Psychology
Vol.5 No.7(2014), Article ID:46175,11 pages DOI:10.4236/psych.2014.57082
Roles of BMP6/7 in Actin Dynamics in Amyloid β-Induced Neurotoxicity
Lin Sun1, Yingjie Zhang2, Shifu Xiao1*
1Department of Geriatric Psychiatry, Shanghai Mental Health Center, Shanghai Jiao Tong University School of Medicine, Shanghai, China
2Department of Psychiatry, Beijing Anding Hospital, Beijing Capital Medical University, Beijing, China
Email: *xiaoshifu@msn.com
Copyright © 2014 by authors and Scientific Research Publishing Inc.
This work is licensed under the Creative Commons Attribution International License (CC BY).
http://creativecommons.org/licenses/by/4.0/


Received 19 February 2014; revised 15 March 2014; accepted 12 April 2014
ABSTRACT
Actin dynamics plays an important role in many physiological functions such as long-term potentiation, neurotransmission, regulatory proteins translocation, ATP cycle, etc. When amyloid β (Aβ)- induced neurotoxicity occurs, the imbalance of actin dynamics leads to dystrophy of dendrites, which is characteristic pathology in Alzheimer’s disease (AD). Transient and persistent Aβ neurotoxicity provokes distinct manifestations of actin dynamics and causes opposing effects in AD. It has been shown that bone morphogenetic protein 6/7 (BMP6/7) protects neuronal morphology against Aβ-induced neurotoxicity, and can directly bind to LIM kinase1 (LIMK1), being one part of the upstream regulatory pathway of actin dynamics. This review aims to discuss a potential mechanism of BMPs underlying maintainance of cytoskeletal stabilization in neurite.
Keywords:Amyloid β, BMP, Actin Dynamics, LIMK1, Alzheimer’s Disease

1. Introduction
Neurons contain three primary kinds of cytoskeleton filaments, i.e. microtubules, actin filaments and intermediate filaments (Guharoy et al., 2013). These three subcellular structures, which interact extensively and intimately with cellular membranes, help to maintain the shape and structure of cells (Doherty et al., 2008). Actin filaments, the thinnest filaments of the cytoskeleton filaments, are the basic cytoskeletal proteins in neurites, especially in dendritic spines (Hotulainen et al., 2010). Actin dynamics contributes to many physiological functions such as long-term potentiation, neurotransmission, enzyme translocation and ATP cycle. Abnormality in actin dynamics is a common feature of many neurodegenerative disorders such as Alzheimer’s disease (AD) (Fulga et al., 2007), which is associated with the accumulation of amyloid β (Aβ) in senile plaque (Sepulveda et al., 2010). Aβ deposition is a typical pathological feature of AD, in which destruction of cytoskeleton occurs in dendritic spines and synapses (Mendoza-Naranjo et al., 2007). However, the mechanism underlying the imbalance of actin dynamics in AD is yet unclear. Thus, elucidating the mechanism underlying neuronal cytoskeletal dysfunction can provide a basis for us to further explore potential drug treatments for patients with AD.
Bone morphogenetic proteins (BMPs), a member of the β-transforming growth factor (TGF-β) subfamily, are synthesized as precursor proteins, proteolytically cleaved at the N-terminus and secreted into the extracellular matrix (Sun et al., 2011). BMPs are originally found as a cartilage anabolic factor, which can induce matrix synthesis and promote fracture healing (Doi et al., 2011). Recent studies have shown that BMP6 and BMP7, as the same subgroup of BMPs family, also are responsible for the development of the nervous system and for protection against brain injuries. BMP6 and BMP7 can protect cerebellar granule cells against low-potassium and serum-free medium-induced apoptosis (Yabe et al., 2002). BMP7 can protect human disc cells against apoptosis through inactivation of caspases-3 (Wei et al., 2009), and reduces lesion volume in injured spinal cord (Enzmann et al., 2005). Our previous experiments have shown that BMP7 protects neuron viability and morphology of dendrites against Aβ-induced neurotoxicity (Sun et al., 2011); however, further study is warranted to better understand the detailed mechanisms. Here we reviewed Aβ neurotoxicity-induced pathological changes in the aspect of actin dynamics, the changes of BMP upstream regulatory pathways, and the mechanism underlying BMP6/7 protecting morphology of dendrites from Aβ-induced neurotoxicity.
2. The Composition of Intracellular Actins
Actin is a major cytoskeletal protein in neurons, and it is uniformly distributed throughout axons and dendrites (Zhang et al., 2013). The actin can be either stable or dynamic. Stable actins underlie specialized cellular structures whereas dynamic actins mediate various types of cellular processes (Hoffman et al., 2011). Actin dynamics is involved in many physiological functions such as spine formation, long-term potentiation, vesicle transport, enzyme translocation, and ATP cycle (Minamide et al., 2000).
Actin has two forms found in neurons: monomer form-globular actin (G-actin) and polymer form-filamentous actin (F-actin) (Suurna et al., 2006). F-actin forms a two-stranded asymmetrical helical structure which allows for different binding specificities at each end. One end is termed the “barbed end” or positive end, while the other end is called “pointed end” or minus end (Evans et al., 2007). The balance of F-actin and G-actin is in a constant flux, which can be attributed to actin treadmilling. New G-actin monomer is preferentially added to the barbed end of F-actin polymer and older G-actin monomer is removed from the pointed end (Ando et al., 2013). The rate of F-actin assembly and disassembly will reach a steady state when the concentration of free G-actin monomers decreases to a critical level (Bennett et al., 2011).
3. The Physiological Functions of Actin
3.1. The Roles of Actin in Long-Term Potentiation
Long-term potentiation (LTP) is a long-lasting enhancement in signal transmission that results from stimulating synchronously. As a typical form of synaptic plasticity, LTP, is required for memory acquisition and learning processes (Whitehead et al., 2013). Actin assembly in dendritic spine plays an essential role in hippocampal LTP process (Chen et al., 2007). Actin provides the only structural basis for cytoskeleton in dendritic spines (Halpain et al., 1998). A rapid increase in F-actin content in dendritic spines is observed 40 seconds after LTP and is maintained for 5 weeks (Fukazawa et al., 2003; Lisman et al., 2003). However, the role of actin in dendritic spines in modulating synaptic plasticity is unclear; Nevertheless, there are several possibilities. First, early phase of LTP is mediated by exocytosis and endocytosis of glutamate receptors at postsynaptic sites, and F-actin serves as an anchor for regulatory proteins and a path for glutamate receptor trafficking. Second, late phase of LTP is dependent on protein synthesis to form new synapses (Fukazawa et al., 2003; Lisman et al., 2003). The actin pool is localized in specialized smooth endoplasmic reticulum, which is a spine apparatus to generate more actins, resulting in protrusion of new dendritic filopodia and spine outgrowth (Vlachos et al., 2013).
