Open Journal of Nephrology
Vol.3 No.1(2013), Article ID:29234,7 pages DOI:10.4236/ojneph.2013.31013
Dimethylarginine Dimethylaminohydrolase 2 Gene Polymorphism and Its Association with Asymmetrical Dimethyl Arginine in Hemodialyzed Patients*
1Cardiology and Vascular Medicine Department, Faculty of Medicine, Airlangga University, Surabaya, Indonesia
2Tropical Disease Center, Airlangga University, Surabaya, Indonesia
3Division of Nephrology, Departement of Internal Medicine, Juntendo University Faculty of Medicine, Tokyo, Japan
4Nephrology-Hypertension Division, Internal Medicine Departement, Faculty of Medicine, Airlangga University, Surabaya, Indonesia
Email: yusuf_505@yahoo.com, yusuf_505@unair.ac.id
Received December 6, 2012; revised February 9, 2013; accepted March 11, 2013
Keywords: ADMA; DDAH; gene polymorphism
ABSTRACT
Introduction: Patients with CKD have elevated plasma levels of Asymmetrical Dimethyl Arginine (ADMA), impaired EDRF/NO responses in isolated resistance vessels, and a marked increase in the frequency of cardiovascular events that are predicated by plasma levels of ADMA. ADMA is considered as a risk factor for endothelial dysfunction, progression of chronic kidney disease and a marked increase in the frequency of cardiovascular events that are predicated by plasma levels of ADMA. Elevated ADMA in CKD have been related to a combination of a reduced renal ADMA excretion and a reduced catabolism of ADMA by dimethylarginine dimethylaminohydrolase (DDAH). The current study was undertaken to determine whether there is a correlation between ADMA and SNPs at −449 DDAH 2. Subjects and Methods: It was a cross sectional analytic study, 56 hemodialysis patients and 30 healthy individuals were enrolled. Based on its etiology, HD patients group was further divided in to hypertension (HT) subgroup and non-HT subgroup. Genotyping of the polymorphisms was performed using PCR-based SNP detection methods based on 5’-exonuclease activity assays for rs805305. Results: Heterozygotes were observed as the most abundant genotypes in both groups, followed by GG genotype in the HD patients (30%) and CC (27%) healthy individuals. Among the HT subgroup, the mean plasma levels of ADMA were sequentially higher from genotypes CC, G/C and GG (p = 0.037). Further multiple comparisons between groups using post hoc test showed results that genotype GG and CC were different at 0.05 level of significance. These findings were not found among non HT subgroup. Conclusion: Genetic variation in the DDAH 2 genes is significantly associated with serum ADMA levels in hypertensive HD patients. We observed that carriage of a G at position −449 in the promoter region of the DDAH 2 gene is associated with higher ADMA levels.
1. Introduction
Atherosclerotic cardiovascular diseases (CVD) are major causes of morbidity and mortality in patients receiving ongoing hemodialysis (HD) [1,2]. Despite its increasing incidence and the accelerated worsening of atherosclerosis in patients on chronic hemodialysis, the proportion of individuals with chronic kidney disease (CKD) receiving appropriate cardiovascular (CV) risk modification treatment is lower than that in the general population [3]. In metabolic disorders associated with atherosclerosis (dyslipidemia, hypertension (HT) and diabetes mellitus), a reduced endothelium-mediated nitric oxide (NO)-dependent vasodilation has been observed, which may contribute to the initiation and progression of atherosclerosis associated with these disorders [4].
The mechanisms of endothelial vasodilator dysfunction (EVD) are likely multi-factorial [5]. Evidences from both animal models and clinical studies suggests that accumulation of the endogenous nitric-oxide synthase (NOS) inhibitors, ADMA and NGmethyl-L-arginine (LNMMA) contributes to reducing nitric oxide (NO) [6,7]. Plasma levels of ADMA are inversely related to the vascular NO bioavailability and ADMA is an independent determinant of the vascular redox-state [8,9]. ADMA is also a risk factor for EVD, CV mortality and progression of CKD [8,10].
