Journal of Diabetes Mellitus
Vol. 2 No. 3 (2012) , Article ID: 22203 , 8 pages DOI:10.4236/jdm.2012.23055
Inhibition of renin activity by aliskiren ameliorates diabetic nephropathy in type 1 diabetes mouse model*
![]()
1Department of Medicine, Division of Biological Sciences, The University of Chicago, Chicago, USA
2Center of System Biomedical Sciences, University of Shanghai for Science and Technology, Shanghai, China
3Laboratory of Metabolic Disease Research and Drug Development, China Medical University, Shenyang, China; †Corresponding Author: cyan@medicine.bsd.uchicago.edu
4Ministry of Health Key Laboratory of Congenital Malformation, Shengjing Hospital, China Medical University, Shenyang, China
Received 17 April 2012; revised 21 May 2012; accepted 3 June 2012
Keywords: Renin Inhibitor; Renin-Angiotensin System; Diabetic Nephropathy; Aliskiren
ABSTRACT
Renin is the rate-limiting enzyme of the reninangiotensin system (RAS). In addition to its enzymatic activity to generate angiotensin I, renin also signals through the (pro)renin receptor to exert angiotensin II-independent effects. In this study we examined the effect of renin inhibition on the development of diabetic nephropathy. Male DBA/2J mice were induced to diabetes with streptozotocin, and the diabetic mice were treated for 16 weeks with saline or aliskiren, a renin enzymatic inhibitor. Aliskiren treatment had little effects on blood glucose and blood pressure in diabetic mice. Saline-treated mice developed progressive albuminuria and glome-rulosclerosis, and aliskiren treatment effectively alleviated albumiuria and glomerulosclerosis. Morphologically aliskiren treatment prevented the thickening of the glomerular basement membrane and reduced podocyte loss. At the molecular levels, aliskiren prevented the decline of slit diaphragm proteins and blocked the synthesis of extracellular matrix and pro-fibrotic factors in the diabetic kidney. Aliskiren treatment results in compensatory renin increase in the glomeruli due to blockade of the negative feedback loop, and also partially suppressed the intracellular signaling mediated by the (pro)renin receptor activated in hyperglycemia. These observations suggest that the therapeutic activity of aliskiren to prevent diabetic renal injury is contributed by inhibition of both the angiotensin II-dependent and -independent pathways. Taken together, it is concluded that inhibition of renin enzymatic activity ameliorates diabetic renal injury in type 1 diabetes, and support the use of aliskiren in diabetes kidney disease.
1. INTRODUCTION
The renin-angiotensin system (RAS) is a major mediator of progressive renal injury. Renin is the rate-limiting enzyme of the RAS cascade, catalyzing the conversion of angiotensinogen to angiotenin (Ang) I. Ang I is converted by angiotensin-converting enzyme (ACE) to Ang II, the effector of the RAS. Renin and its precursor, prorenin, can also bind to and activate a membrane receptor, namely (pro)renin receptor. Binding of renin or prorenin to the receptor not only enhances the enzymatic activity of renin and prorenin, but also activates MAP kinases ERK1/2 pathway [1].
Diabetic nephropathy is the most common renal complication of diabetes mellitus and a leading cause for endstage renal disease. The local RAS in the kidney plays a major damaging role in diabetic nephropathy. Intrarenal renin and angiotensinogen levels are elevated in diabetic animals [2,3], leading to an increase in intrarenal Ang II concentration. Ang II promotes the progression of renal injury via multiple pathways, including induction of oxidative stress and stimulation of profibrotic and proinflammatory factors and extracellular matrix proteins [4,5]. These molecular pathways promote the development of proteinuria, glomerulosclerosis and tubulointerstitial fibrosis. On the other hand, renin may also induce renal injury in an Ang II-independent manner via activation of the (pro)renin receptor. For example, renin via binding to its receptor in mesangial cells stimulates the production of TGF-b [6], a potent pro-fibrotic factor. Therefore, inhibition of renin may affect both the Ang II-dependent and -independent pathways.
Given the critical role of the RAS in diabetic kidney injury, inhibition of the RAS has been the mainstay therapy for diabetic nephropathy. Large clinical trials have demonstrated that ACE inhibitors and Ang II receptor blockers (ARBs) are able to attenuate the progression of nephropathy in diabetic patients [7-11]. Inhibition of renin with a decoy peptide or with vitamin D analogs has also been shown to block the development of diabetic nephropathy in diabetic animal models [12-14]. Aliskiren, the first renin inhibitor approved for clinical use, is a small molecule competitive inhibitor that specifically inhibits the enzymatic activity of rennin [15]. In clinical trials, aliskiren and losartan combination reduces albuminuria in type 2 diabetes patients, and the renoprotective effect appears to be independent of its blood-pressure-lowering effect [16,17]. Given these observations in humans, however, the renoprotective mechanism of aliskiren remains elusive. In the present study we explored the renoprotective action of aliskiren using a type 1 diabetic mouse model. Our data suggest that aliskiren alone is able to ameliorate diabetic nephropathy by blockade of Ang IIdependent and (pro)renin receptor-dependent signal pathways.