3.2. The Roles of Actin in Neurotransmission
The abilities of nervous system to orchestrate complex behaviors and deal with complex concepts depend mainly upon neurotransmission between two neurons (Chotibut et al., 2013). Actin dynamics plays an important role in the process of neurotransmission. There is an active zone in the portion of the presynaptic membrane which is the site of synaptic vesicle docking and neurotransmitter releasing (Kreis et al., 2013). When synaptic vesicles are close to presynaptic membrane, actin is recruited to assemble and form an extensive F-actin network, which surrounds vesicle clusters and promotes vesicles movement. Vesicles move directionally along these network connections and reach the presynaptic membrane for neurotransmission in a random and flexible manner (Schuh et al., 2011).
3.3. The Roles of Actin in the ATP Cycle
Actin has a subdomain that binds to ATP and is involved in ATP cycle (Baker et al., 2013). ATP-bound G-actin is available for further assembly to form F-actin. F-actin can hydrolyze its bound ATP to ADP + Pi. The release of Pi causes a conformational change in F-actin that promotes actin depolymerization, leading to ADP-bound G-actin being released from F-actin. ADP released from G-actin can bind ATP through the ADP/ATP exchanger, which usually occurs in neurons at high concentration of ATP (Becker et al., 2006). The brain consumes a disproportionate (20%) of total oxygen, most of which is used to produce ATP for electrical activity (Becker et al., 2006; Ravera et al., 2009). ATP hydrolysis is required for actin dynamics in synaptic regions (Kneussel et al., 2013). Typically, actin dynamics in cultured embryonic neurons consumes approximately 50% of total cellular ATP (Becker et al., 2006). Thus, slowing actin dynamics is an effective way to preserve ATP, which can significantly decrease energy consumption and protects neuronal viability under stress (Barbara et al., 2003).
3.4. The Roles of Actin in Translocation of Regulatory Proteins in Cytoplasm
F-actin plays an important role in the translocation of intracellular regulatory proteins to complete signaling pathway. In response to extracellular stimulus, the F-actin cytoskeleton can be used for transmitting target proteins from integrins to intracellular organelles (Rustom et al., 2004). Typically, in process of sterol depletion, sterol regulatory element binding proteins (SREBPs) as key regulators of cellular sterol and lipid homeostasis, are needed to transported from plasm membrane to endoplasmic reticulum (ER) or ER to Golgi body (Lin et al., 2003). The transmitting forces of active proteins to move organelles are attributed to enhanced F-actin activity, which provides a scaffolding on which regulatory molecules can translocate (Janmey et al., 1999).
4. The Change of Actin Dynamics in Aβ-Induced Pathology
Abnormality in cytoskeletal organization in dendritic spines is a common feature of many neurodegenerative disorders, including Alzheimer’s disease (AD) (Iijima-Ando et al., 2012). The characteristic of senile plaque deposition in AD appears to be linked to the accumulation of amyloid β (Aβ) in the brain and exacerbates the loss of dendritic spines, which causes dramatic cognitive decline in patients (Mendoza-Naranjo et al., 2007). Aβ1-42 is the full length of Aβ and the most toxic form, which exists as monomers, oligomers, fibrils and insoluble aggregates. The soluble monomeric form is non-toxic, and soluble oligomers are more neurotoxic than fibrils and insoluble aggregates (Heredia et al., 2006).
Imbalance in actin dynamics is believed to contribute to the formation of actin rods, which is found in neurodegeneration diseases (Davis et al., 2009). Brain sections of AD display rod-like inclusions, especially in areas surrounding Aβ deposition, supporting the idea that Aβ can induce the turbulence of actin dynamics (Mendoza-Naranjo et al., 2007). The rods are the precursors of Hirano bodies which are found in the neurons of the brains of patients with certain neurodegenerative disorders, such as AD and Creutzfeldt-Jakob disease (Minamide et al., 2000; Davis et al., 2011). Besides Aβ, other rod inducers, such as glutamate, peroxide, and anoxia, initially impair mitochondria and then induce ATP depletion (Maloney et al., 2005). Rods formation has been initially observed in organotypic slice cultures within 10 minutes (min) of ATP depletion, and rods are generated in more than 80% of cultured hippocampal neurons after 20 - 30 minutes of ATP depletion (Davis et al., 2011; Bernstein et al., 2006).
Rods contain actin and actin depolymerizing factor/cofilin (ADF/cofilin) in a 1:1 complex, and they form in tandem arrays within neurites (Davis et al., 2011). As we known, ADF and cofilin are both actin-binding proteins with similar amino acid sequences and functions (Marsick et al., 2012). The binding of ADF/cofilin to G-actin leads to sequestering actin monomers, while the binding of ADF/cofilin to F-actin leads to severing actin polymers (Jang et al., 2005). ADF/cofilin prefers binding the ADP form of F-actin, inducing a twist in the filament that will destabilize interaction between actin monomers and enhances the dissociation of actin at the pointed end of F-actin (Minamide et al., 2000; Suurna et al., 2006). Filament severing by ADF/cofilin is a “Janus-faced” process, since the process can cause either an increase or a decrease in polymerized actin. ADF/cofilin causes fast and extensive severing which leads to actin depolymerization at the slow-growing pointed ends, and eventually, filament loss (Ono et al., 2007). Meanwhile, filament severing creates new barbed ends on actin fragments, which facilitates actin reassembly and elongation if the rate of monomer association overrides monomer dissociation (Marsick et al., 2011). Therefore, ADF/cofilin proteins are critical modulators in actin filament turnover and maintain a balance in cytoskeletal dynamics (Reymann et al., 2011) (Figure 1).
In acute period of ATP depletion, the rapid drop of ATP levels leads to an activation of ADF/cofilin pool through dephosphorylation (Maloney et al., 2005). Reactive ADF/cofilin which binds ADP-bound form of F-actin or G-actin with a higher affinity than the ATP form, can inhibit ADP/ATP exchange, depolymerize actin filaments, and reserve more ATP for important organelles activities (Sinha et al., 2011). Mitochondria are recovered within many of the neurites, proximal to rods, because local ATP levels have been restored to near normal levels (Minamide et al., 2000). The formation of rods during times of transient neurodegenerative stress represents an early neuroprotective mechanism (Maloney et al., 2005). Rapid reassembly and elongation of actin filaments can be seen during cell recovery in the early period of neurodegenerative stimulation. First, less F-actin destruction is observed following 30 min of ATP depletion, then the concentration of F-actin increases at 60 minutes of ATP depletion, leading to promote actin assembly and form a very fine actin network (Suurna et al., 2006). Incubating Aβ with primary cultured cortical neurons for 60 minutes also induces actin polymerization, which can form stress fibers and exert a protective role (Song et al., 2002) (Figure 1).
