Advances in Biological Chemistry
Vol. 2 No. 2 (2012) , Article ID: 18980 , 15 pages DOI:10.4236/abc.2012.22022
Recent progress and concerns in dementia: Distinguishing Alzheimer’s disease and dementia with Lewy bodies via biochemical markers in the cerebrospinal fluid
![]()
1The Division of Neuroscience, Oregon National Primate Research Center, Oregon Health and Science University, Beaverton, USA
2The Department of Public Health and Preventive Medicine, The Knight Cancer Institute, Oregon Health and Science University, Portland, USA
Email: maop@ohsu.edu
Received 16 March 2012; revised 19 April 2012; accepted 25 April 2012
Keywords: Dementia; Alzheimer’s dementia; Dementia with Lewy body; Biomarker; Oxidative stress; Differential diagnosis
ABSTRACT
Dementia is mainly a neurodegenerative disorder involved in several systems, including central nervous system, endocrinology/metabolism system and circulatory system. Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB) are the most common forms of the dementia, accounting for 60% - 80% and 10% - 20% of all cases, respectively. DLB is defined by widespread neocortical, limbic and brainstem Lewy bodies but frequently accompanied by variable levels of AD pathology. This pathological and clinical overlap makes their differential diagnosis complicated. Recent advances in the identification of disease biomarkers now make it possible to detect and distinguish their pathology in the early or preclinical stage of the diseases, even in cognitively normal individuals. In addition to the key biomarkers (amyloid β or Aβ, tau and α-synuclein), neurotrophins such as cocaineand amphetamine-regulated transcript (CART) have also drawn attention due to their expressions and functions. This article summarizes the progress in the definition, pathology and diagnosis of dementia, with a focus on potential biochemical markers in the cerebrospinal fluid (CSF) in the differential diagnosis of the main forms of dementia. To prediction or early diagnosis of dementia, the role of specific and sensitive CSF biomarkers seems to be crucial in a routine clinical setting. The concerns and challenges in the biomarker field are also discussed.
1. INTRODUCTION
Dementia is a multi-system related neurodegenerative disorder. There are several forms of dementia, including mainly Alzheimer’s dementia, dementia with Lewy bodies (DLB), corticobasal degeneration/dementia (CBD), frontotemporal dementias (FTD) known also as frontotemporal lobar degenerations (FTLD), vascular dementia (VAD) and prion diseases or Creutzfeldt-Jakob disease (CJD) [1,2].
Dementia with Lewy bodies (DLB) is the second most frequent cause of hospitalization for dementia, after Alzheimer’s disease (AD), characterized by a dysexecutivevisuoperceptual dementia frequently accompanied by visual hallucinations, fluctuating attention, and Parkinsonism. Strategic α-synuclein aggregates, neuronal loss, and variable degrees of amyloid deposition constitute the key pathological features [1,3-5]. Therefore, these pathological features overlap with AD, and make their differential diagnosis difficult. The disease progression and treatment of DLB are different from AD hence the differential diagnosis is a basic procedure in clinical practice.
In addition, cumulative oxidative stress, disrupted mitochondrial respiration, and mitochondrial damage are involved in various neurodegenerative disorders, including AD and DLB [6-8]. With increased longevity and increased prevalence of obesity, diabetes, hypertension and other chronic diseases, dementia is likely to raise significantly [9,10]. Aging related functional and structural declines in multiple systems, especially mitochondria dysfunction and less blood supply in the brain may be the fundamental events for this complex disorder. CART is one of the latest hormones, expressed in the nervous system and circulatory system with multiple functions. We recently found that CART is a mitochondrial booster and a novel endogenous antioxidant [11,12], suggesting its potential relationship with neurodegenerative diseases.
Disease-related biochemical markers can provide diagnostic, prognostic and therapeutic targets, and it also can address some etiological questions and important problems in clinical and translational medical research. In this article, I attempt to highlight the progress in pathology and diagnosis of dementia, with a focus on the cerebrospinal fluid (CSF) biomarkers in the main forms of dementia, AD and DLB, including their possible correlations with CART peptide. This review provides valuable information that may help to identify new clues and targets for the etiology, diagnosis, prevention and treatment of dementia.