2. MATERIALS AND METHODS
2.1. Animals and Treatment
Male DBA/2J mice, which are susceptible to hyperglycemia-induced renal injury, were used in this study. Eight-week old male DBA/2J mice (Jackson Laboratory, Bar Harbor, ME) were made diabetic by intraperitoneal (i.p.) injection of freshly prepared streptozotocin (STZ, dissolved in 10 mM citrate buffer, pH 4.2), given at a dose of 35 mg/kg/day for five consecutive days. Three weeks after STZ injection (week 0), the mice were randomly separated into two groups and treated with saline (vehicle) or aliskiren (25 mg/kg, i.p. injection, 3x/week; kindly provided by Novatis), respectively. This dose was chosen based on a previous dose-dependent study [18]. Non-diabetic mice served as control. Blood glucose levels were monitored with a glucometer (Bayer). Spot urine was collected at various time points, and urinary albumin and creatinine levels were determined using commercial kits as reported previously [19]. Blood pressure of the mice was measured at week 16 using the carotid artery cannulation method as described previously [20]. All mice were killed at 16 weeks following STZ treatment, and serum and kidneys were immediately harvested for analyses. The animal study protocols were approved by the Institutional Animal Care and Use Committee of the University of Chicago.
2.2. Histology and Immunostaining
Freshly dissected kidneys were fixed overnight with 4% formaldehyde in PBS (pH 7.2), processed, embedded in paraffin, and cut into 3-mm sections. Renal sections were stained with periodic acid-Schiff (PAS). Semiquantitative scoring of glomerular sclerosis in PASstained kidney sections was performed using a five-grade method described previously [19,21]. Immunostaining of kidney sections was carried out as described previously [19].
2.3. Electron Microscopy
Tissue blocks of kidney cortex were fixed in 2% glutaraldehyde in 0.1 M phosphate buffer, pH 7.4 and then post-fixed in 1% osmium tetroxide. Electron microscopic analyses and measurement of the GBM thickness were performed as described previously [14].
2.4. Glomerular Purification
Mouse glomeruli were isolated based on a previously published method [22]. Briefly, after mice were perfused with deactivated Dynabeads M-450, the perfused kidneys were collected and cut into small pieces, which were ground on a 100-mesh sterile stainless sieve. The beadstrapping glomeruli were collected with a magnetic bar. After digestion with collagenase and washes, glomeruli were recollected with a magnetic bar. The purity of glomeruli obtained by this method was >95% with no renal tubule contamination. The purified glomeruli were used for RNA isolation or lysed for protein analyses.
2.5. Western Blot
Total kidney or purified glomeruli were homogenized in Laemmli buffer (Boston Bioproducts, Worcester, MA), followed by 5 min boiling and centrifugation to obtain the supernatant. Protein concentrations were determined using a BioRad Protein Assay kit (BioRad, Hercules, CA). Proteins were separated by SDS-PAGE and transferred onto Immobilon membranes. Western blotting was carried out as previously described [23].
2.6. RT-PCR
Total RNAs were isolated using TRIzol reagents (Invitrogen, Carlsbad, CA). First-strand cDNAs were synthesized from 2 mg of total RNAs in 20 ml reaction using MML-V reverse transcriptase (Invitrogen) and hexanucleotide random primers. The first strand cDNAs served as the template for PCR. Regular PCR was performed using a BioRad DNA Engine. Real time RT-PCR quantification was carried out in an Applied Biosystems 7900 Real Time PCR System using a SYBR green PCR reagent kit (Applied Biosystems, Foster City, CA) as described previously [19]. GAPDH and b-2 microglobulin (B2M) served as the internal controls. The PCR primers used in this study were as described previously [13,19].
2.7. Statistical Analysis
Data values were presented as means ± SEM. Statistical comparisons were made using two-tailed unpaired Student’s t-test and Mann-Whitney U test, with P < 0.05 being considered statistically significant.
3. RESULTS
Three weeks after STZ treatment (week 0), the mice were randomized into 2 groups (n = 7 in each group) and treated respectively with saline or aliskiren for 16 weeks. Blood glucose levels in diabetic mice rose to above 300 mg/dl within three weeks of STZ treatment and continued to rise over time to >600 mg/dl, and aliskiren treatment had no effects on blood glucose levels (Table 1). These mice survived relatively well through the experimental period without the need of insulin supplementation. The diabetic mice were mostly normotensive. At the end of the treatment (week 16), the mean blood pressure of aliskiren-treated diabetic mice was slightly lower compared to untreated diabetic mice, but the difference was not statistically significant (Table 1). Thus aliskiren did not significantly affect glucose metabolism or blood pressure in this animal model.
In contrast to the non-diabetic mice, all STZ-treated mice failed to gain weight during the 19-week experimental period, regardless of treatment with saline vehicle or aliskiren (Figure 1(a)). As expected, vehicle treated diabetic mice developed progressive albuminuria, with the urinary albumin to creatinine ratio (ACR) increased by more than 6-fold over the baseline at week 16. Aliskiren treatment effectively blocked the increase in ACR in the entire treatment period (Figure 1(b)). Serum creatinine and BUN levels were significantly increased in the vehicle treated diabetic mice, and aliskiren virtually normalized these serum parameters (Figures 1(c) and (d)).