The co-localization of DDAH 2 and NOS at several sites supports the hypothesis that DDAH may regulate NOS activity by controlling the metabolism of ADMA [11]. As DDAH 2 regulates ADMA levels, it is believable that functional DDAH 2 gene polymorphisms may account for the variation in ADMA levels [12]. ADMA levels may be genetically determined by a promoter polymorphism in a regulatory gene encoding DDAH 2 [13]. However, the functional significance has not been determined previously in chronic HD patients. Therefore, the current study was undertaken to determine whether or not there is a correlation between single nucleotide polymorphisms (SNPs) at −449 (promoter region) DDAH 2 with ADMA plama levels in HD patients.
2. Patients and Methods
2.1. Study Population and Dialysis Protocol
Fifty-six patients with a creatinine clearance of less than 15 ml/min per 1.73 m2 who had been on chronic HD treatment for more than 3 months were recruited for the present study. Thirty-four HD patients (60.7%) were male. Clinical practice was not changed or modified for the purpose of the study. As the control group, 30 healthy individuals paramedics who had no cardiovascular clinical risk factors (HT, diabetes, dyslipidemia, smoking, family history of premature coronary artery disease or CKD), were enrolled. Five healthy individuals (16.7%) as controls were male.
All participants (patients and controls) signed the Free and Informed Consent Form and the study was approved by the Ethics Committee of Al Irsyad Hospital, Surabaya, Indonesia. It was an analytic cross-sectional study. Clinical and demographic details were also collected.
All patients underwent dialysis twice a week for 4 weeks using a commercially available dialyser (type F6-8 HPS polysulfone semi-synthetic, fresenius GMBH, Germany). Each HD session lasted 4 hours.
Blood pressure (BP) was measured using standard mercury sphygmomanometers after participants had been sitting for at least 10 min. Hypertension was defined as blood pressure ≥ 140/90 mmHg at the time of recruitment, history of hypertension before HD, or the use of anti-hypertensive medication to have BP ≤ 130/80 mmHg. Hypertension was considered as an etiology of CKD if a patient had chronic hypertension before HD, and still had hypertension after HD.
The clinical features of HD patients are shown in Table 1. The predominant causes of CKD were hyperten-
Table 1. Clinical features of HD patients.
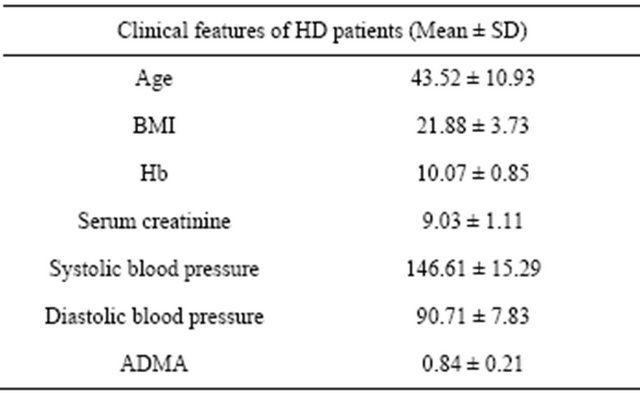
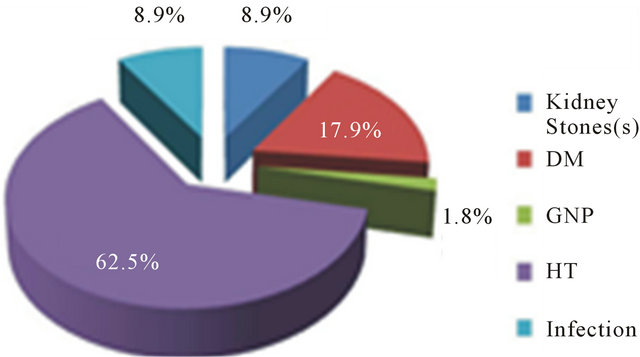
Figure 1. Etiologies of CKD in HD patients.
sion (35 patients, 62,5%) and diabetes (10 patients, 17.9%). Other etiologies such as infection, glomerulonephritis and kidney stone were less than 10% (Figure 1).
2.2. Blood Sampling
Venous blood samples were drawn from the arterialvenous fistula 10 minutes before the first HD session in a week. Peripheral whole blood was obtained from 30 healthy individuals. Blood samples were 3 ml for each participant without additives. Plasma was immediately separated by centrifugation at 800 g for 15 minutes, and then was stored at −20˚C prior to use.