2. RECENT PROGRESS IN DEMENTIA
2.1. Definition of Dementia
According the DSM-IIIR (the Diagnostic and Statistical Manual, 3rd edition, revised) states, the essential feature of dementia is impairment in shortand long-term memory, associated with impairment in abstract thinking, impaired judgment, other disturbances of higher cortical function, or personality change. The disturbance is severe enough to interfere significantly with work or usual social activities or relationships with others. The diagnosis of dementia is not made if these symptoms occur in delirium. The DSM-IIIR definition of dementia, which was identical to the DSM-IV definition, was reliable and used routinely as guideline [1].
Recently, National Institute on Aging-Alzheimer’s Association (NIA-AA) proposed new, detailed guidelines for dementia [13]. According the new guidelines, dementia is diagnosed when there are cognitive or behavioral (neuropsychiatric) symptoms that are: 1) Interfere with the ability to function at work or at usual activities; and 2) Represent a decline from previous levels of functioning and performing; and 3) Are not explained by delirium or major psychiatric disorder; 4) Cognitive impairment is detected and diagnosed through a combination of a) history-taking from the patient and a knowledgeable informant and b) an objective cognitive assessment, either a “bedside” mental status examination or neuropsychological testing. The cognitive or behavioral impairment involves a minimum of two of the following domains: 1) Impaired ability to acquire and remember new information; 2) Impaired reasoning and handling of complex tasks, poor judgment; 3) Impaired visuospatial abilities; 4) Impaired language functions (speaking, reading, writing); 5) Changes in personality, behavior, or comportment.
Among all forms of dementia, Alzheimer’s dementia is the most common dementia, accounting for an estimated 60% - 80% of recorded cases; and dementia with lewy bodies is accounting for 10% - 20% of the total number of recorded cases of dementia [14-16]. Therefore, this review will focus on these two forms of dementia.
2.2. New Definition and Pathology of Alzheimer’s Dementia
AD is a fast growing world-wide epidemic and the most common neurodegenerative disease, characterized by slowly progressive cognitive deterioration, together with declining activities of daily living and by neuropsychiatric symptoms or behavioral changes. The most striking early symptom is memory loss (amnesia), usually manifest as minor forgetfulness that becomes steadily denser with illness progression, with relative preservation of older memories. The cause of Alzheimer’s is currently unknown. First, genetic factors are clearly indicated as evidenced by dominant mutations in different genes (such as the gene encoding the amyloid precursor protein APP, genes encoding presenilins PS1 and PS2). However, it only accounts for a small number of cases of AD, the familial, early-onset AD. Other gene mutations or variations (such as ApoE) may increase the risk of the most common form of AD, the late onset AD (LOAD) or sporadic AD affecting people usually over the age of 65. The etiology of LOAD is much complicated, and age has a significant impact on its development. In pathology, extracellular fibrillar amyloid-β (Aβ), especially long form 42 amino acids of Aβ (Aβ42) deposits in the brain [8,17], intracellular neurofibrillary tangles (NFT) and neuronal as well as axonal degeneration are the main features of the disease.
Even fairly accurate, the clinical diagnosis of probable AD based on traditional diagnostic criteria does not take the long preclinical and prodromal course of AD into account. In fact, AD-related pathophysiological changes occur several years and even decades before the onset of dementia. Importantly, the latest criteria proposed last year and published at early this year for the neuropathologic assessment of AD differ from the traditional criteria, which require a history of dementia [13]. From the clinical perspective, the concept of AD has evolved to cover patients with milder symptoms, including the proposition that there is a preclinical phase of the illness. Indeed, substantial evidence exists to indicate that the pathophysiological processes of AD are present in the brain well in advance of subjective or objective deficits. Therefore, recent NIA-AA-sponsored consensus reports on three defined stages in a clinical continuum that includes preclinical, mild cognitive impairment, and dementia. It refers to the presence and extent of neuropathologic changes of AD observed at autopsy, regardless of the clinical setting [13].
The clinical loss of learning and memory abilities is mainly due to a significant decrease in brain cholinergic activity, and marked glutamatergic excitotoxicity that results in neuronal death [18-20]. The cholinergic neurotoxicity of Aβ and the regulatory role of cholinergic receptor in the processing of APP have been summarized recently [21-23].