It has been shown that total ADF/cofilin recovery, being slower than total actin recovery, is consistent with ADF/cofilin being more effectively sequestered in rods and eliminating the blockage of actin turnover and assembly (Bernstein et al., 2006). ADF/cofilin in rods is exposed and regulated by upstream molecules, unless the actin in rods are separated and reassembled in an orderly fashion (Bernstein et al., 2006). With time, prolonged depletion of ATP occurs, the activities of ADF/cofilin upstream regulators begin to decline, with the rods
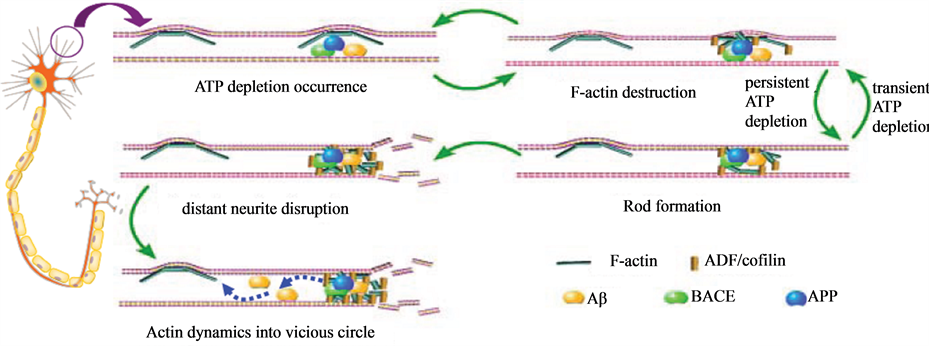
Figure 1. The imbalance of actin dynamics in dendrites induced by Aβ. As an ATP depletion inducer, Aβ can lead to an activation of ADF/cofilin pool through dephosphorylation by phosphatase SSH. Reactive ADF/cofilin can depolymerize F-actin, inhibit ADP/ATP exchange, and reserve more ATP for important organelles activities. The inhibitory effect of reactive ADF/cofilin results in formation of rods which contain actin and ADF/cofilin in a 1:1 complex. When Aβ is removed and just induces transient ATP depletion, ATP levels are restored to near normal levels and lead to inactivation of ADF/cofilin through phosphorylation by LIMK1. F-actin concentration increases secondary to phospho-ADF/cofilin, tending to promote actin assembly and form a stable actin dynamics. When prolonged depletion of ATP occurs, the rods become large enough to completely occlude and disrupt distant neurite. The accumulation of Aβ protein precursor (APP) and Aβ converting enzyme (BACE) assemble at rods, resulting in enhanced production of Aβ. Therefore, the actin dynamics enter a deteriorative cycle. An increase in rods can block and disrupt the neurite, leading to more Aβ peptide generation and the induction of rod formation.
becoming large enough to completely occlude the neurites (Koyama et al., 1996). This phenomenon can disrupt the distal cytoskeleton and inhibit vesicular transport. The blockage in transport leads to the swelling of distant neurites and synapse loss, which are typical pathological features of AD (Davis et al., 2011). The accumulation of amyloid precursor protein (APP) and β-site APP cleaving enzyme (BACE) assemble at rods, resulting in enhanced production of Aβ peptides (Maloney et al., 2005). Therefore, with extensive neurodegenerative stress, the actin dynamics enter a deteriorative cycle, whereby an increase in rods can block and disrupt the neurite cytoskeleton, leading to more Aβ peptide generation and the induction of rods formation (Figure 1).
5. The Possible Mechanism of BMP6/7 Protecting Actin Cytoskeleton against Aβ-Induced Neurodegenerative Stress
BMPs, members of β-transforming growth factor (TGF-β) subfamily, have been shown to act as a cartilage anabolic factor due to their abilities to induce matrix synthesis and promote cartilage repair (Odabas et al., 2013). Recently, it has been shown that BMP6/7 can affect neurogenesis, axonal pathfinding, and dendritic branching during development of nervous system (Garred et al., 2011; Di Liddo et al., 2010). On the other hand, BMP6/7 can protect mature neurons against Aβ-induced neurodegenerative stress (Sun et al., 2011).
Bone morphogenetic protein receptors (BMPRs) are transmembrane serine/threonine protein kinases that belong to the receptors of TGF-β family and consist of BMP receptor type I (BMPRI) and BMP receptor type II (BMPRII). BMP ligand binding leads to phosphorylation and activation of BMPRII (Miyagi et al., 2011). BMPRII, in turn, phosphorylates one of receptor regulating small mothers against decapentaplegic (R-Smad) proteins including Smad1, Smad5 and Smad8, then combined with common mediator Smad (co-Smad)-Smad4 which specially transduces extracellular signals from TGF-β to the nucleus and elicits genes transcription, including actin genes (Sartori et al., 2013). Whereas, inhibitory Smad (I-Smad) including Smad6 and Smad7 could negatively regulate BMP-Smad signaling pathway (Sartori et al., 2013). Endoplasmic reticulum produces actin monomers that supply the material basis for actin dynamics (Ramabhadran et al., 2013).
As shown in Figure 2, BMPRII has a large cytoplasmic tail of approximately 600 amino acids C-terminal to the kinase domain (Breen et al., 2013). As one of ADF/cofilin upstream molecules, LIM kinase 1 (LIMK1) interacts specifically via its LIM domains to the tail of BMPRII and this interaction results in negative regulation of LIMKI pathway (Foletta et al., 2003; Podkowa et al., 2013). The reason is that LIMKI binding with BMPRII can reduce C-terminal availability in the cytoplasm and limit their accesses to the upstream activators (Foletta et al., 2003). However, this inhibitory effect appears to be alleviated by BMPs ligand stimulation. Because upon engagement of BMPs, followed by formation of BMPs-BMPR complex, LIMKI is dissociated from the C-terminal of BMPRII, bringing about more free interaction with the upstream activators to phosphorylate LIMKI pathway (Miyagi et al., 2011). ADF/cofilin is the sole substrate of LIMK1 and is inactivated by LIMK1 through phosphorylation. Phosphorylated ADF/cofilin is unable to bind F-actin, consequently, leading to actin polymerization (Zhao et al., 2008).
