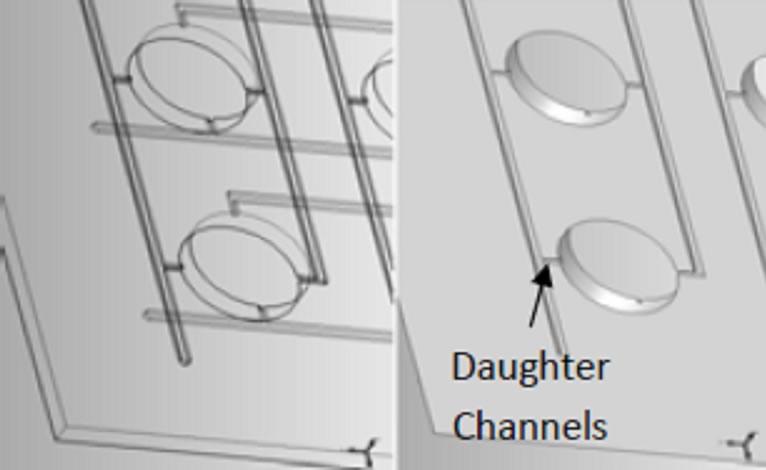 (b)
(b)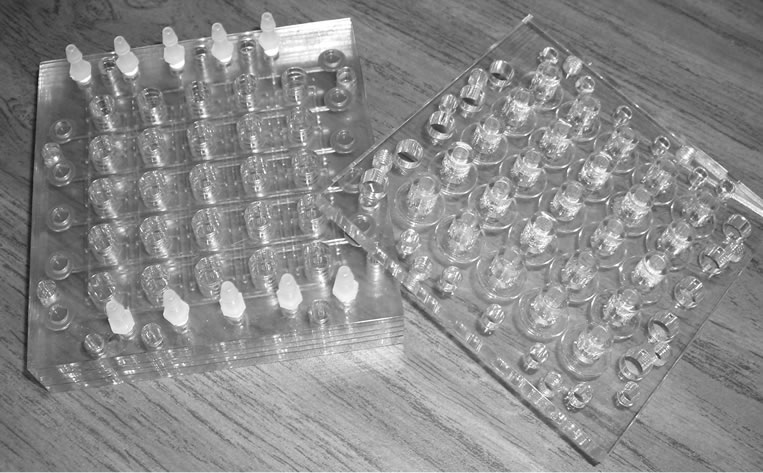 (c)
(c)
Figure 1. Incubator design: (a) Solid model assembly drawing of micro liter incubator array plate design; (b) Enlarge view of the plates with channels; and (c) photo of final assembled unit.
mm by 101.6 mm by 25.4 mm high. The wells are 8 mm in diameter with a maximum liquid depth of 6.2 mm— yielding a maximum allowable volume of 312 µL per well. Clear polycarbonate was chosen because of its resistance to a number of chemical agents and for its overall biocompatibility with cells. Additionally, the transparency facilitates in-well, real-time spectroscopy data acquisition.
The gas/nutrient supply channels in this device are laid out in a grid pattern on two layers that are rotated 90˚ to one another. The bottom of each channel layer consists of two primary, or parent, channels on opposing sides of each row of wells for a total of 10 parent channels per layer. The top of each channel layer contains two secondary, or daughter, channels that align as extensions from the parent channels on opposite sides of each well and open directly in to each well, for a total of 50 daughter channels per layer. The parent channels and daughter channels are connected by small through-holes to facilitate equal filling of the wells. Because of the difficulty associated with equal filling, vertical potential energy, and specific geometric restrictions, in through-hole orifice dimension were implemented for optimum delivery of equal volumes to all wells.
Once the layers are fully assembled, a total of four daughter channels feed the different gas and/or nutrient combinations simultaneously to each well. The parent channels are accessed from the exterior of plate via needle or flexible plastic tubing. Because there are 10 parent channels on each of the two layers, a total of 20 entrance points are located around the top perimeter of the incubator. Ten of these entrances are penetrable septa to be used in conjunction with a needle and the other ten are barbed hose fittings that accommodate connection with flexible tubing.