Since the important roles of mitochondria in cellular energy production, metabolism and cell death, it is considered to be the center of some degenerative diseases [24,25]. Recent progress in AD, especially Aβ, mitochondria and oxidative stress related pathology has been reviewed [8,26]. The role of the microtubule-associated protein tau, another hallmark protein, in the pathogenesis of Alzheimer’s disease, including tau as a crucial partner of Aβ, as well as their interactions with mitochondria, has also been summarized recently [27,28]. Notably, oxidative damage is thought to be the earliest event in Alzheimer’s disease [8,29]. Aβ directly induces reactive oxygen species (ROS) [8], and toxic soluble Aβ oligomers appear to have synaptic receptors colocalizing with PSD-95 (postsynaptic density protein 95), and Aβ42 accumulates in dendrites in AD patients where it may cause oxidative damage and caspase activation and eventually apoptosis [30]. On the other hand, mitochondrial dysfunction and mitochondria-derived ROS lead to enhanced Aβ formation [8,26,31]. Therefore, a vicious cycle starting either from mitochondrial dysfunction or Aβ toxicity is triggered that contributes to the pathogenesis of the most AD and the mitochondrial cascade hypothesis is increasingly recognized, especially for the most common form, later onset of AD.
Interestingly, AD has been considered to be a neuroendocrine disorder, even referred to by some as type III diabetes [32-34], indicating the importance of energy metabolism and insulin signaling in the development of Alzheimer’s disease. Metabolic syndrome, prediabetes, and type II diabetes are putative risk factors in the development of AD. More importantly diabetes and AD have signs of increased oxidative stress in common. For example, neurotoxins, coined amyloid β-derived diffusible ligands (ADDLs), disrupt signal transduction at synapses, contribute to oxidative damage, and cause AD-type tau hyperphosphorylation [32,34]. Furthermore, Aβ and human amylin, the diabetes pathogen, share a common toxicity pathway through mitochondrial dysfunction [35].
In addition, Alzheimer and vascular pathology can exist together more often than in isolation, in fact, autopsy data show that 60% - 90% of AD cases exhibit variable cerebrovascular pathology, hence cerebral ischemia is probably a causal factor for AD [36,37]. Furthermore, aging is thought to be the single most important risk factor for AD, and available data indicate that memory disturbances in normal elderly and early AD patients are intimately related to hypoxia, a reduction in blood supply, and glucose hypometabolism in the hippocampus and a number of key brain areas [38-40]. Notably, Aβ is generated from APP through sequential cleavages by BACE1 (the major β-secretase in the brain) and γ-secretase, the hypoxia condition increases Aβ production via the limiting enzyme BACE1 and γ-secretase, furthermore hypoxia stimulates ROS production, especially the radicals came from mitochondrial complex II [8,41,42].
Biomarkers that are related to the pathophysiology of AD may thus help detect the preclinical stages of disease, and improve early and differential diagnosis. Thus, core AD biomarkers, Aβ peptides (especially Aβ42) and phosphor-tau (p-tau) in CSF, PET and MRI imaging are considered to be the most useful approaches on their diagnostic and predictive value in preclinical and clinical stages of AD [8,43-48]. In addition, plasma biomarkers for AD are currently controversial, and CSF is the most economically effective approach and the main topic of this article.
2.3. Definition and Pathology of Dementia with Lewy Bodies
DLB has been defined clinically by the presence of dementia, gait/balance disorder, prominent hallucinations and delusions, sensitivity to traditional antipsychotics, and fluctuations in alertness [49]. The main points of this guideline were followed by recent workshops [13,15].
Filamentous protein inclusions in neurons (Lewy bodies, LB) and dystrophic neuritis (Lewy neuritis, LN) containing pathologic alpha (α)-synuclein (aSyn) are the morphologic hallmarks of dementia with Lewy bodies and sporadic Parkinson disease (PD). Their formation runs through several phases from initial dust-like particles cross-linked with aSyn to aggregation of ubiquitinated dense filaments, formation of LBs, in which aSyn is the major component and hundreds of proteins may be included [50], finally degradation and death of the afflicted neurons [4]. Studies from biochemistry and biophysics have suggested that the oligomeric, protofibrillary aSyn is cytotoxic [51,52], whereas LB and LN composed of fibrillary aSyn, which are typically observed in the brain at autopsy. In post-mortem cortical tissue, significant reduction of the a-subunit of the 20S proteasome was observed in DLB, but not AD, in comparison to controls, and elevated levels of soluble aSyn oligomers in post-mortem brain extracts from DLB patients suggest that these aSyn oligomers and the proteasome are involved in the pathogenesis of DLB [53].