Diabetic renal injury usually accompanies podocyte loss. Immunostaining with podocyte-specific anti-WT1 antibody showed that WT1-positive cells were markedly reduced in the glomerulus in vehicle-treated diabetic mice compared to non-diabetic mice, and aliskiren treatment significantly prevented the loss of glomerular WT1-positive cells (Figures 2(a) and (b)). Electron microscopic examination revealed that, compared to the non-diabetic control, the vehicle-treated diabetic mice had a marked increase in the thickness of the glomerular basement membrane (GBM), a marker of impaired glomerular filtration, and aliskiren treatment prevented the thickening of GBM (Figures 2(c) and (d)). The slit diaphragm controls protein filtration through the glomerular filtration barrier. We quantified expression of several key proteins involved in the formation of the slit diaphragm by real time RT-PCR. Relative to the non-diabetic control, mRNA levels of ZO-1, Neph-1, nephrin and FAT-1 were significantly reduced in vehicle treated diabetic mice, and aliskiren effectively prevented the decline of ZO-1, Neph-1 and FAT-1 (Figure 2(e)).
As expected, vehicle treated diabetic mice developed glomerulosclerosis at week 16, with marked accumulation of extracellular matrix (ECM) in the mesangium. Aliskiren treatment significantly attenuated glomerulosclerosis and reduced the glomerulosclerotic score (Figures 3(a) and (b)). Consistently, the mRNA and protein levels of fibronectin (FN), a key ECM protein, were markedly enhanced in vehicle treated diabetic mice compared to non-diabetic mice, as assessed by real time qRTPCR (Figure 3(c)), Western blotting (Figure 3(d)) and immunostaining (Figure 3(e)). The increase in FN transcript and protein levels was attenuated in aliskirentreated mice (Figures 3(c)-(e)). Similarly, levels of profibrotic factors TGF-b, VEGF and CTGF in the kidney were induced in vehicle-treated diabetic mice, and aliskiren significantly blocked the expression of TGF-b (Figure 3(f)).
We also examined the components of the RAS in the purified glomeruli at week 16, including renin, (pro)renin receptor, ACE, angiotensinogen and AT1 receptor. As shown in Figure 4, there was a moderate increase in renin expression in vehicle-treated diabetic mice relative to non-diabetic controls at both mRNA (Figure 4(a)) and protein levels (Figures 4(a) and (c)), and renin was further increased to a much higher levels in aliskiren-treated mice (Figures 4(a)-(c)), due to a compensatory increase
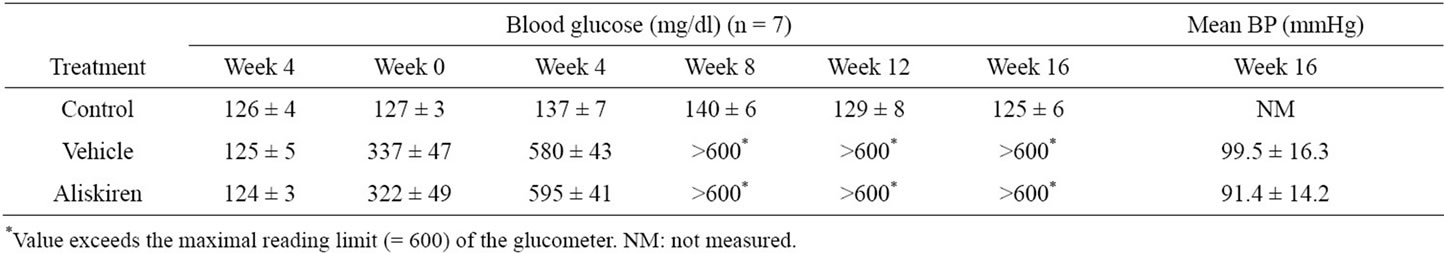
Table 1. Levels of blood glucose and mean blood pressure in control mice and diabetic mice treated with vehicle or aliskiren during the experimental period.
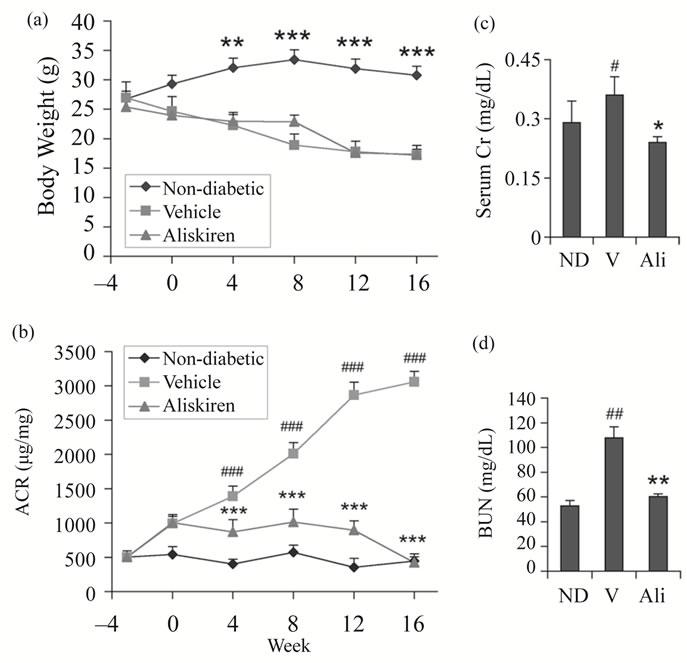
Figure 1. Effect of aliskiren on body weight, serum and urinary parameters in STZ-induced diabetic mice. Eight-week old male DBA/2J mice were injected i.p. with STZ for five consecutive days in the first week (week 3). Three weeks after STZ injection (week 0), the mice were treated with vehicle (V) or aliskiren (Ali) for 16 weeks. Non-diabetic (ND) mice served as controls. (a) Body weight curves. ***P < 0.001 vs. vehicle and aliskiren; (b) Changes in urinary albumin to creatinine ratio (ACR) over time; (c) Serum creatinine (Cr) levels; and (d) BUN levels. Note the dramatic inhibition of albuminuria by aliskiren. #P < 0.05; ##P < 0.01, ###P < 0.001 vs. ND control; *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle; n = 6 - 7.