Furthermore, the device incorporates a filtration layer for easy collection of extracellular materials for off-line detailed characterization. The filtration layer is the top most layer of the device when oriented for incubation. It is designed such that the wells are isolated from one another through the use of individual Q-rings, and each well is aligned with a separate membrane of porosity <1 µm. When filtration is desired, the device can be flipped over and placed on an array of Eppendorf tubes to collect the extracellular media. This is the only removable layer of the device so that both the membranes and Q-rings can be replaced when necessary. Extraction of extracellular media, and the subsequent infusion of fresh media (a process time scale of minutes), can be done without opening the device; allowing for separate analyses at several time points throughout the cycle if desired.
In many infectious disease pathologies, a key environmental factor of interest is temperature. It is important to be able to incubate microorganisms at a specific temperature, as well as have the ability to alter the temperature in real-time for various experimental reasons. Some examples include the induction of the expression of a temperature sensitive mutation or to recapitulate the optimal temperature requirements of a particular microorganism. This device is designed for such situations, being heated by a small, fluid-fed hot plate, thus allowing for the easy exploration of time varying temperature profiles as well as multiple temperature conditions. To demonstrate the functionality of our prototype device, and to serve as a proof-of-concept, we chose yeast as a model organism.
3. MATERIALS AND METHODS
Before There is precedent in the literature for the use of yeast to observe growth characteristics in high-throughput microfluidic systems [11,12] The device was used to observe the cellular growth of yeast cells as an indicator of cellular stress. The cells were induced by 2% galactose to express C99, a precursor peptide for the generation of toxic amyloid peptides. Previous studies using this yeast model system demonstrated that the processing of C99 in the absence of a proteasome leads to the generation of multiple amyloidogenic fragments that have since been shown to induce cellular stress (induction of the unfolded protein response-UPR) and death (apoptosis-TUNEL assay). Hence, growth of the cells in galactose induces the expression of C99 [13]. Glucose and raffinose are both used as carbon sources in which yeast grow well, with glucose being the better of the two media. Additionally, raffinose does not result in the inhibition of the galactose inducible promoter. In this proof-ofconcept study, the cell density of yeast grown in Synthetic Complete (SC) media lacking uracil (-ura) and leucine (-leu), supplemented with 2% of all of these carbon sources was measured and cells were subsequently examined for structural changes as indicators of toxicity.
Wild-type yeast cells (PRE1PRE2) expressing C99 were grown on standard media, utilizing common handling procedures as previously described [13]. One colony of PRE1PRE2 cells containing C99 and the unfolded protein response element (UPRE) construct (KCZY) were grown in SC-ura-leu dropout medium supplemented with 2% raffinose until an adequate cell density was reached to complete the study. These cells were then utilized to seed all wells in the incubator array. Log phase cells were required for the device; therefore, a desired starting optical density (OD) of 0.2 ODs/ml stock solutions was prepared accordingly. Absorbence was read at 600 nm for all samples.
Stocks of 10 ml of sugar solutions were pre-mixed by combining the same SC dropout medium described above with the appropriate percentages of glucose, galactose, and raffinose. A total of no more than 2% sugar concentration by volume was used when making each solution. Galactose was varied along one set of vertical channels in the device and glucose was varied along one set of horizontal channels in the device. Concentrations of both began with 0% and increased by 0.5% until 2% was reached. Since a concentration of less than 2% sugar (or carbon source) may lead to cell starvation, raffinose was used as a filler sugar. In this way, it could be assured that each channel was receiving a total of 2% sugar (e.g. 0.5% glucose was mixed with 1.5% raffinose).
To achieve the best results, a total liquid volume of 200 µL (195 µL of stock sugar solution and 5 µL cells) per well was used. Once the cells and the appropriate sugar solutions were infused into the device, the filtration layer was carefully assembled to the bonded layers and a humidifier was connected. The incubator was placed on a rocking platform (VWR Model 200); the air supply line was turned on to approximately 0.014 MPa and the cells were grown for 24 hours. The set up time for the experiment was less than one hour.
4. RESULTS
Normal yeast growth is characterized by the budding of cells in twos and threes, and numerous individual cells. Prior to conducting this experiment with varying sugar percentage combinations, the incubator was tested to see if it was a viable growing environment using only 2% raffinose as the medium. Results of that preliminary test indicated that the device was indeed capable of simply allowing normal and healthy yeast growth (data not shown).