2.3. Determination of ADMA
ADMA was measured using an enzyme-linked immunosorbent assay (DLD Diagnostika, Hamburg, Germany) as previously reported [14]. The plasma HCY concentration was measured using The AxSYM HCY assay (Abbott USA), that is a fluorescence polarization immunoassay in the accordance with the procedure previously published [15,16].
2.4. Genomic DNA
Genomic DNA was extracted from whole blood with a commercially available DNA isolation kit (QIAmp DNA blood Mini kit, Qiagen GmBH, Crawley, West Sussex, UK). Genotyping of the polymorphisms was performed using PCR-based SNP detection methods (Applied Biosystems, Carlsbad, USA) based on 5’-exonuclease activity assays for rs805305 (−449 G/C) [17,18]. Allelic variation was assessed in each participant.
3. Statistics
All variables were examined for normal distribution using the Kolmogorov-Smirnov test. Normally distributed variables were presented as mean ± SD. Pearson correlation was undertaken for associations of clinical covariates with ADMA levels. Distribution differences of gene polymorphism between the groups were analyzed using Mann-Whitney Test. One way Anova and post hoc test were also used to analyze signicant difference between the genotypes within subgroups.
4. Results
4.1. Correlation of ADMA and eGFR in HD Patients
Before performing a parametric correlation test (Pearson), a Kolmogorov-Smirnov test was performed, and the results were normal distribution (ADMA: 0.95; eGFR: 1.05). The Pearson correlation test showed a weak (p = 0.134) non-significant correlation between estimated glomerular filtration rate (eGFR) and ADMA in HD patients.
4.2. Gene Polymorphism Distribution HD Patients and Healthy Individuals
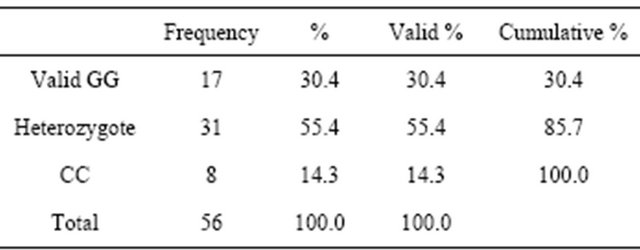
Gene polymorphisms were assesed among healthy individuals and HD patients. Heterozygotes were observed as the most abundant genotypes in both groups (70% in healthy individuals and 55% in HD patients), followed by GG genotype in the HD patients (30%), while CC (27%) were the second most common genotype polymorphism in the healthy individuals (Tables 2 and 3). Using the Mann-Whitney U test, a significant polymorphism distribution between both groups was sought, and the result was 0.006. There was a significant difference in polymorphism distribution among both groups.
4.3. Subgroups within HD Patients
The HD patients were divided into two subgroups. The subgroups were categorized based on the etiology of the CKD in HD patients. One subgroup was a hypertension subgroup (35 of 56 HD patients samples). The other was a non-HT subgroup, a subgroup of HD patients in which the etiologies of CKD were DM, infection, glomerulonephritis and kidney stone (21 of 56 HD patients samples). There was no significant difference in the distribution of DDAH gene polymorphism among subgroups, as the
Table 2. Polymorphism in healthy individuals.

Table 3. Polymorphism in HD patients.
results of Mann-Whitney test was 0.364.
4.3.1. Subgroup Hypertension Analysis
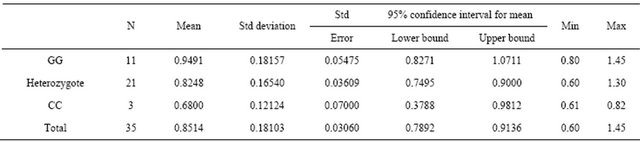
As the most frequent cause of CKD in this study, DDAH gene polymorphisms and the levels of plasma ADMA were analyzed, including as to whether or not there were differences between genotypes. The distribution of polymorphism is shown in Table 4.
The table also reveals that the mean plasma levels of ADMA were sequentially higher from genotypes CC, G/C and GG. These different findings between genotypes were confirmed statistically using a one-way anova test with the results of 0.037. Further multiple comparisons between groups using post hoc test showed results that genotype GG and CC were different at 0.05 level of significance (Table 5).