as a result of blockade of negative feedback regulation [24,25]. Angiotensinogen was highly induced in vehicle-treated diabetic mice, which was attenuated by aliskiren treatment (Figure 4(a)). The other RAS components (renin receptor, ACE and AT1 receptor) were only moderately or not altered in vehicleor aliskiren-treated diabetic mice relative to the non-diabetic control (Figures 4(a)-(c)).
To address whether aliskiren blocked the (pro)renin receptor-mediated signaling, we examined ERK1/2 phosphorylation in the glomerular lysates. ERK activation was the best known signaling pathway of (pro)renin receptor activation [1]. ERK1/2 phosphorylation was increased in vehicle-treated diabetic mice, indicating the activation of the ERK1/2 pathway in diabetic state. Aliskiren partially suppressed ERK1/2 phosphorylation (Figures 4(d) and (e)), suggesting that this drug reduced diabetic renal injury in part by inhibiting the ERK signaling pathway activated by renin via the (pro)renin receptor.
4. DISCUSSION
The local RAS plays a key role in diabetic renal com-
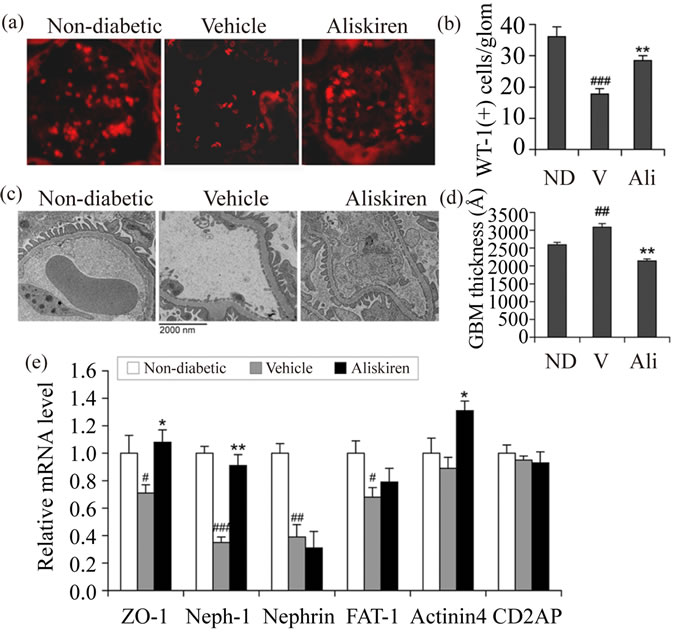
Figure 2. Glomerular filtration barrier and podocytes. (a) Immunostaining with podocyte-specific anti-WT1 antibody. Shown are representative glomeruli from non-diabetic (ND) and diabetic mice treated with vehicle (V) or aliskiren (Ali); (b) Average podocyte number per glomerulus (glom). WT1-positive cells were counted in 30 glomeruli randomly selected from immunostained sections in each group of mice; (c) Transmission electron microscopic images of the glomerular filtration barrier. Note the increased thickness of the glomerular basement membrane (GBM) in vehicle-treated diabetic mice; (d) Quantitation of GBM thickness from each group of mice as indicated; (e) Quantitation of slit diaphragm proteins. Transcript levels of ZO-1, Neph-1, nephrin, FAT-1, a-actinin-4 and CD2AP were determined by real time RT-PCR. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. ND control; *P < 0.05, **P < 0.01, ***P < 0.001 vs. vehicle.