BMPs-BMPR complex has the ability to maintain synaptic actin stability and dendrite outgrowth, via both slow Smad-dependent and fast LIMK1-dependent pathway (Eaton et al., 2005). It has been reported that the protective effects of BMP7 on Aβ-induced neurotoxicity involve decreasing cell morphology damage and maintaining dendrites outgrowth (Sun et al., 2011), which are mainly attributed to the stability of intracellular actin filaments. As shown in Figure 2, the possible mechanism underlying BMPs reducing Aβ-induced cytoskeleton impairment in dendrites may involve release of BMPRII C-terminal and reactivation of LIMK1, leading to phosphorylation of ADF/cofilin and reassembly of F-actin. BMPs can mediate early protective signalling of actin filaments via indirectly regulating the activity of the LIMK1 pathway.
Similar to LIMK1, LIM kinase 2 (LIMK2) is also an actin binding serine/threonine kinase and serve as a main negative regulator, which can phosphorylate and inactivate its sole substrate-ADF/cofilin (Wang et al., 2013). There are two upstream kinases including p21 activated kinase (PAK) and Rho associated coiled-coil kinase (ROCK) to activate LIMK1 and 2 by phosphorylation in the kinase domain, respectively (Aleksic et al., 2009). Rho GTPases, which belong to Ras superfamily of small GTPases, translate transmembrane signaling to regulate actin cytoskeleton (Rex et al., 2009). RhoA and CDC42/Rac, as members of Rho GTPases, are the upstream activators of ROCK and PAK, respectively, and both can phosphorylate LIMK pathway (Hocking et al., 2009). By switching between a GDP-bound inactive form and a GTP-bound active form, Rho GTPases can regulate the cytoskeleton construction guided by many actin dynamic cues (Lin et al., 2003). Guanine nucleotide exchange

Figure 2. The possible mechanisms underlying maintaining the stabilization of actin dynamics in dendrites by BMPs. BMP ligand transduces downstream signaling pathway including SMAD and non-SMAD through bingding to BMP receptor. BMPRII phosphorylates one of receptor regulating small mothers against decapentaplegic (R-Smad) proteins including Smad1, Smad5 and Smad8, then combines with common mediator Smad (co-Smad)-Smad4 which specially transduces extracellular signals from TGF-β to the nucleus and elicits genes transcription, including actin genes. Whereas, inhibitory Smad (I-Smad) including Smad6 and Smad7 can negatively regulate BMP-Smad signaling pathway. Futhermore, BMPRII through its large cytoplasmic tail can bind and inactivate LIMK1. When BMP binds BMPR to form BMP-BMPR complex, LIMK1 is dissociated from the cytoplasmic tail of BMPRII, leading to LIMK1 activated by the upstream molecules such as PAK and CDC42/Rac. LIMK1 can phosphorylate and inactivate ADF/cofilin. Phosphorylated ADF/cofilin is unable to sever F-actin, which finally leads to actin polymerization, actin organization and then F-actin stabilization. We speculate that the mechanism of BMPs maintaining actin dynamics in dendrites is LIMK1-dependent. Compared to LIMK1, SSH plays an opposite role which can dephosphorylate and activate ADF/cofilin, at the same time, SSH can also dephosphorylate and inactivate LIMK1. As an auxiliary regulatory molecule, 14-3-3ζ can cooperate with LIMK1 activities. Additionally, LIMK2 and its upstream activators including ROCK and RhoA can be an inhibitory pathway for ADF/cofilin as well. However, the precise role of BMPs in actin dynamics is unclear. Besides LIMK1 pathway, BMPs probably can activate Smad-dependent pathway via BMPR to elicit actin gene transcription. Endoplasmic reticulums produce actin monomers to supply the material basis for actin polymerization.
factors (GEFs) activate the switch by catalyzing the exchange of GDP for GTP, whereas GTPase activating proteins (GAPs) inactivate the switch and increase intrinsic GTPase activity (Hall et al., 2010). These upstream cascades of LIMK pathway converge on the ADF/cofilin protein that directly regulates the organization of actin cytoskeleton. The constitutively active forms of RhoA-ROCK-LIMK2 and CDC42/Rac-PAK-LIMK1 induce phosphorylated and inactive form of ADF/cofilin, which promotes actin polymerization and stress fiber formation (Ng et al., 2008).
In contrast, re-activation of ADF/cofilin protein occurs by dephosphorylation, which has been reported to be mediated by Slingshot (SSH) phosphatase (Soosairajah et al., 2005). Apart from dephosphorylating ADF/cofilin, SSH also serves as a phosphatase toward LIMK1 to inhibit its activity. The effect of SSH on LIMK1 pathway appears to decrease the levels of both ADF/cofilin and F-actin, consequently, inhibiting LIMK1-induced actin reorganization (Soosairajah et al., 2005). At the same time, the phosphatase activity of SSH toward LIMK1- ADF/cofilin is suppressed by phosphorylation of PAK (Ishibashi et al., 2008). There is another regulatory molecule, 14-3-3ζ scaffolding proteins, which can bind to phospho-ADF/cofilin and cooperate with LIMK. The role of 14-3-3ζ scaffolding proteins is to protect phospho-ADF/cofilin from dephosphorylation, resulting in increased levels of F-actin (Foletta et al., 2003; Soosairajah et al., 2005). Furthermore, 14-3-3ζ scaffolding proteins have the property of phospho-SSH binding and reduce its effects on F-actin, serving as a negative regulator of SSH activity (Soosairajah et al., 2005). Therefore, multiple and complex regulatory molecules are required to maintain the balance of intracellular actin dynamics (Figure 2).
ADF/cofilin activity depends on the net balance of kinase LIMK1 and phosphatase SSH (Kousaka et al., 2008). There are very complex molecule signals involving the regulation of LIMK1 and SSH. Previous experiments have demonstrated that CDC42/Rac considerably overlaps with early cytoskeleton abnormalities which were observed in AD brain sections, the defects in P21-activated kinase (PAK) activity can lead to actin pathology during development of AD, and high levels of LIMK1 coexist with delayed F-actin cytoskeleton destruction in transient ATP depletion (Mendoza-Naranjo et al., 2007; Suurna et al., 2006; Heredia et al., 2006). Thus, it can be assumed that reactive CDC42/Rac-PAK-LIMK1 pathway is promoted to counteract over hyper-dephosphorylation of ADF/cofilin and rescue cytoskeleton damage. It has been reported that CDC42/Rac can synergize the interaction of LIMK1 with BMPRII and active CDC42/Rac can enhance the activity of LIMK1 (Matsuura et al., 2007; Liu et al., 2009; Ng et al., 2004), however, the exact correlation between BMPRII and CDC42/Rac is unknown. Previous experiments have demonstrated that there are inhibitory effects on PAK-SSH pathway and SSH-LIMK1 pathway respectively (Kousaka et al., 2008; Ng et al., 2004). Alternatively, PAK can downregulate the activities of SSH, and SSH can downregulate the activities of LIMK1 (Maloney et al., 2005).