4.3.2. Subgroup Non-Hypertension Analysis
The gene polymorphism distributions and their correlation with plasma levels of ADMA were also analyzed in non-hypertension HD patients. Compared to the HT subgroup, there was a similar composition of gene polymorphism frequency among Non-HT (Table 6). But, when statistical tests were used to define association between mean plasma levels of ADMA and DDAH gene polymorphism, there were no significant differences found between groups of genotypes; either by using the Anova test (p = 0.322), or using the multiple comparison post hoc test (p > 0.05).
5. Discussion
There are considerable data on the role of ADMA in hemodialysis patients and likewise on the association of DDAH gene polymorphism and ADMA in certain popu-
Table 4. ADMA levels among HT subgroups.
ADMA.
Table 5. Multiple comparison of ADMA levels among DDAH gene polymorphisms.
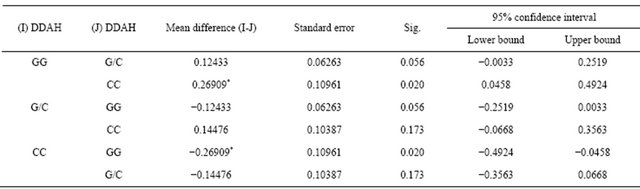
Dependent Variable: ADMA; LSD. *The mean difference is significant at the 0.05 level.
Table 6. ADMA levels among Non-HT & HD patients.
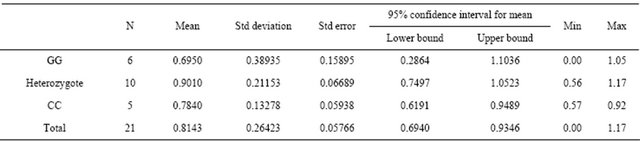
ADMA.
lations. Nonetheless, later studies have not only drawn different conclusions (DDAH isoforms, invitro/invivo, human/animal), but also have found opposite results. In this study, we described the correlation between a novel genetic polymorphism DDAH 2 and ADMA in hypertensive HD patients. The study also revealed a significant distribution difference in DDAH polymorphism between healthy individuals and hemodialysis patients. We believe that this is the first study that has reported such correlations among HD patients.
In the first evaluation, we examined the correlation of estimated glomerular filtration rate (eGFR) and ADMA. There was no significant correlation between eGFR and ADMA. An inverse relationship between plasma ADMA and glomerular filtration rate (GFR) was apparent in patients with coronary artery disease, which has been implicated in their increased cardiovascular risk [19]. However, it has been questioned whether elevated circulating levels of ADMA are merely a marker of kidney disease, coinciding with serum creatinine, or part of the pathophysiological process [20]. Indeed, plasma levels of SDMA correlated more closely with parameters of renal function than did plasma levels of ADMA [21].
ADMA is a naturally-occurring non-selective inhibitor of NOS, and Free ADMA ≥ 80% is actively metabolized by the intracellular enzyme DDAH [11,22]. DDAH is most likely a key determinant of plasma ADMA concentration [14]. Thus, DDAH plays a crucial role in the regulation of NO synthesis via modulating endogenous ADMA levels [23]. Alterations in DDAH activity may effect vascular structure, as well as vascular reactivity [24]. As research has progressed, DDAH has emerged as an important regulator of NO bioavailability and the integrity of renal and vascular function [10].
In higher organisms, including humans, two isoforms of DDAH exist which have distinct tissue distributions but similar enzymatic activity [25]. Based on tissue messenger ribonucleic acid (mRNA) levels, it has been suggested that the expression pattern of DDAH 1 overlaps more closely with the expression pattern of neuronal NOS, whereas DDAH 2 expression has greater similarities with endothelial NOS expression [26,27]. However, it is still unclear which DDAH isoform represents the principal methylarginine-metabolizing enzyme.