plications. Ang II, the central effector of the RAS, activates multiple pathways in the kidney to induce renal injury [5]. Inhibition of Ang II production with ACE inhibitors and blockade of AT1 receptor with ARBs have been shown to ameliorate diabetic nephropathy in animals and humans [7-11]. In this report we demonstrated that inhibition of renin activity with aliskiren alleviated diabetic renal injury, confirming the critical role of the reninangiotensin cascade in the development of diabetic nephropathy. Aliskiren markedly suppressed albuminuria, prevented podocyte loss, improved glomerular filtration barrier structure and reduced glomerulosclerosis in diabetic mice. As aliskiren did not alter blood glucose levels, the therapeutic effect is unlikely by improving hyperglycemia. Aliskiren slightly lowered blood pressure in diabetic mice, but the effect was not statistically significant. This is consistent with previous observation that aliskiren does not alter blood pressure in normotensive subjects [26] (STZ-induced diabetic mice were normotensive). Previous studies suggest that aliskiren can protect the kidney through both blood pressure-dependent and -in-
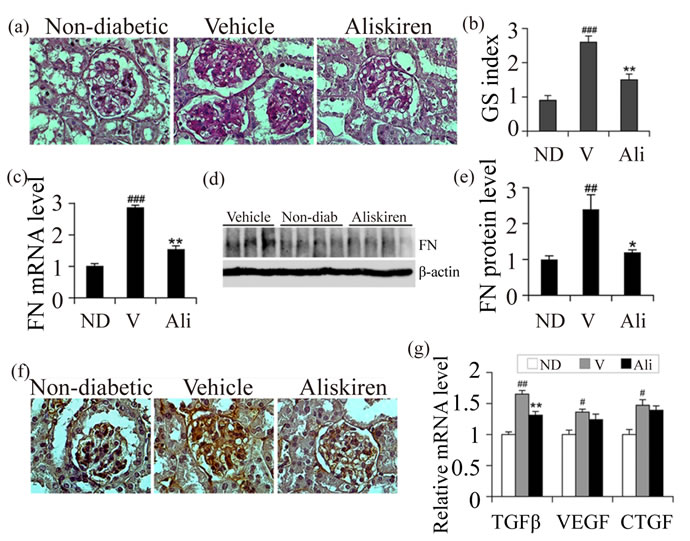
Figure 3. Effect of aliskiren on glomerulosclerosis. (a) Periodic acid-Schiff (PAS) staining. Shown are representative glomerular morphology of non-diabetic (ND) control and diabetic mice treated with vehicle (V) or aliskiren (Ali); (b) Glomerulosclerotic (GS) scores, counting >50 glomeruli in each group of mice; (c) Real time RT-PCR quantittation of fibronectin (FN) mRNA levels in the kidney of the three groups of mice; (d) Western blot analysis of FN protein levels; (e) Semi-quantitative data for FN protein levels; (f) Glomerular FN accumulation. Kidney sections were immunostained with anti-FN antibody; (g) Real time RT-PCR quantification of TGF-b, VEGF and CTGF transcripts in the three groups of mice. #P < 0.05, ##P < 0.01, ###P < 0.001 vs. ND control; *P < 0.05, **P < 0.01 vs. vehicle. N = 6 - 7.
dependent mechanisms [16,17,27]; therefore, it is possible that in this model aliskiren protects the kidney by targeting the local intrarenal RAS as well as by altering renal hemodynamics. Further studies are needed to clarify this issue.
Renin is the rate-limiting enzyme of the RAS cascade. As such, it is considered as an ideal drug target for RAS blockade. The development of direct renin inhibitors (e.g. aliskiren), however, is much slower than that of ACE inhibitors and AT1 receptor blockers [25]. Whether aliskiren is therapeutically better than ACE inhibitors or ARBs remains to be determined. Blockade of the RAS with all these RAS inhibitors inevitably disrupts the negative feedback loop that is critical for maintaining renin homeostasis, leading to compensatory induction of renin [28]. In fact, renin induction was considered as an excellent indicator for aliskiren actions [29]. In this study, we observed a clear and dramatic renin induction by aliskiren, but not reduction of (pro)renin receptor as reported previously in transgenic rats [27]. Aliskiren induction of renin is known to be more profound compared to ARBs [29], thus it is interesting to compare the renoprotective efficacy between aliskiren and ACE inhibitors or ARBs in future studies.
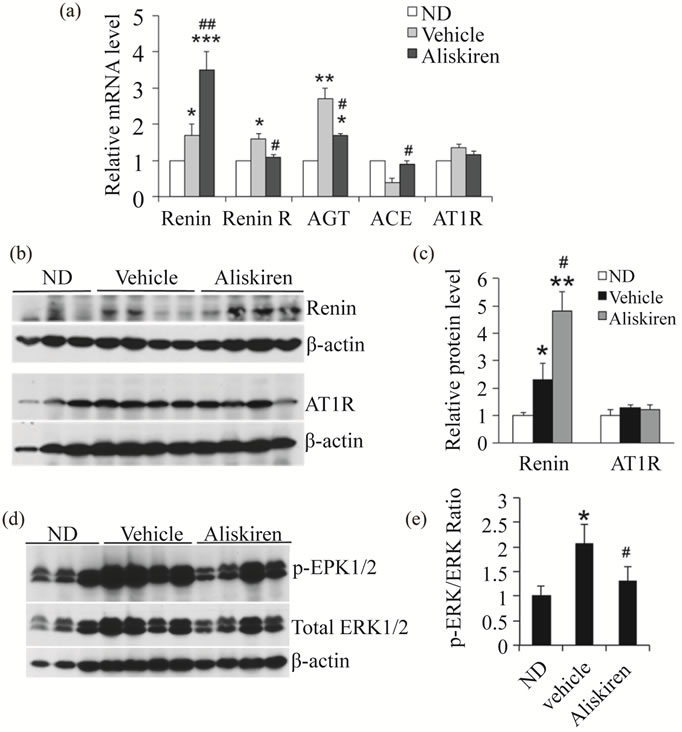
Figure 4. Effect of aliskiren on the renin-angiotensin system in the glomeruli. (a) Transcript levels of renin, (pro)renin receptor (Renin R), angiotensin-converting enzyme (ACE), angiotensinogen (AGT), and AT1 receptor (AT1R), quantified by real time RT-PCR in non-diabetic (ND) mice or diabetic mice treated with vehicle or aliskiren. *P < 0.05, **P < 0.01, ***P < 0.001 vs. ND control; #P < 0.05; ##P < 0.01 vs. Vehicle. (b and c) Western blotting (b) and semi-quantitation (c) for renin protein levels in purified glomerular lysates from the three groups of mice. Note renin is highly induced by aliskiren. *P < 0.05, **P < 0.01 vs. ND; #P < 0.05 vs. Vehicle; (d) Western blot analysis of ERK1/2 phosphorylation and total ERK levels in purified glomerular lysates; (e) Phospho-ERK1/2 to total ERK1/2 ratio. *P < 0.05 vs. ND; #P < 0.05 vs. Vehicle. Each lane represents one mouse.