Interestingly, the ubiquitin ligase Nedd4, which functions in the endosomal-lysosomal pathway, robustly ubiquitinates aSyn and purified Nedd4 recognizes the carboxyl terminus of aSyn and attaches ubiquitin chains. Overexpression of Nedd4 enhances aSyn ubiquitination and clearance by a lysosomal process in human cells. In human brains, Nedd4 is present in pigmented neurons and is expressed especially strongly in neurons containing Lewy bodies. Thus, ubiquitination by Nedd4 targets aSyn to the endosomal-lysosomal pathway and, by reducing α-Syn content, may help reduce α-synucleinopathies [54].
Marked cholinergic deficits accompanied by inclusions positive for aSyn and Ubi suggest that proteasomal abnormalities are present in cortical LBD [55]. On the other hand, serine 129 phosphorylation of aSyn as its dominant modification in LB disorders [56] induces unfolded protein response-mediated cell death [57,58]. While the duration and severity of motor function, the corresponding decrease of dopamine, tyrosine hydroxylase (TH), dopamine transporter (DAT), and vesicular monoamine transporter-2 (VMAT-2) immunoreactivity in the striatum are negatively correlated with total substantia nigra (SN) aSyn burden and neuronal loss [4,59].
In addition, not just in SN, oxidative damage, particularly lipoxidation, advanced glycation (AGE), and AGE receptors (RAGE) in other structures, including the cerebral cortex, also been seen in early stages of diseases with Lewy bodies [60]. Moreover, it demonstrates that aSyn lipoxidation is an early event in LBDs which precedes aSyn solubility modification and aggregation, and formation of Lewy bodies and neuritis [61]. It also been found that the human brain cortex, mitochondrial oxygen uptake and complex I activity were significantly lower in DLB, comparing to healthy controls, whereas oxidative damage and mtNOS activity, cytochrome content, expression of Mn-SOD and mitochondrial mass were significantly higher in the frontal cortex in DLB [62]. The decreases in tissue and mitochondrial oxygen uptake and complex I activity are considered the consequences of mitochondrial oxidative damage and mitochondrial dysfunction. The increases in mtNOS activity and mitochondrial mass are interpreted as an adaptive response of the frontal cortex that involves increased NO signaling for mitochondrial biogenesis. The adaptive response would partially compensate for mitochondrial dysfunction in these neurodegenerative diseases and would afford a human evolutionary response to shortage of ATP in the frontal cortex. This compensation also can be found in the brain of aged non-human primates [63].
Telomeres, the ends of linear chromosomes, repetitive DNA regions of hexanucleotide repeats, protect chromosomal ends from deterioration during physiological and pathological conditions. Telomere shortening represents a cell-intrinsic mechanism leading to DNA damage accumulation in aged cells [8]. In fact, telomere length was significantly shorter in both Alzheimer’s disease and DLB than in the non-demented elderly controls [64], indicating an increased oxidative stress in patients with dementia.
Heavy metals have been implicated as the causative agents for the pathogenesis of the most prevalent neurodegenerative diseases. Various mechanisms have been proposed to explain the toxic effects of metals ranging from metal-induced oxidation of protein to metal-induced changes in the protein conformation. Aggregation of aSyn is a key mechanism in PD and DLB, and various metals, including copper, constitute a prominent group of aSyn aggregation enhancers. The aSyn-Cu21 binding sites have been systematically characterized, and the possible role of metal binding in aSyn fibrillation has been analyzed using biophysical techniques [65].
On the other hand, lipoxidative/protein damage of aldolase A, enolase 1, and glyceraldehyde dehydrogenase (GAPDH) was found in the frontal cortex in the majority of cases of incidental Parkinson’s disease (iPD), PD, and dementia with LB. Densitometric studies have shown that the ratio of oxidized protein per spot is higher in iPD, PD, and DLB than controls. These findings show oxidation of three enzymes linked with glycolysis and energy metabolism in the adult human brain as well as increased oxidation of aldolase A, enolase 1, and GAPDH in the frontal cortex in Lewy body diseases [66].
Taken together, these findings further support the idea that oxidative stress is involved in the pathological process of neurodegenerative diseases, and oxidative stressassociated biomarkers may be useful for the disease diagnosis, as well as antioxidant therapy is reasonable treatment for neurodegenerative diseases [67].