Both groups, the HD patients and the healthy individuals, showed a significant difference in the distribution DDAH 2 gene polymorphism. They shared the same most abundant genotype, which was G/C, followed by GG in the HD group and CC in the healthy indivduals group. The polymorphisms were in the non-coding part of the gene, putatively affecting promoter activity, designated DDAH 2 −449. Although our study may not have been adequately powered to detect outcome variations, these findings implied possible functionality polymerphism in the DDAH 2 promoter. As DDAH 2 regulates ADMA levels, it is plausible to think that functional DDAH 2 gene polymorphisms may account for variation in ADMA levels [12]. Data suggest that expression of DDAH 2 may be affected by polymorphisms of the promoter region [17].
For the primary analysis, the HD patients group was divided into a HT subgroup and a Non-HT subgroup. In the subgroup HT, we demonstrated correlations of ADMA with the carriage of the DDAH 2 −449 G allele, where plasma level of ADMA in GG > G/C > CC. Average blood pressure of containing −449 G allele ( GG and G/C) was also higher than that of CC allele. O’Dwyer et al. recently reported that the −449G allele of the DDAH 2 gene was associated with increased ADMA concentrations [13]. Higher prevalence of hypertension with an odds ratio of 1.80 (95%CI: 1.29 - 2.49, p < 0.001) for individuals homozygous for the −449G allele was also reported [18]. In the secondary analysis, we could not find any correlation of ADMA and DDAH 2 gene polymorphism among non-hypertensive HD patients.
Oxidative stress is defined as tissue damage caused by the disequilibrium between pro-oxidants and anti-oxidants [28]. Vascular oxidative stress is a major factor in the pathogenesis of atherosclerosis [29]. One of the main effects of oxidative stress is the decrease in the biological activity of NO [28]. Among the mechanisms for impaired NO synthesis is the accumulation of the endogenous NOS inhibitor asymmetric ADMA [7]. ADMA inhibits all three isoforms of nitric oxide synthase and therefore has the potential to produce diverse biological effects, particularly in the cardiovascular system [26,30]. Some studies of human subjects with essential HT report elevated plasma levels of ADMA [31,32]. Investigations of ADMA concentration by independent methods in patients with renal failure, have identified plasma ADMA concentration as a strong independent predictor of disease progression. Of particular interest was the finding that a 0.1 µmol/L increase in plasma ADMA was associated with a 20% increase in cardiovascular event rate [7,33].
The enzyme DDAH specifically hydrolyzes these asymmetrically methylated arginine residues to citrulline and methylamines [34,35]. Several groups have demonstrated that modulating DDAH activity can have a profound effect on endothelial NO production [7,36,37]. Two isoforms of DDAH have been identified. Since DDAH 2 is the predominant isoform expressed in the cardiovascular system and since hypercholesterolemia impaired endothelial DDAH activity while DDAH 1 protein expression remained unchanged [23,38,39], DDAH 2 gene polymorphism was studied in the present study. The finding that the gene for DDAH is evolutionarily well conserved suggests that the enzyme confers an important survival advantage [10]. And it can be postulated that genetic variations in DDAH that affect the ability of the enzyme to metabolize ADMA might increase cardiovascular risk in patients with renal failure and might also determine, in part, the rate of disease progression [26].
Growing and irresistable epidemiological evidence links ED with cardiorenal syndrome. Recently, a human study, using artery and vein graft suggests that ADMA may be directly implicated in the regulation of the vascular redox state of atherosclerosis by affecting superoxide generation and NO bioavailability [8]. Therefore, any therapeutic modulation of ADMA levels, via genetic modification of DDAH expression or activity, may represent a new strategy for the treatment of cardiovascular disorders, including in HD patients.
6. Conclusions
Genetic variation in the DDAH 2 genes is significantly associated with serum ADMA levels in hypertensive HD patients. We observed that carriage of a G at position −449 in the promoter region of the DDAH 2 gene is associated with higher ADMA levels.
Further studies are required to determine the pathophysiological significance of elevated serum ADMA in HD patients and to better understand how DDAH gene variation influences ADMA levels. And we propose that a polymorphism at position −449 in the DDAH 2 may be functional and that it has the potential to be used as a marker for susceptibility to cardiovascular events among hypertensive hemodialyzed patients.
7. Limitations
This study has some limitations. The number of patients was relatively small, we used only one marker of oxidative stress and we did not evaluate other factors potentially involved in endothelial dysfunction. Furthermore, our study was limited by its cross-sectional design, allowing no causal interferences.