6. Conclusion
Taken together, we can speculate that CDC42/Rac, and its downstream effector PAK, contribute to activating LIMK1 and inhibiting SSH. As the upstream activators of CDC42/Rac, GEFs, GAPs, or other proteins, may increase CDC42/Rac activities to modulate BMPs protective signaling; however, future studies are necessary to verify this assumption. The future studies will focus on how to avoid or decrease the impairment of dendritic actin caused by Aβ and explore the changes of different signaling pathways including BMP ligand, BMP receptor, downstream signaling of the BMP-LIMK1 pathway, protein associated with LIMK1-SSH, and upstream signalings of the BMP-Rho-GTPase pathway. Moreover, it is essential to search the targets which can alleviate or inhibit the impairment of dendritic actin induced by Aβ. The study of roles of BMP6/7 in dendritic actin dynamics could provide us with a better understanding of Aβ-induced degenerative diseases, which may be beneficial for the clinical treatment of AD.
References
- Aleksic, M., Walcher, D., Giehl, K., Bach, H., Grüb, M., Durst, R., Hombach, V., & Marx, N. (2009). Signalling Processes Involved in C-Peptide-Induced Chemotaxis of CD4-Positive Lymphocytes. Cellular and Molecular Life Sciences, 66, 1974-1984. http://dx.doi.org/10.1007/s00018-009-9057-y
- Ando, K., Fukuhara, S., Moriya, T., Obara, Y., Nakahata, N., & Mochizuki, N. (2013) Rap1 Potentiates Endothelial Cell Junctions by Spatially Controlling Myosin II Activity and Actin Organization. The Journal of Cell Biology, 202, 901-616. http://dx.doi.org/10.1083/jcb.201301115
- Baker, J. L., & Voth, G. A. (2013) Effects of ATP and Actin-Filament Binding on the Dynamics of the Myosin II S1 Domain. Biophysical Journal, 105, 1624-1634. http://dx.doi.org/10.1016/j.bpj.2013.08.023
- Barbara, J. G., Auclair, N., Roisin, M. P., Otani, S., Valjent, E., Caboche, J., Soubrie, P., & Crepel, F. (2003). Direct and Indirect Interactions between Cannabinoid CB1 Receptor and Group II Metabotropic Glutamate Receptor Signalling in Layer V Pyramidal Neurons from the Rat Prefrontal Cortex. European Journal of Neuroscience, 17, 981-990. http://dx.doi.org/10.1046/j.1460-9568.2003.02533.x
- Becker, E. W. (2006). The Roles of ATP in the Dynamics of the Actin Filaments of the Cytoskeleton. The Journal of Biological Chemistry, 387, 401-406.
- Bennett, M. R., Farnell, L., & Gibson, W. G. (2011). A Model of NMDA Receptor Control of F-Actin Treadmilling in Synaptic Spines and Their Growth. Bulletin of Mathematical Biology, 73, 2109-2131. http://dx.doi.org/10.1007/s11538-010-9614-4
- Bernstein, B. W., Chen, H., Boyle, J. A., & Bamburg, J. R. (2006). Formation of Actin-ADF/Cofilin Rods Transiently Retards Decline of Mitochondrial Potential and ATP in Stressed Neurons. Cell Physiology—American Journal of Physiology, 291, C828-C839. http://dx.doi.org/10.1152/ajpcell.00066.2006
- Breen, M. J., Moran, D. M., Liu, W., Huang, X., Vary, C. P., & Bergan, R. C. (2013). Endoglin-Mediated Suppression of Prostate Cancer Invasion Is Regulated by Activin and Bone morphogenetic Protein Type II Receptors. PLoS One, 8, e72407. http://dx.doi.org/10.1371/journal.pone.0072407
- Enzmann, G. U., Benton, R. L., Woock, J. P., Howard, R. M., Tsoulfas, P., & Whittemore, S. R. (2005). Consequences of Noggin Expression by Neural Stem, Glia, and Neuronal Precursor Cells Engrafted into the Injured Spinal Cord. Experimental Neurology, 195, 293-304. http://dx.doi.org/10.1016/j.expneurol.2005.04.021
- Chen, L. Y., Rex, C. S., Casale, M. S., Gall, C. M., & Lynch, G. (2007). Changes in Synaptic Morphology Accompany Actin Signaling during LTP. Journal of Neuroscience, 27, 5363-5372. http://dx.doi.org/10.1523/JNEUROSCI.0164-07.2007
- Chotibut, T., Davis, R. W., Arnold, J. C., Frenchek, Z., Gurwara, S., Bondada, V., Geddes, J. W., & Salvatore, M. F. (2013). Ceftriaxone Increases Glutamate Uptake and Reduces Striatal Tyrosine Hydroxylase Loss in 6-OHDA Parkinson’s Model. Molecular Neurobiology, 12. [Epub ahead of print].
- Davis, R. C., Maloney, M. T., Minamide, L. S., Flynn, K. C., Stonebraker, M. A., & Bamburg, J. R. (2009). Mapping Cofilin-Actin Rods in Stressed Hippocampal Slices and the Role of cdc42 in Amyloid-Beta-Induced Rods. Journal of Alzheimers Disease, 18, 35-50.