The yeast structure observed in Figure 2, as a result of the varying sugars experiment, exhibited some differences from those seen under traditional growth conditions. Although a majority of the well array showed typical, healthy, dividing cells, some wells contained populations of oversized yeast cells. Generally, the average cell size is altered only when the cell division cycle is inhibited; as protein synthesis progresses, the cells continue to grow and larger average sizes are observed. Definitive conclusions as to why some wells are dominated by populations of oversized cells are beyond the scope of this work. However, the results provide preliminary information on the culture conditions that may have caused these changes. Examples of oversized cells are pointed out in well 13 (0.5% galactose, 1.0% raffinose, 0.5% glucose), well 18 (0.5% galactose, 0.75% raffinose, 0.75% glucose), and well 25 (1.0% galactose, 0% raffinose, 1.0% glucose).
Figure 3 shows results of OD measurements from wells initially populated with the same volume of yeast cells after 24-hours of incubation and 25 different nutrient conditions. The figure uses color to show a population gradient as a function of nutrient conditions. Several wells demonstrated high OD measurements. It is critical to realize that under a single or similar conditions OD measurements are being compared on samples containing similar cell sizes. It is well understood from light scattering theory that larger particles scatter greater amounts
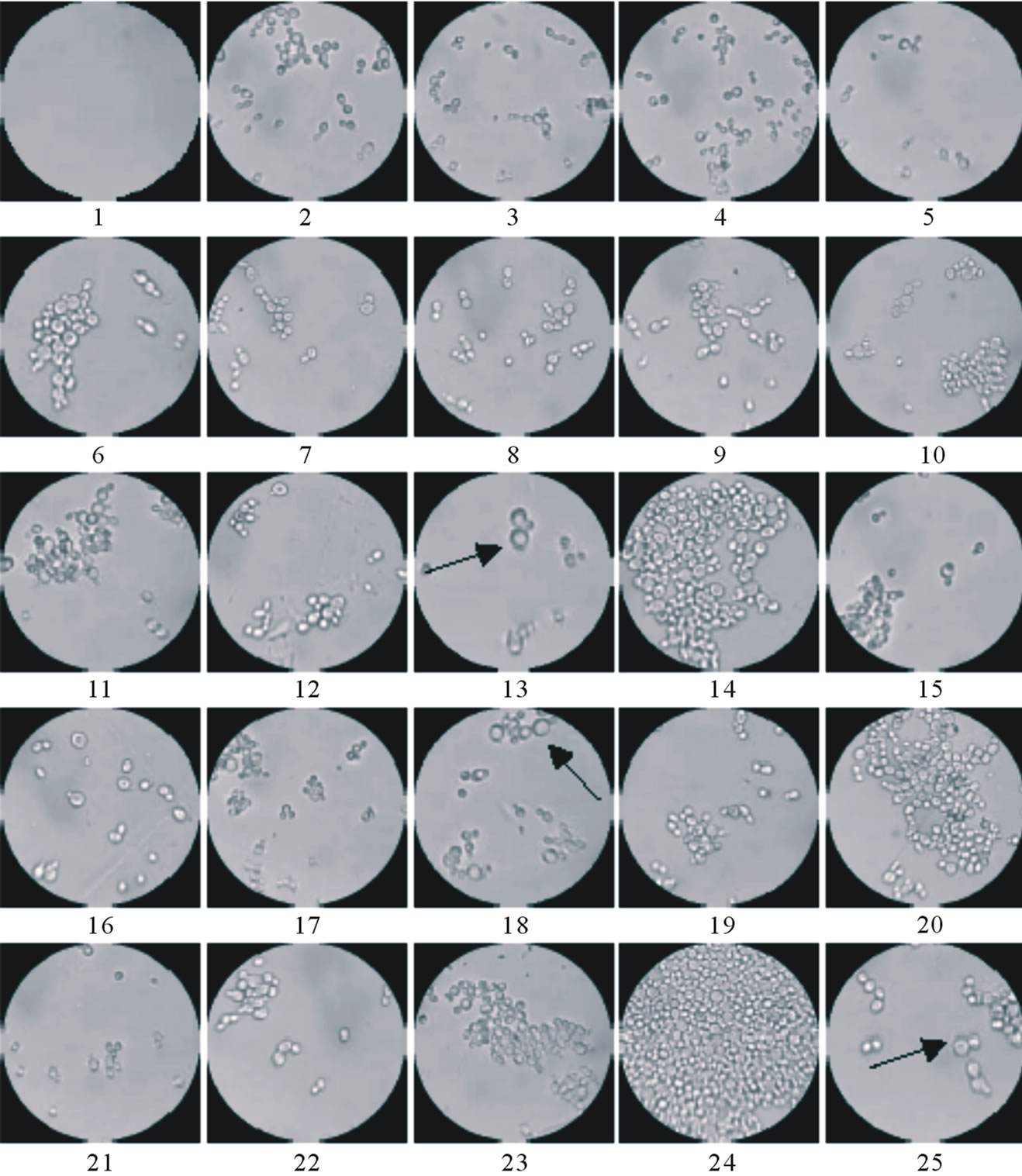
Figure 2. Characteristic growth patterns observed after the 24 hr of incubation in the micro liter incubator array. Arrows indicate examples of larger cells.