REFERENCES
- M. Fujisawa, R. Haramaki, H. Miyazaki, T. Imaizumi and S. Okuda, “Role of Lipoprotein (a) and TGF-β1 in Atherosclerosis of Hemodialysis Patients,” Journal of the American Society of Nephrology, Vol. 11, No. 10, 2000, pp. 1889-1895.
- A. Lindner, B. Charra, D. Sherrard and B. Scribner, “Accelerated Atherosclerosis in Prolonged Maintenance Hemodialysis,” New England Journal of Medicine, Vol. 290, No. 13, 1974, pp. 697-701.
- K. Ma, E. Greene and L. Raij, “Cardiovascular Risk Factors in Chronic Renal Failure and Hemodialysis Populations,” American Journal of Kidney Diseases, Vol. 19, No. 6, 1992, pp. 505-513.
- J. Cooke and V. Dzau, “Derangements of the Nitric Oxide Synthase Pathway, L-Arginine, and Cardiovascular Diseases,” Circulation, Vol. 96, No. 2, 1997, pp. 379-382.
- A. Ito, P. S. Tsao, S. Adimoolam, M. Kimoto, T. Ogawa and J. P. Cooke, “Novel Mechanism for Endothelial Dysfunction: Dysregulation of Dimethylarginine Dimethylaminohydrolase,” Circulation, Vol. 99, No. 24, 1999, pp. 3092-3095.
- R. M. J. Palmer, A. G. Ferrige and S. Moncada, “Nitric Oxide Release Accounts for the Biological Activity of Endothelium-Derived Relaxing Factor,” Nature, Vol. 327, 1987, pp. 524-526. doi:10.1038/327524a0
- A. J. Pope, K. Karrupiah, P. N. Kearns, Y. Xia and A. J. Cardounel, “Role of Dimethylarginine Dimethylaminohydrolases in the Regulation of Endothelial Nitric Oxide Production,” Journal of Biological Chemistry, Vol. 284, No. 51, 2009, pp. 35338-35347.
- C. Antoniades, C. Shirodaria, P. Leeson, A. Antonopoulos, N. Warrick, T. Van-Assche, et al., “Association of Plasma Asymmetrical Dimethylarginine (ADMA) with Elevated Vascular Superoxide Production and Endothelial Nitric Oxide Synthase Uncoupling: Implications for Endothelial Function in Human Atherosclerosis,” European Heart Journal, Vol. 30, No. 9, 2009, pp. 1142-1150.
- R. Maas, K. Quitzau, E. Schwedhelm, L. Spieker, W. Rafflenbeul, A. Steenpass, et al., “Asymmetrical Dimethylarginine (ADMA) and Coronary Endothelial Function in Patients with Coronary Artery Disease and Mild Hypercholesterolemia,” Atherosclerosis, Vol. 191, No. 1, 2007, pp. 211-219.
- F. Palm, M. L. Onozato, Z. Luo and C. S. Wilcox, “Dimethylarginine Dimethylaminohydrolase (DDAH): Expression, Regulation, and Function in the Cardiovascular and Renal Systems,” Heart and Circulatory Physiology: American Journal of Physiology, Vol. 293, No. 6, 2007, pp. H3227-H3245.
- C. Tran, J. Leiper and P. Vallance, “The DDAH/ADMA/ NOS Pathway,” Atherosclerosis Supplements, Vol. 4, No. 4, 2003, pp. 33-40.
- R. Ryan, J. Thornton, E. Duggan, E. McGovern, M. J. O’Dwyer, A. W. Ryan, et al., “Gene Polymorphism and Requirement for Vasopressor Infusion after Cardiac Surgery,” Annals of Thoracic Surgery, Vol. 82, No. 3, 2006, pp. 895-901.
- M. O’Dwyer, F. Dempsey, V. Crowley, D. Kelleher, R. McManus and T. Ryan, “Septic Shock Is Correlated with Asymmetrical Dimethyl Arginine Levels, Which May Be Influenced by a Polymorphism in the Dimethylarginine Dimethylaminohydrolase II Gene: A Prospective Observational Study,” Critical Care, Vol. 10, No. 5, 2006, p. R139.