- Davis, R. C., Marsden, I. T., Maloney, M. T., Minamide, L. S., Podlisny, M., Selkoe, D. J., & Bamburg, J. R. (2011). Amyloid Beta Dimers/Trimers Potently Induce Cofilin-Actin Rods That Are Inhibited by Maintaining Cofilin-Phosphorylation. Molecular Neurodegeneration, 6, 10. http://dx.doi.org/10.1186/1750-1326-6-10
- Di Liddo, R., Grandi, C., Venturini, M., Dalzoppo, D., Negro, A., Conconi, M. T., & Parnigotto, P. P. (2010). Recombinant Human TAT-OP1 to Enhance NGF Neurogenic Potential: Preliminary Studies on PC12 Cells. Protein Engineering Design and Selection, 23, 889-897. http://dx.doi.org/10.1093/protein/gzq067
- Doherty, G. J., & McMahon, H. T. (2008). Mediation, Modulation and Consequences of Membrane-Cytoskeleton Interactions. Annual Review of Biophysics, 37, 65-95. http://dx.doi.org/10.1146/annurev.biophys.37.032807.125912
- Doi, Y., Miyazaki, M., Yoshiiwa, T., Hara, K., Kataoka, M., & Tsumura, H. (2011). Manipulation of the Anabolic and Catabolic Responses with BMP-2 and Zoledronic Acid in a Rat Femoral Fracture Model. Bone, 49, 777-782. http://dx.doi.org/10.1016/j.bone.2011.07.005
- Eaton, B. A. & Davis, G. W. (2005). Lim Kinase1 Controls Synaptic Stability Downstream of the Type II BMP Receptor. Neuron, 47, 695-708. http://dx.doi.org/10.1016/j.neuron.2005.08.010
- Evans, J. H., & Falke, J. J. (2007). Ca2+ Influx Is an Essential Component of the Positive-Feedback Loop That Maintains Leading-Edge Structure and Activity in Macrophages. Proceedings of the National Academy of Sciences of the United States of America, 104, 16176-16181. http://dx.doi.org/10.1073/pnas.0707719104
- Foletta, V. C., Lim, M. A., Soosairajah, J., Kelly, A. P., Stanley, E. G., Shannon, M., He, W., Das, S., Massague, J., & Bernard, O. (2003). Direct Signaling by the BMP Type II Receptor via the Cytoskeletal Regulator LIMK1. Journal of Cell Biology, 162, 1089-1098. http://dx.doi.org/10.1083/jcb.200212060
- Fukazawa, Y., Saitoh, Y., Ozawa, F., Ohta, Y., Mizuno, K., & Inokuchi, K. (2003). Hippocampal LTP Is Accompanied by Enhanced F-Actin Content within the Dendritic Spine That Is Essential for Late LTP Maintenance In Vivo. Neuron, 38, 447-460. http://dx.doi.org/10.1016/S0896-6273(03)00206-X
- Fulga, T. A., Elson-Schwab, I., Khurana, V., Steinhilb, M. L., Spires, T. L., Hyman, B. T., & Feany, M. B. (2007). Abnormal Bundling and Accumulation of F-Actin Mediates Tau-Induced Neuronal Degeneration in Vivo. Nature Cell Biology, 9, 139-148. http://dx.doi.org/10.1038/ncb1528
- Garred, M. M., Wang, M. M., Guo, X., Harrington, C. A., & Lein, P. J. (2011). Transcriptional Responses of Cultured Rat Sympathetic Neurons during BMP-7-Induced Dendritic Growth. PLoS ONE, 6, e21754. http://dx.doi.org/10.1371/journal.pone.0021754
- Guharoy, M., Szabo, B., Martos, S. C., Kosol, S., & Tompa, P. (2013). Intrinsic Structural Disorder in Cytoskeletal Proteins. Cytoskeleton (Hoboken), 70, 550-571. http://dx.doi.org/10.1002/cm.21118
- Hall, A., & Lalli, G. (2010). Rho and Ras GTPases in Axon Growth, Guidance, and Branching. Cold Spring Harbor Perspectives in Biology, 2, a001818. http://dx.doi.org/10.1101/cshperspect.a001818
- Halpain, S., Hipolito, A., & Saffer, L. (1998). Regulation of F-Actin Stability in Dendritic Spines by Glutamate Receptors and Calcineurin. Journal of Neuroscience, 18, 9835-9844.
- Heredia, L., Helguera, P., de Olmos, S., Kedikian, G., Solá Vigo, F., LaFerla, F., Staufenbiel, M., De Olmos, J., Busciglio, J., Cáceres, A., & Lorenzo, A. (2006). Phosphorylation of Actin-Depolymerizing Factor/Cofilin by LIM-Kinase Mediates Amyloid Beta-Induced Degeneration: A Potential Mechanism of Neuronal Dystrophy in Alzheimer’s Disease. Journal of Neuroscience, 26, 6533-6542. http://dx.doi.org/10.1523/JNEUROSCI.5567-05.2006
- Hocking, J. C., Hehr, C. L., Bertolesi, G., Funakoshi, H., Nakamura, T., & McFarlane, S. (2009). LIMK1 Acts Downstream of BMP Signaling in Developing Retinal Ganglion Cell Axons but Not Dendrites. Developmental Biology, 330, 273-285. http://dx.doi.org/10.1016/j.ydbio.2009.03.027
- Hoffman, B. D., Grashoff, C., & Schwartz, M. A. (2011). Dynamic Molecular Processes Mediate Cellular Mechanotransduction. Nature, 475, 316-323. http://dx.doi.org/10.1038/nature10316
- Hotulainen, P., & Hoogenraad, C. C. (2010). Actin in Dendritic Spines: Connecting Dynamics to Function. Journal of Cell Biology, 189, 619-629. http://dx.doi.org/10.1038/nature10316
- Iijima-Ando, K., Sekiya, M., Maruko-Otake, A., Ohtake, Y., Suzuki, E., Lu, B., & Iijima, K. M. (2012). Loss of Axonal Mitochondria Promotes Tau-Mediated Neurodegeneration and Alzheimer’s Disease-Related Tau Phosphorylation via PAR-1. PLoS Genetics, 8, e1002918. http://dx.doi.org/10.1371/journal.pgen.1002918
- Ishibashi, F. (2008). High Glucose Increases Phosphocofilin via Phosphorylation of LIM Kinase Due to Rho/Rho Kinase Activation in Cultured Pig Proximal Tubular Epithelial Cells. Diabetes Research and Clinical Practice, 80, 24-33. http://dx.doi.org/10.1016/j.diabres.2007.11.004
- Jang, D. H., Han, J. H., Lee, S. H., Lee, Y. S., Park, H., Lee, S. H., Kim, H., & Kaang, B. K. (2005). Cofilin Expression Induces Cofilin-Actin Rod Formation and Disrupts Synaptic Structure and Function in Aplysia Synapses. Proceedings of the National Academy of Sciences of the United States of America, 102, 16072-16077.
- Janmey, P. A., Kas, J., Shah, J. V., Allen, P. G., & Tang, J. X. (1999). Cytoskeletal Networks and Filament Bundles: Regulation by Proteins and Polycations. The Biological Bulletin, 194, 334-335. http://dx.doi.org/10.2307/1543105
- Kneussel, M., & Wagner, W. (2013). Myosin Motors at Neuronal Synapses: Drivers of Membrane Transport and Actin Dynamics. Nature Reviews Neuroscience, 14, 233-247. http://dx.doi.org/10.1038/nrn3445
- Kousaka, K., Kiyonari, H., Oshima, N., Nagafuchi, A., Shima, Y., Chisaka, O., & Uemura, T. (2008). Slingshot-3 Dephosphorylates ADF/Cofilin but Is Dispensable for Mouse Development. Genesis, 46, 246-255. http://dx.doi.org/10.1002/dvg.20389
- Koyama, T., Boston, D., Ikenouchi, H., & Barry, W. H. (1996). Survival of Metabolically Inhibited Ventricular Myocytes Is Enhanced by Inhibition of Rigor and SR Ca2+ Cycling. American Journal of Physiology, 271, H643-650.