2.4. Classification of Dementia with Lewy Bodies
Pathologic accumulation of aSyn/LBs and classification of DLB have been proposed by Braak et al. [68], with lesions from medullary and olfactory nuclei to the cortex. Further, the consensus pathologic guidelines of DLB [15], by semiquantitative scoring to aSyn pathology (LB density and distribution) in different brain regions, distinguish three phenotypes (brainstem, transitional/limbic, and diffuse neocortical), and also consider concomitant Alzheimer-related pathology.
Recently proposed modified criteria added amygdala as one of predominant regions since aSyn pathology in the amygdala is often associated with Alzheimer disease [4,69]. Furthermore, based on examination of a standard set of 10 brain regions in a large number of autopsy cases, a unified staging system for LB disorders has been reported [70] that suggested four stages of LB pathology: (I) olfactory bulb only; (IIa) brainstem predominant; (IIb) limbic predominant; (III) brainstem and limbic; (IV) neocortical. Progression of these stages was correlated with nigrostriatal degeneration, cognitive impairment and motor dysfunction. This proposed staging system was suggested to improve classification of LB disorders [4,70]. NIA-AA recently recommends that LBD (Lewy body diseases, including Parkinson disease and dementia with Lewy bodies) be classified as no LBs, brainstem predominant, limbic (transitional), neocortical (diffuse), or amygdala predominant [13,71].
Although Braak notions and revised criteria present the essential basis for further developments, current staging conceptualizations seem inadequate to comply with the progress on the field [72]. For example, the contributions of various biomarkers of disease progression, such as Aβ, and neurotransmitter system imbalances, e.g., dopamine receptor supersensitivity and interactive propensities, as well as CART, await their incorporation into the existing staging models thereby underlining the ongoing, dynamic feature of the staging of the brain disorder, it is probably more valuable for the early diagnosis.
3. PROGRESS IN CSF BIOCHEMICAL MARKERS IN DEMENTIA
Physiological processes and pathological changes in the brain can be monitored by analysis of cerebrospinal fluid (CSF). It has been shown that CSF markers reflected neuropathology in 94% [2]. Since about 20 years ago, there has been intense interest in developing markers related to the neuropathology of AD in CSF. Several key CSF biomarkers of pathological processes in the brain are now available. CSF levels of total tau (t-tau) reflect cortical axonal degeneration, levels of phosphorylated tau (p-tau) reflect tangle pathology and levels of Aβ42 reflect brain amyloid pathology [73]. Most reports focused on these Aβ peptides and tau, showing mainly CSF Aβ42 is reduced, but CSF t-tau and p-tau are increased in AD patients.
3.1. CSF Aβ42 and Tau in AD and Mild Cognitive Impairment
Aβ42 is the most toxic amyloid peptide, and the key component of amyloid plaques [8,17]. Reduced levels of Aβ42 in CSF of patients with AD compared with normal elderly controls have been observed repeatedly. Using post-hoc cut-points, moderate sensitivities (78% to 92%) and specificities (81% to 83%) have been achieved in distinguishing patients with AD from normal elderly controls [1]. CSF tau level is also useful in supporting the diagnosis of AD early in the course of dementia [74,75]. CSF tau level was shown to be significantly elevated in patients with AD compared with normal controls [76,77]. However, tau alone may lack diagnosis power since elevated CSF tau level has also been detected in patients with other neurodegenerative diseases [73,78]. The diagnostic yield may be improved by the simultaneous measurement of CSF Aβ42 and tau.
Interestingly, it is going clear that CSF levels of Aβ42 retain diagnostic usefulness in patients with very mild AD and even mild cognitive impairment (MCI), and a combination of CSF t-tau and Aβ42 at baseline yielded a sensitivity of ~95% and a specificity of ~83% for detection of incipient AD in patients with MCI [75,79-81]. Recently, a large-scale multicenter study also showed that CSF Aβ42, t-tau, and p-tau identify incipient AD with good accuracy, even less accurately than reported from single-center studies [82].
Subjects with MCI are at a high risk of developing clinical AD. The levels of CSF tau were increased, whereas levels of Aβ42 were decreased in MCI subjects. In a multiple Cox regression analysis within the MCI group, low baseline CSF levels of Aβ42, but not other predictor variables (tau protein, gender, age, apolipoprotein E (APOE) epsilon4 carrier status, Mini Mental Status Examination score, observation time, antidementia therapy), correlated with conversion status (P < 0.05) [79], indicating the fundamental role of Aβ42 in the pathology of AD.