In addition to converting angiotensinogen to Ang I, renin and prorenin can activate the (pro)renin receptor [1] and induce intracellular ERK phosphorylation independent of Ang II. This pathway has been shown to promote TGF-b production [6]. Prorenin is usually converted to the enzymatically active renin via proteolytic cleavage of the prosegment; however, binding of prorenin to the (pro)renin receptor causes a conformation change that leads to nonproteolytic activation of prorenin, which also promotes Ang I production. Intrarenal renin and prorenin are induced in hyperglycemia and diabetes [2,3]. When exposed to high glucose, mesangial cells and podocytes can increase renin production [30-32]. As inhibition of renin ameliorates diabetic nephropathy, one interesting question is which of these two renin pathways is responsible for renal damage. We have demonstrated that vitamin D analogs inhibit the development of diabetic nephropathy by targeting the intrarenal RAS [13,14]; however, because vitamin D suppresses renin gene expression [2033], those studies cannot distinguish the effect of these two renin pathways. Ichihara et al. used a decoy peptide corresponding to the “handle” region to block nonproteolytic activation of prorenin, and found that this competition inhibited diabetic nephropathy [34]. This peptide actually blocks the Ang II-dependent pathway of prorenin. Feldman et al. showed that aliskiren treatment of transgenic Ren-2 rats led to reduction in blood pressure and albuminuria [27], whereas Feldt et al. demonstrated that aliskiren could not inhibit (pro)renin receptor-mediated ERK1/2 activation in cell cultures [35]. Here our data from this study showed that aliskiren partially inhibited the (pro)renin receptor-mediated ERK1/2 activation in the glomerulus. These data suggest that aliskiren protects the kidney from diabetic injury by inhibition of both the Ang II-dependent and -independent pathways of renin activities. The Ang II-dependent pathway signals through the AT1 receptor, and the Ang II-independent pathway is mediated by the (pro)renin receptor and transduces via ERK phosphorylation. Given the broad functionalities of the RAS, drugs that target the RAS have a wide range of therapeutic potentials. As the first direct renin inhibitor approved for clinical use, interests in the therapeutic capacities of aliskiren are rising. In this context, demonstration of renoprotective effects of aliskiren in diabetic nephropathy has important clinical implications.
Taken together, this study demonstrates that blockade of renin activity ameliorates diabetic renal injury in a diabetic animal model, reaffirming the critical role of the RAS in the development of diabetic nephropathy. The renal protective actions of aliskiren appear to be dependent on both the Ang II-dependent pathway and the (pro)- renin receptor-mediated pathway.
5. AUTHORS’ CONTRIBUTIONS
YZ and YW performed the study; YC, DKD and TS provided material and technical assistance; YCL supervised the project, designed the experiment and wrote the manuscript.
6. ACKNOWLEDGEMENTS
We thank Novatis for providing aliskiren. This work was supported in part by NIH grant R01HL085793.
![]()
![]()
REFERENCES
- Nguyen, G., Delarue, F., Burckle, C., Bouzhir, L., Giller, T. and Sraer, J.D. (2002) Pivotal role of the renin/prorenin receptor in angiotensin II production and cellular responses to renin. Journal of Clinical Investigation, 109, 1417-1427.
- Anderson, S., Jung, F.F. and Ingelfinger, J.R. (1993) Renal renin-angiotensin system in diabetes: Functional, immunohistochemical, and molecular biological correlations. American Journal of Physiology, 265, 477-486.