- Kreis, P., Hendricusdottir, R., Kay, L., Papageorgiou, I. E., van Diepen, M., Mack, T., Ryves, J., Harwood, A., Leslie, N. R., Kann, O., Parsons, M., & Eickholt, B. J. (2013). Phosphorylation of the Actin Binding Protein Drebrin at S647 Is Regulated by Neuronal Activity and PTEN. PLoS ONE, 8, e71957. http://dx.doi.org/10.1371/journal.pone.0071957
- Lin, T., Zeng, L., Liu, Y., DeFea, K., Schwartz, M. A., Chien, S., & Shyy, J. Y. (2003). Rho-ROCK-LIMK-Cofilin Pathway Regulates Shear Stress Activation of Sterol Regulatory Element Binding Proteins. Circulation Research, 92, 1296-1304. http://dx.doi.org/10.1161/01.RES.0000078780.65824.8B
- Lisman, J. (2003). Actin’s Actions in LTP-Induced Synapse Growth. Neuron, 38, 361-362. http://dx.doi.org/10.1016/S0896-6273(03)00257-5
- Liu, Y., Ren, W., Warburton, R., Toksoz, D., & Fanburg, B. L. (2009). Serotonin Induces Rho/ROCK-Dependent Activation of Smads1/5/8 in Pulmonary Artery Smooth Muscle Cells. The FASEB Journal, 23, 2299-2306. http://dx.doi.org/10.1096/fj.08-127910
- Maloney, M. T., Minamide, L. S., Kinley, A. W., Boyle, J. A., & Bamburg, J. R. (2005). Beta-Secretase-Cleaved Amyloid Precursor Protein Accumulates at Actin Inclusions Induced in Neurons by Stress or Amyloid Beta: A Feedforward Mechanism for Alzheimer’s Disease. The Journal of Neuroscience, 25, 11313-11321. http://dx.doi.org/10.1523/JNEUROSCI.3711-05.2005
- Marsick, B. M., & Letourneau, P. C. (2011). Labeling F-Actin Barbed Ends with Rhodamine-Actin in Permeabilized Neuronal Growth Cones. Journal of Visualized Experiments, 17, Article ID 2409.
- Marsick, B. M., Miguel-Ruiz, J. E. S., & Letourneau, P. C. (2012). Activation of Ezrin/Radixin/ Moesin Mediates Attractive Growth Cone Guidance through Regulation of Growth Cone Actin and Adhesion Receptors. Journal of Neuroscience, 32, 282-296. http://dx.doi.org/10.1523/JNEUROSCI.4794-11.2012
- Matsuura, I., Endo, M., Hata, K., Kubo, T., Yamaguchi, A., Saeki, N., & Yamashita, T. (2007). BMP Inhibits Neurite Growth by a Mechanism Dependent on LIM-Kinase. Biochemical Biophysical Research Communications, 360, 868-873. http://dx.doi.org/10.1016/j.bbrc.2007.06.157
- Mendoza-Naranjo, A., Gonzalez-Billault, C., & Maccioni, R. B. (2007) Abeta1-42 Stimulates Actin Polymerization in Hippocampal Neurons through Rac1 and Cdc42 Rho GTPases. Journal of Cell Science, 120, 279-288. http://dx.doi.org/10.1242/jcs.03323
- Minamide, L. S., Striegl, A. M., Boyle, J. A., Meberg, P. J., & Bamburg, J. R. (2000). Neurodegenerative Stimuli Induce Persistent ADF/Cofilin-Actin Rods That Disrupt Distal Neurite Function. Nature Cell Biology, 2, 628-636. http://dx.doi.org/10.1038/35023579
- Miyagi, M., Mikawa, S., Hasegawa, T., Kobayashi, S., Matsuyama, Y., & Sato, K. (2011). Bone Morphogenetic Protein Receptor Expressions in the Adult Rat Brain. Neuroscience, 176, 93-109. http://dx.doi.org/10.1016/j.neuroscience.2010.12.027
- Ng, J. (2008). TGFβ Signals Regulate Axonal Development through Distinct Smad-Independent Mechanisms. Development, 135, 4025-4035. http://dx.doi.org/10.1242/dev.028209
- Ng, J., & Luo, L. (2004). Rho GTPases Regulate Axon Growth through Convergent and Divergent Signaling Pathways. Neuron, 44, 779-793. http://dx.doi.org/10.1016/j.neuron.2004.11.014
- Odabas, S., Feichtinger, G. A., Korkusuz, P., Inci, I., Bilgic, E., Yar, A. S., Cavusoglu, T., Menevse, S., Vargel, I., & Piskin, E. (2013). Auricular Cartilage Repair Using Cryogel Scaffolds Loaded with BMP-7-Expressing Primary Chondrocytes. Journal of Tissue Engineering and Regenerative Medicine, 7, 831-840.