- R. Maas, “Pharmacotherapies and Their Influence on Asymmetric Dimethylargine (ADMA),” Vascular Medicine, Vol. 1, Suppl. 1, 2005, pp. S49-S57.
- M. Thaha, Widodo, M. Yogiantoro and Y. Tomino, “Intravenous N-Acetylcysteine during Hemodialysis Reduces Asymmetric Dimethylarginine Levels in End-Stage Renal Disease Patients,” Clinical Nephrology, Vol. 69, No. 1, 2008, pp. 24-32.
- M. Thaha, M. Yogiantoro and Y. Tomino, “Intravenous N-Acetylcysteine during Haemodialysis Reduces the Plasma Concentration of Homocysteine in Patients with End-Stage Renal Disease,” Clinical Drug Investigation, Vol. 26, 2006, pp. 195-202.
- L. Jones, C. Tran, J. Leiper, A. Hingorani and P. Vallance, “Common Genetic Variation in a Basal Promoter Element Alters DDAH 2 Expression in Endothelial Cells,” Biochemical and Biophysical Research Communications, Vol. 310, No. 3, 2003, pp. 836-843.
- R. Maas, J. Erdmann, N. Luneburg, J. Stritzke, E. Schwedhelm, C. Meisinger, et al., “Polymorphisms in the Promoter Region of the Dimethylarginine Dimethylaminohydrolase 2 Gene Are Associated with Prevalence of Hypertension,” Pharmacological Research, Vol. 60, No. 6, 2009, pp. 488-493.
- J. Wang, A. Sim, X. Wang, C. Salonikas, D. Naidoo and D. Wilcken, “Relations between Plasma Asymmetric Dimethylarginine (ADMA) and Risk Factors for Coronary Disease,” Atherosclerosis, Vol. 184, No. 2, 2006, pp. 383-388.
- P. Vallance and J. Leiper, “Asymmetric Dimethylarginine and Kidney Disease—Marker or Mediator?” Journal of the American Society of Nephrology, Vol. 16, No. 8, 2005, pp. 2254-2256.
- J. T. Kielstein, S. R. Salpeter, S. M. Bode-Boeger, J. P. Cooke and D. Fliser, “Symmetric Dimethylarginine (SDMA) as Endogenous Marker of Renal Function—A Meta-Analysis,” Nephrology Dialysis Transplantation, Vol. 21, No. 9, 2006, pp. 2446-2451.
- V. Achan, M. Broadhead, M. Malaki, G. Whitley, J. Leiper, R. MacAllister, et al., “Asymmetric Dimethylarginine Causes Hypertension and Cardiac Dysfunction in Humans and Is Actively Metabolized by Dimethylarginine Dimethylaminohydrolase,” Arteriosclerosis, Thrombosis, and Vascular Biology, Vol. 23, No. 8, 2003, pp. 1455-1459.
- M. Feng, L. Liu, Z. Guo and Y. Xiong, “Gene Transfer of Dimethylarginine Dimethylaminohydrolase-2 Improves the Impairments of DDAH/ADMA/NOS/NO Pathway in Endothelial Cells Induced by Lysophosphatidylcholine,” European Journal of Pharmacology, Vol. 584, No. 1, 2008, pp. 49-56.
- H. Dayoub, V. Achan, S. Adimoolam, J. Jacobi, M. C. Stuehlinger, B.-Y. Wang, et al., “Dimethylarginine Dimethylaminohydrolase Regulates Nitric Oxide Synthesis,” Circulation, Vol. 108, No. 24, 2003, pp. 3042-3047.
- J. Leiper, J. Santa Maria, A. Chubb, R. MacAllister, I. Charles, G. Whitley, et al., “Identification of Two Human Dimethylarginine Dimethylaminohydrolases with Distinct Tissue Distributions and Homology with Microbial Arginine Deiminases,” Biochemical Journal, Vol. 341, 1999, pp. 209-214. doi:10.1042/0264-6021:3430209
- J. Leiper and M. Nandi, “The Therapeutic Potential of Targeting Endogenous Inhibitors of Nitric Oxide Synthesis,” Nature Reviews Drug Discovery, Vol. 10, No. 4, 2011, pp. 277-291.