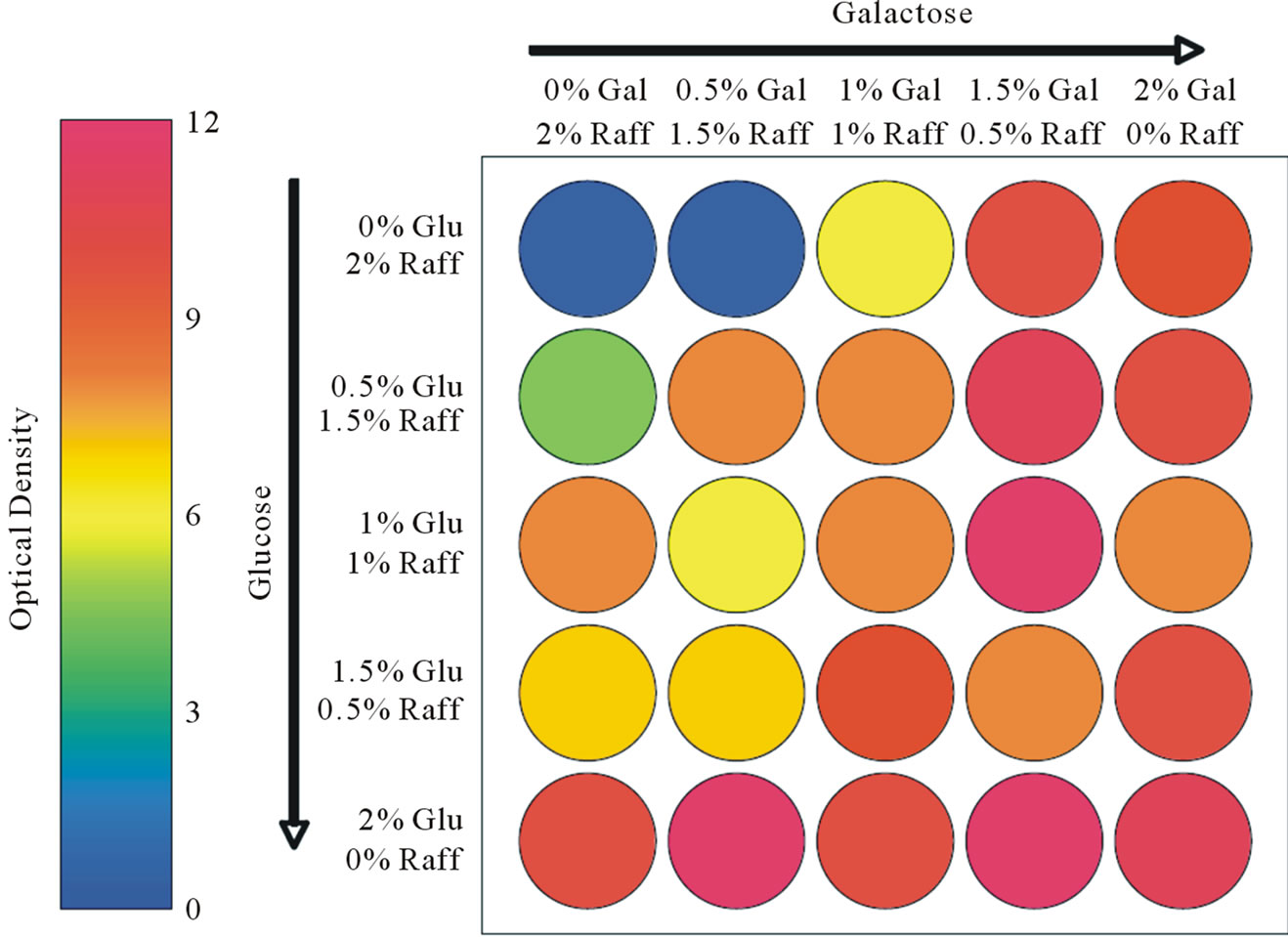
Figure 3. Optical or population density measurements in proof-of-concept yeast study.