- Zimpelmann, J., Kumar, D., Levine, D.Z., Wehbi, G., Imig, J.D., Navar, L.G. and Burns, K.D. (2000) Early diabetes mellitus stimulates proximal tubule renin mRNA expression in the rat. Kidney International, 58, 2320- 2330. doi:10.1046/j.1523-1755.2000.00416.x
- Gilbert, R.E., Krum, H., Wilkinson-Berka, J. and Kelly, D.J. (2003) The renin-angiotensin system and the longterm complications of diabetes: Pathophysiological and therapeutic considerations. Diabetic Medicine, 20, 607- 621. doi:10.1046/j.1464-5491.2003.00979.x
- Carey, R.M. and Siragy, H.M. (2003) The intrarenal renin- angiotensin system and diabetic nephropathy. Trends in Endocrinology & Metabolism, 14, 274-281. doi:10.1016/S1043-2760(03)00111-5
- Huang, Y., Wongamorntham, S., Kasting, J., McQuillan, D., Owens, R.T., Yu, L., Noble, N.A. and Border, W. (2006) Renin increases mesangial cell transforming growth factor-beta1 and matrix proteins through receptor-mediated, angiotensin II-independent mechanisms. Kidney International, 69, 105-113. doi:10.1038/sj.ki.5000011
- Chan, J.C., Ko, G.T., Leung, D.H., Cheung, R.C., Cheung, M.Y., So, W.Y., Swaminathan, R., Nicholls, M.G., Critchley, J.A. and Cockram, C.S. (2000) Long-term effects of angiotensin-converting enzyme inhibition and metabolic control in hypertensive type 2 diabetic patients. Kidney International, 57, 590-600. doi:10.1046/j.1523-1755.2000.00879.x
- Lewis, E.J., Hunsicker, L.G., Clarke, W.R., Berl, T., Pohl, M.A., Lewis, J.B., Ritz, E., Atkins, R.C., Rohde, R., and Raz, I. (2001) Renoprotective effect of the angiotensinreceptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. The New England Journal of Medicine, 345, 851-860. doi:10.1056/NEJMoa011303
- Brenner, B.M., Cooper, M.E., de Zeeuw, D., Keane, W.F., Mitch, W.E., Parving, H.H., Remuzzi, G., Snapinn, S.M., Zhang, Z. and Shahinfar, S. (2001) Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. The New England Journal of Medicine, 345, 861-869. doi:10.1056/NEJMoa011161
- Parving, H.H., Lehnert, H., Brochner-Mortensen, J., Gomis, R., Andersen, S., and Arner, P. (2001) The effect of irbesartan on the development of diabetic nephropathy in patients with type 2 diabetes. The New England Journal of Medicine, 345, 870-878. doi:10.1056/NEJMoa011489
- Andersen, S., Tarnow, L., Rossing, P., Hansen, B.V. and Parving, H.H. (2000) Renoprotective effects of angiotensin II receptor blockade in type 1 diabetic patients with diabetic nephropathy. Kidney International, 57, 601-606. doi:10.1046/j.1523-1755.2000.00880.x
- Ichihara, A., Hayashi, M., Kaneshiro, Y., Suzuki, F., Nakagawa, T., Tada, Y., Koura, Y., Nishiyama, A., Okada, H., Uddin, M.N., Nabi, A.H., Ishida, Y., Inagami, T. and Saruta, T. (2004) Inhibition of diabetic nephropathy by a decoy peptide corresponding to the “handle” region for nonproteolytic activation of prorenin. Journal of Clinical Investigation, 114, 1128-1135. doi:10.1172/JCI21398
- Zhang, Z., Zhang, Y., Ning, G., Deb, D.K., Kong, J. and Li, Y.C. (2008) Combination therapy with AT1 blocker and vitamin D analog markedly ameliorates diabetic nephropathy: Blockade of compensatory renin increase. Proceedings of the National Academy of Sciences USA, 105, 15896-15901. doi:10.1073/pnas.0803751105
- Zhang, Y., Deb, K.D., Kong, J., Ning, G., Wong, Y., Li, G., Chen, Y., Zhang, Z., Strugnell, S., Sabbagh, Y., Arbeeny, C. and Li, Y.C. (2009) Long-term therapeutic effect of vitamin D analog doxercalciferol on diabetic nephropathy: Strong synergism with AT1 receptor antagonist. American Journal of Physiology Renal Physiology, 297, F791- F801. doi:10.1152/ajprenal.00247.2009
- Wood, J.M., Maibaum, J., Rahuel, J., Grutter, M.G., Cohen, N.C., Rasetti, V., Ruger, H., Goschke, R., Stutz, S., Fuhrer, W., Schilling, W., Rigollier, P., Yamaguchi, Y., Cumin, F., Baum, H.P., Schnell, C.R., Herold, P., Mah, R., Jensen, C., O’Brien, E., Stanton, A. and Bedigian, M.P. (2003) Structure-based design of aliskiren, a novel orally effective renin inhibitor. Biochemical and Biophysical Research Communications, 308, 698-705. doi:10.1016/S0006-291X(03)01451-7
- Parving, H.H., Persson, F., Lewis, J.B., Lewis, E.J. and Hollenberg, N.K. (2008) Aliskiren combined with losartan in type 2 diabetes and nephropathy. The New England Journal of Medicine, 358, 2433-2446. doi:10.1056/NEJMoa0708379
- Persson, F., Lewis, J.B., Lewis, E.J., Rossing, P., Hollenberg, N.K. and Parving, H.H. (2011) Aliskiren in combination with losartan reduces albuminuria independent of baseline blood pressure in patients with type 2 diabetes and nephropathy. Clinical Journal of the American Society of Nephrology, 6, 1025-1031. doi:10.2215/CJN.07590810
- Lu, H., Rateri, D.L., Feldman, D.L., Fukamizu, A., Ishida, J., Oesterling, E.G., Cassis, L.A. and Daugherty, A. (2008) Renin inhibition reduces hypercholesterolemia-induced atherosclerosis in mice. Journal of Clinical Investigation, 118, 984-993.
- Zhang, Z., Sun, L., Wang, Y., Ning, G., Minto, A.W., Kong, J., Quigg, R.J. and Li, Y.C. (2008) Renoprotective role of the vitamin D receptor in diabetic nephropathy. Kidney International, 73, 163-171. doi:10.1038/sj.ki.5002572
- Li, Y.C., Kong, J., Wei, M., Chen, Z.F., Liu, S.Q. and Cao, L.P. (2002) 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. Journal of Clinical Investigation, 110, 229-238.