In other report, the relative risk of progression to Alzheimer’s disease was substantially increased in patients with MCI who had pathological concentrations of t-tau and Aβ42 at baseline (hazard ratio 17.7, p < 0.0001). The association between pathological CSF and progression to Alzheimer's disease was much stronger than, and independent of, established risk factors including age, sex, education, APOE genotype, and plasma homocysteine. The combination of t-tau and Aβ42/p-tau181 ratio yielded closely similar results (sensitivity 95%, specificity 87%, hazard ratio 19.8). Interpreted concentrations of t-tau, p-tau181, and Aβ42 in CSF are strongly associated with future development of Alzheimer's disease in patients with MCI [80]. Importantly, CSF tau/Aβ42 ratios show strong promise as antecedent (preclinical) biomarkers that predict future dementia in cognitively normal older adults [81]. This AD-pattern in CSF was also already detectable in patients with MCI in a new approach, called capillary-electrophoresis coupled to massspectrometry (CE-MS) [83].
Such changes in CSF biomarker levels over time in healthy older adults are associated with a concurrent decline in cognitive performance [84]. The latest report showed that approximately 90% of patients with MCI and pathologic CSF biomarker levels at baseline develop AD within 9 to 10 years. Levels of Aβ42 are already fully decreased at least 5 to 10 years before conversion to AD type dementia, whereas t-tau and p-tau seem to be later markers. These results provide direct evidence supporting the view that altered Aβ metabolism precedes tau-related pathology and neuronal degeneration [85].
Similarly, in a routine clinical setting with 4-year follow-up, at the end of the follow-up, 31 MCI patients (56%) did not progress to dementia (stable MCI), while 24 (44%) developed a dementia condition. At baseline, the mean levels of CSF Aβ42, t-tau, and p-tau were significantly altered in MCI patients who were converting to dementia with respect to those with stable MCI. All MCI patients with the three altered CSF biomarkers developed dementia within 1 year. Among the stable MCI patients, none showed all three pathological values [86]. These data clearly indicate the complexity of the pathology of dementia. Therefore, early diagnosis of dementia and, specifically, a correct prediction of MCI outcome represent a primary goal for prevent and treatment of dementia. To this respect, the role of pathological-associated CSF biomarkers seems to be crucial in a routine clinical setting.
On the other hand, oligomers of the Aβ peptide are more toxic than monomers and are linked to the development of AD. Santos et al. used a flow cytometric approach for the detection and assessment of oligomers in CSF from AD patients and other neurological disorders. CSF Aβ-oligomer levels in AD patients showed an elevation in trend (p = 0.073) and the ratio Aβ-oligomers/ Aβ42 was significantly elevated compared to non-AD subjects (p = 0.001). Most important, there was a negative correlation between the amount of Aβ-oligomers and the Mini-Mental Status Exam score (r = −0.65; p = 0.013) in AD patients. The detection of Aβ-oligomers using flow cytometry analysis seems to be useful in assessing the stage of AD. This is a novel and important finding as none of the currently used CSF biomarkers are clearly associated with dementia severity [87]. The baseline of the oligomers in the healthy adults should be established for further comparison.
Further, a novel misfolded protein assay for the detection of soluble oligomers composed of Aβ x-40 and x-42 peptide from CSF has been developed. Preliminary validation of this assay with 36 clinical samples demonstrated the presence of aggregated Aβx-40 in the CSF of AD patients. Together with measurements of total Aβ42, diagnostic sensitivity and specificity were greater than 95% and 90%, respectively. Although larger sample populations are needed to confirm this diagnostic sensitivity, these studies demonstrate a sensitive method of detecting circulating Aβx-40 oligomers from AD CSF and suggest that these oligomers could be a powerful new biomarker for the early detection of AD [88].
Putative core markers are summarized in the Table 1. Briefly Aβ42 as the earliest biomarker is significantly decreased in AD as well as MCI, while tau and p-tau are significantly increased in AD, comparing to healthy control adults.