of light, thus larger cells will remove more light from the transmission path and inappropriately register a high optical density reading. In this specific case where cellular sizes are known to be changing that conclusion on optimal growth conditions should be drawn from results obtained via multiple techniques. For example, micrographs of cellular density and morphology, Figure 2, show abnormal cellular growth and lower populations for some of the high OD wells (well # 22 is a good example of this particular circumstance). The best cellular number densities achieved in this experiment, based on corroborating evidence from OD measurements and optical images, occurred in wells 14 (0.75% galactose, 0.75% raffinose, 0.5% glucose) and 24 (0.75% galactose, 0.25% raffinose, 1.0% glucose).
5. CONCLUSION
The design of a microliter incubator array for understanding culture condition selectivity is presented. Results show that cells are able to sufficiently grow in this incubator and a few optimal growth conditions for yeast are presented. In summary the effort demonstrates this is an ideal tool to increase the ease and speed of performing assays where many nutrient conditions need to be varied.
6. ACKNOWLEDGEMENTS
This work was supported by a grant from the Keck Futures Initiative through the National Academies of Science and Engineering.
REFERENCES
- Lederberg, J. (2000) Infectious history. Science, 288, 287-293. doi:10.1126/science.288.5464.287
- Hammerschmidt, S., Hacker, J. and Klenk, H.-D. (2005) Threat of infection: Microbes of high pathogenic potential—Strategies for detection, control and eradication. International Journal of Medical Microbiology, 295, 141- 151. doi:10.1016/j.ijmm.2005.03.004
- Fauci, A.S. (2006) Emerging and re-emerging infectious diseases: Influenza as a prototype of the host-pathogen balancing act. Cell, 124, 665-670. doi:10.1016/j.cell.2006.02.010
- Rino, R. (2001) Reverse vaccinology, a genome-based approach to vaccine development. Vaccine, 19, 2688- 2691. doi:10.1016/S0264-410X(00)00554-5
- Raoult, D., et al. (2000) Cultivation of the bacillus of Whipple’s Disease. New England Journal of Medicine, 342, 620-625. doi:10.1056/NEJM200003023420903
- Mora, M., et al. (2003) Reverse vaccinology. Drug Discovery Today, 8, 459-464. doi:10.1016/S1359-6446(03)02689-8
- Mäkelä, P.H. (2000) Vaccines, coming of age after 200 years. FEMS Microbiology Reviews, 24, 9-20.
- Rappuoli, R. and Covacci, A. (2003) Reverse vaccinology and genomics. Science, 302, 602. doi:10.1126/science.1092329
- Baron, M.D., et al. (2005) The Plowright vaccine strain of Rinderpest virus has attenuating mutations in most genes. Journal of General Virology, 86, 1093-1101. doi:10.1099/vir.0.80751-0
- Roop, R.M. (1999) Identification of secondary mutations which enhance and stabilize the attenuation of brucella HTRA Mutants: Improving brucella HTRA-based strains as vaccine. University Medical Center, Louisiana State University, Shreveport.
- Cookson, S., et al. (2005) Monitoring dynamics of single-cell gene expression over multiple cell cycles. Molecular Systems Biology, 1, 1-6. doi:10.1038/msb4100032
- Groisman, A., et al. (2005) A microfluidic chemostat for experiments with bacterial and yeast cells. Nature Methods, 2, 685-689. doi:10.1038/nmeth784
- Sparvero, L.J., et al. (2007) Proteomic analysis of the amyloid precursor protein fragment C99: Expression in yeast. Analytical Biochemistry, 370, 162-170. doi:10.1016/j.ab.2007.07.033
NOTES
*μl Incubator Array for Culture Selectivity.