- Ono, S. (2007). Mechanism of Depolymerization and Severing of Actin Filaments and Its Significance in Cytoskeletal Dynamics. International Review of Cytology, 258, 1-82. http://dx.doi.org/10.1016/S0074-7696(07)58001-0
- Podkowa, M., Christova, T., Zhao, X., Jian, Y., & Attisano, L. (2013). P21-Activated Kinase (PAK) Is Required for Bone Morphogenetic Protein (BMP)-Induced Dendritogenesis in Cortical Neurons. Molecular Cell Neuroscience, 57, 83-92. http://dx.doi.org/10.1016/j.mcn.2013.10.005
- Ramabhadran, V., Hatch, A. L., & Higgs, H. N. (2013). Actin Monomers Activate Inverted Formin 2 by Competing with Its Autoinhibitory Interaction. The Journal of Biology Chemistry, 288, 26847-26855. http://dx.doi.org/10.1074/jbc.M113.472415
- Ravera, S., Panfoli, I., Calzia, D., Aluigi, M. G., Bianchini, P., Diaspro, A., Mancardi, G., & Morelli, A. (2009). Evidence for Aerobic ATP Synthesis in Isolated Myelin Vesicles. The International Journal of Biochemistry & Cell Biology, 41, 1581-1591. http://dx.doi.org/10.1016/j.biocel.2009.01.009
- Rex, C. S., Chen, L. Y., Sharma, A., Liu, J., Babayan, A. H., Gall, C. M., & Lynch, G. (2009). Different Rho GTPase-Dependent Signaling Pathways Initiate Sequential Steps in the Consolidation of Long-Term Potentiation. Journal of Cell Biology, 186, 85-97. http://dx.doi.org/10.1083/jcb.200901084
- Reymann, A. C., Suarez, C., Guérin, C., Martiel, J. L., Staiger, C. J., Blanchoin, L., & Boujemaa-Paterski, R. (2011). Turnover of Branched Actin Filament Networks by Stochastic Fragmentation with ADF/ Cofilin. Molecular Biology Cell, 22, 2541-2550. http://dx.doi.org/10.1091/mbc.E11-01-0052
- Rustom, A., Saffrich, R., Markovic, I., Walther, P., & Gerdes, H. H. (2004). Nanotubular Highways for Intercellular Organelle Transport. Science, 303, 1007-1010. http://dx.doi.org/10.1126/science.1093133
- Sartori, R., Schirwis, E., Blaauw, B., Bortolanza, S., Zhao, J., Enzo, E., Stantzou, A., Mouisel, E., Toniolo, L., Ferry, A., Stricker, S., Goldberg, A.L., Dupont, S., Piccolo, S., Amthor, H., & Sandri, M. (2013). BMP Signaling Controls Muscle Mass. Nature Genetics, 45, 1309-1318. http://dx.doi.org/10.1038/ng.2772
- Schuh, M. (2011). An Actin-Dependent Mechanism for Long-Range Vesicle Transport. Nature Cell Biology, 13, 1431-1436. http://dx.doi.org/10.1038/ncb2353
- Sepulveda, F. J., Parodi, J., Peoples, R. W., Opazo, C., & Aguayo, L. G. (2010). Synaptotoxicity of Alzheimer Beta Amyloid Can Be Explained by Its Membrane Perforating Property. PLoS ONE, 5, e11820. http://dx.doi.org/10.1371/journal.pone.0011820
- Sinha, B., Köster, D., Ruez, R., Gonnord, P., Bastiani, M., Abankwa, D., Stan, R. V., Butler-Browne, G., Vedie, B., Johannes, L., Morone, N., Parton, R. G., Raposo, G., Sens, P., Lamaze, C., & Nassoy, P. (2011). Cells Respond to Mechanical Stress by Rapid Disassembly of Caveolae. Cell, 144, 402-413. http://dx.doi.org/10.1016/j.cell.2010.12.031
- Song, C., Perides, G., Wang, D., & Liu, Y. F. (2002). Beta-Amyloid Peptide Induces Formation of a Ctin Stress Fibers through p38 Mitogen-Activated Protein Kinase. Journal of Neurochemistry, 83, 828-836. http://dx.doi.org/10.1046/j.1471-4159.2002.01182.x
- Soosairajah, J., Maiti, S., Wiggan, O., Sarmiere, P., Moussi, N., Sarcevic, B., Sampath, R., Bamburg, J. R., & Bernard, O. (2005). Interplay between Components of a Novel LIM Kinase-Slingshot Phosphatase Complex Regulates Cofilin. The EMBO Journal, 24, 473-486. http://dx.doi.org/10.1038/sj.emboj.7600543
- Sun, L., Guo, C., Liu, D., Zhao, Y., Zhang, Y., Song, Z., Han, H., Chen, D., & Zhao, Y. (2011). Protective Effects of Bone Morphogenetic Protein 7 against Amyloid-Beta Induced Neurotoxicity in PC12 Cells. Neuroscience, 184, 151-163. http://dx.doi.org/10.1016/j.neuroscience.2011.03.059
- Suurna, M. V., Ashworth, S. L., Hosford, M., Sandoval, R. M., Wean, S. E., Shah, B. M., Bamburg, J. R., & Molitoris, B. A. (2006). Cofilin Mediates ATP Depletion-Induced Endothelial Cell Actin Alterations. American Journal of Physiology, Renal Physiology, 290, F1398-F1407. http://dx.doi.org/10.1152/ajprenal.00194.2005
- Vlachos, A., Ikenberg, B., Lenz, M., Becker, D., Reifenberg, K., Bas-Orth, C., & Deller, T. (2013). Synaptopodin Regulates Denervation-Induced Homeostatic Synaptic Plasticity. Proceedings of the National Academy of Sciences of the United States of America, 110, 8242-8247. http://dx.doi.org/10.1073/pnas.1213677110
- Wang, Y., Dong, Q., Xu, X. F., Feng, X., Xin, J., Wang, D. D., Yu, H., Tian, T., & Chen, Z. Y. (2013). Phosphorylation of Cofilin Regulates Extinction of Conditioned Aversive Memory via AMPAR Trafficking. Journal of Neuroscience, 33, 6423-6433. http://dx.doi.org/10.1523/JNEUROSCI.5107-12.2013
- Wei, A., Brisby, H., Chung, S. A., & Diwan, A. D. (2008). Bone Morphogenetic Protein-7 Protects Human Intervertebral Disc Cells in Vitro from Apoptosis. The Spine Journal, 8, 466-474. http://dx.doi.org/10.1016/j.spinee.2007.04.021
- Whitehead, G., Jo, J., Hogg, E. L., Piers, T., Kim, D. H., Seaton, G., Seok, H., Bru-Mercier, G., Son, G. H., Regan, P., Hildebrandt, L., Waite, E., Kim, B. C., Kerrigan, T. L., Kim, K., Whitcomb, D. J., Collingridge, G. L., Lightman, S. L., & Cho, K. (2013). Acute Stress Causes Rapid Synaptic Insertion of Ca2+-Permeable AMPA Receptors to Facilitate LongTerm Potentiation in the Hippocampus. Brain, 136, 3753-3765. http://dx.doi.org/10.1093/brain/awt293
- Yabe, T., Samuels, I., & Schwartz, J. P. (2002). Bone Morphogenetic Proteins BMP-6 and BMP-7 Have Differential Effects on Survival and Neurite Outgrowth of Cerebellar Granule Cell Neurons. Journal of Neuroscience Research, 68, 161-168. http://dx.doi.org/10.1002/jnr.10210
- Zhang, X. L., Pöschel, B., Faul, C., Upreti, C., Stanton, P. K., & Mundel, P. (2013). Essential Role for Synaptopodin in Dendritic Spine Plasticity of the Developing Hippocampus. Journal of Neuroscience, 33, 12510-12518. http://dx.doi.org/10.1523/JNEUROSCI.2983-12.2013
- Zhao, R., Du, L., Huang, Y., Wu, Y., & Gunst, S. J. (2008). Actin Depolymerization Factor/Cofilin Activation Regulates Actin Polymerization and Tension Development in Canine Tracheal Smooth Muscle. The Journal of Biology Chemistry, 283, 36522-36531. http://dx.doi.org/10.1074/jbc.M805294200
NOTES

*Corresponding author.