- Taneda, S., Pippin, J.W., Sage, E.H., Hudkins, K.L., Takeuchi, Y., Couser, W.G. and Alpers, C.E. (2003) Amelioration of diabetic nephropathy in SPARC-null mice. Journal of the American Society of Nephrology, 14, 968- 980. doi:10.1097/01.ASN.0000054498.83125.90
- Takemoto, M., Asker, N., Gerhardt, H., Lundkvist, A., Johansson, B.R., Saito, Y. and Betsholtz, C. (2002) A new method for large scale isolation of kidney glomeruli from mice. American Journal of Pathology, 161, 799-805. doi:10.1016/S0002-9440(10)64239-3
- Li, Y.C., Bolt, M.J.G., Cao, L.-P. and Sitrin, M.D. (2001) Effects of vitamin D receptor inactivation on the expression of calbindins and calcium metabolism. American Journal of Physiology—Endocrinology and Metabolism, 281, 558-564.
- Qi, Z., Fujita, H., Jin, J., Davis, L.S., Wang, Y., Fogo, A.B. and Breyer, M.D. (2005) Characterization of susceptibility of inbred mouse strains to diabetic nephropathy. Diabetes, 54, 2628-2637. doi:10.2337/diabetes.54.9.2628
- Li, Y.C. (2007) Inhibition of renin: An updated review of the development of renin inhibitors. Current Opinion in Investigational Drugs, 8, 750-757.
- Nussberger, J., Wuerzner, G., Jensen, C. and Brunner, H.R. (2002) Angiotensin II suppression in humans by the orally active renin inhibitor Aliskiren (SPP100): Comparison with enalapril. Hypertension, 39, 1-8. doi:10.1161/hy0102.102293
- Feldman, D.L., Jin, L., Xuan, H., Contrepas, A., Zhou, Y., Webb, R.L., Mueller, D.N., Feldt, S., Cumin, F., Maniara, W., Persohn, E., Schuetz, H., Jan Danser, A.H. and Nguyen, G. (2008) Effects of aliskiren on blood pressure, albuminuria, and (pro)renin receptor expression in diabetic TG(mRen-2)27 rats. Hypertension, 52, 130-136. doi:10.1161/HYPERTENSIONAHA.107.108845
- Muller, D.N. and Luft, F.C. (2006) Direct renin inhibition with aliskiren in hypertension and target organ damage. Clinical Journal of the American Society of Nephrology, 1, 221-228. doi:10.2215/CJN.01201005
- Azizi, M., Menard, J., Bissery, A., Guyenne, T.T., BuraRiviere, A., Vaidyanathan, S. and Camisasca, R.P. (2004) Pharmacologic demonstration of the synergistic effects of a combination of the renin inhibitor aliskiren and the AT1 receptor antagonist valsartan on the angiotensin II-renin feedback interruption. Journal of the American Society of Nephrology, 15, 3126-3133. doi:10.1097/01.ASN.0000146686.35541.29
- Durvasula, R.V. and Shankland, S.J. (2008) Activation of a local renin angiotensin system in podocytes by glucose. American Journal of Physiology—Renal Physiology, 294, 830-839. doi:10.1152/ajprenal.00266.2007
- Vidotti, D.B., Casarini, D.E., Cristovam, P.C., Leite, C.A., Schor, N. and Boim, M.A. (2004) High glucose concentration stimulates intracellular renin activity and angiotensin II generation in rat mesangial cells. American Journal of Physiology—Renal Physiology, 286, 1039-1045. doi:10.1152/ajprenal.00371.2003
- Yoo, T.H., Li, J.J., Kim, J.J., Jung, D.S., Kwak, S.J., Ryu, D.R., Choi, H.Y., Kim, J.S., Kim, H.J., Han, S.H., Lee, J.E., Han, D.S. and Kang, S.W. (2007) Activation of the renin-angiotensin system within podocytes in diabetes. Kidney International, 71, 1019-1027. doi:10.1038/sj.ki.5002195
- Yuan, W., Pan, W., Kong, J., Zheng, W., Szeto, F.L., Wong, K.E., Cohen, R., Klopot, A., Zhang, Z. and Li, Y.C. (2007) 1,25-Dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP Response element in the renin gene promoter. The Journal of Biological Chemistry, 282, 29821-29830. doi:10.1074/jbc.M705495200
- Ichihara, A., Suzuki, F., Nakagawa, T., Kaneshiro, Y., Takemitsu, T., Sakoda, M., Nabi, A.H., Nishiyama, A., Sugaya, T., Hayashi, M. and Inagami, T. (2006) Prorenin receptor blockade inhibits development of glomerulosclerosis in diabetic angiotensin II type 1a receptor-deficient mice. Journal of the American Society of Nephrology, 17, 1950-1961. doi:10.1681/ASN.2006010029
- Feldt, S., Batenburg, W.W., Mazak, I., Maschke, U., Wellner, M., Kvakan, H., Dechend, R., Fiebeler, A., Burckle, C., Contrepas, A., Jan Danser, A.H., Bader, M., Nguyen, G., Luft, F.C. and Muller, D.N. (2008) Prorenin and renin-induced extracellular signal-regulated kinase 1/2 activation in monocytes is not blocked by aliskiren or the handle-region peptide. Hypertension, 51, 682-688. doi:10.1161/HYPERTENSIONAHA.107.101444
ABBREVIATIONS:
RAS: renin-angiotensin system;
ACE: angiotensin-converting enzyme;
ARB: angiotensin receptor blocker;
STZ: streptozotocin;
ACR: albumin creatinine ratio;
PAS: periodic acid-Schiff;
GBM: glomerular basement membrane.
NOTES
*Conflicts of interest: None.
#These two authors contribute equally to this work.