- A. Tojo, W. Welch, V. Bremer, M. Kimoto, K. Kimura, M. Omata, et al., “Colocalization of Demethylating Enzymes and NOS and Functional Effects of Methylarginines in Rat Kidney,” Kidney International, Vol. 52, No. 6, 1997, pp. 1593-1601.
- M. E. Widlansky, N. Gokce, J. F. Keaney Jr. and J. A. Vita, “The Clinical Implications of Endothelial Dysfunction,” Journal of the American College of Cardiology, Vol. 42, No. 7, 2003, pp. 1149-1160.
- N. Dhalla, R. Temsah and T. Netticadan, “Role of Oxidative Stress in Cardiovascular Diseases,” Journal of Hypertension, Vol. 18, No. 6, 2000, pp. 655-673.
- C. Tran, M. Fox, P. Vallance and J. Leiper, “Chromosomal Localization, Gene Structure, and Expression Pattern of DDAH 1: Comparison with DDAH 2 and Implications for Evolutionary Origins,” Genomics, Vol. 68, No. 1, 2000, pp. 101-105.
- A. Ito, K. Egashira, T. Narishige, K. Muramatsu and A. Takeshita, “Renin-Angiotensin System Is Involved in the Mechanism of Increased Serum Asymmetric Dimethylarginine in Essential Hypertension,” Japanese Circulation Journal, Vol. 65, No. 9, 2001, pp. 775-778.
- F. Perticone, A. Sciacqua, R. Maio, M. Perticone, R. Maas, R. H. Boger, et al., “Asymmetric Dimethylarginine, L-Arginine, and Endothelial Dysfunction in Essential Hypertension,” Journal of the American College of Cardiology, Vol. 46, No. 3, 2005, pp. 518-523.
- J. Leiper and P. Vallance, “New Tricks from an Old Dog : Nitric Oxide-Independent Effects of Dimethylarginine Dimethylaminohydrolase,” Arteriosclerosis, Thrombosis, and Vascular Biology, Vol. 26, No. 7, 2006, pp. 1419- 1420.
- T. Ogawa, M. Kimoto and K. Sasaoka, “Purification and Properties of a New Enzyme, NG,NG-Dimethylarginine Dimethylaminohydrolase, from Rat Kidney,” Journal of Biological Chemistry, Vol. 264, No. 17, 1989, pp. 10205- 10209.
- T. Teerlink, “ADMA Metabolism and Clearance,” Vascular Medicine, Vol. 1, Suppl. 1, 2005, pp. S73-S81.
- J. Jacobi, K. Sydow, G. von Degenfeld, Y. Zhang, H. Dayoub, B. Wang, et al., “Overexpression of Dimethylarginine Dimethylaminohydrolase Reduces Tissue Asymmetric Dimethylarginine Levels and Enhances Angiogenesis,” Circulation, Vol. 111, No. 11, 2005, pp. 1431- 1438.
- C. Smith, G. Birdsey, S. Anthony, F. Arrigoni, J. Leiper and P. Vallance, “Dimethylarginine Dimethylaminohydrolase Activity Modulates ADMA Levels, VEGF Expression, and Cell Phenotype,” Biochemical and Biophysical Research Communications, Vol. 308, No. 4, 2003, pp. 984-989.
- V. Achan, C. T. L. Tran, F. Arrigoni, G. S. J. Whitley, J. M. Leiper and P. Vallance, “All-Trans-Retinoic Acid Increases Nitric Oxide Synthesis by Endothelial Cells,” Circulation Research, Vol. 90, No. 7, 2002, pp. 764-769.
- R. H. Böger, S. M. Bode-Böger, K. Sydow, D. D. Heistad and S. R. Lentz, “Plasma Concentration of Asymmetric Dimethylarginine, an Endogenous Inhibitor of Nitric Oxide Synthase, Is Elevated in Monkeys with Hyperhomocyst(e)inemia or Hypercholesterolemia,” Arteriosclerosis, Thrombosis, and Vascular Biology, Vol. 20, No. 6, 2000, pp. 1557-1564.
NOTES
*Potential conflict of interest: No potential conflict of interest relevant to this article was reported.