3.2. Other Biomarkers for AD and MCI
No other biomarkers that had been extensively studied and had promising detection abilities for AD or other
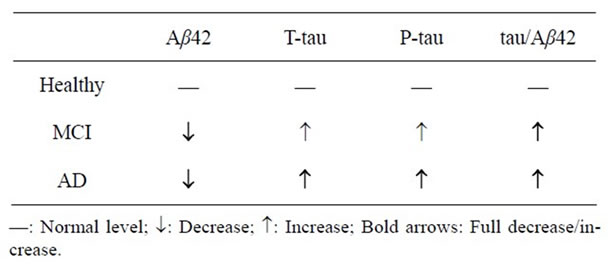
Table 1. Changes of the core CSF biomarkers in MCI and AD.
dementias were uncovered in the literature search. However, a few trophic factors such as CART and glial cellline derived neurotrophic factor (GDNF) may be hopeful biomarkers.
As neurotrophic factors play a pivotal role in development and maintenance of global central nervous system (CNS) function, GDNF, which has been extensively studied for its survival promoting effects especially concerning catecholaminergic neurons, also plays a significant role in Alzheimer’s disease characterized mainly by damage of cholinergic CNS neurons. Straten G. et al. compared GDNF concentrations in CSF and serum of patients with AD and normal controls (NC). While GDNF concentrations in CSF were significantly increased in patients with AD compared with NC subjects, GDNF concentration of AD patients in serum were significantly decreased compared with the NC group. Increased GDNF in CSF of AD might be due to an upregulated expression in CNS as an adaptive process of the impaired brain to enhance neurotrophic support at least in early stages of disease and/or impairment of CSF turnover [89]. It is currently unclear why serum GDNF is significantly decreased in AD patients, but it may be an important signal for the disease.
Neurotransmitter CART is widely expressed in the brain including hippocampus and cortex and has a general role of cytoprotection this will be further discussed in Section 4. Interestingly, CART level in CSF is decreased in AD patients comparing to healthy controls [90], indicating CART may be involved in the pathology or etiology of AD, it is worth further pursuing for their connection. Recently it has been shown that icariin, a flavonoid extracted from a Chinese herbal medicine Yin Yang Huo, also called Xian Ling Pi that has a protective role against Aβ42 neurotoxicity via up-regulation of CART in both mRNA and protein levels [91], suggesting that low levels of CART may be related to Aβ42 toxicity that is the main pathological event associated with Alzheimer’s disease.
Cytokines are important mediators in AD development. TNF-α converting enzyme (TACE/ADAM-17) does not only cleave pro-TNF-α but also TNF receptors, it has been shown that TACE activity was significantly elevated in the CSF from AD patients compared with healthy controls. Furthermore, AD patients had higher levels of both TACE cleaved soluble TNFR1 (sTNFR1) and TNFR2 (sTNFR2) in the CSF compared to ageand gender-matched healthy controls. To examine if changes in TACE activity and levels of cleaved soluble TNFRs are an early event in the course of AD, they measured these molecules in the CSF from 47 subjects with MCI, which is considered as a preclinical stage of AD. Unexpectedly, significantly higher levels of TACE activity and soluble TNFRs in the MCI group than that in AD patients. These results suggest that TACE activity and soluble TNF receptors may be potential diagnostic candidate biomarkers in AD and especially in MCI [92].
In addition, macrophage colony-stimulating factor (MCSF) is a hematopoietic growth factor that activates microglial cells, involved in phagocytosis of Aβ in the brain. MCI patients showed significantly decreased M-CSF levels in CSF compared to AD patients and patients with other non-inflammatory neurological disease. M-CSF CSF levels < or = 357.8 pg/ml yielded 73.7% sensitivity and 75.0% specificity for diagnosing MCI patients in comparison with control subjects. In conclusion, these data indicate that M-CSF in CSF could be a useful biomarker for MCI [93].
To assess the potential biomarkers of early diagnosis of AD, a new method using capillary-electrophoresis coupled to mass-spectrometry (CE-MS) was developed. Using CSF measurements of Aβ42, t-tau, and p-tau, AD-diagnosis had a sensitivity of 88% and a specificity of 67% in the CSF sample. Sequence analysis of the discriminating biomarkers identified fragments of synaptic proteins like proSAAS, apolipoprotein J, neurosecretory protein VGF, phospholemman, and chromogranin A [83]. These newly identified protein/peptide candidates have to be further examined in the future to verify the usefulness as a biomarker. Certainly, MS-based methods will provide more biomarker candidates for diagnosis of AD and related disorders in the feature.
3.3. Distinguishing AD and DLB in CSF Biomarkers
The clinical diagnosis of dementia with Lewy bodies (DLB) is made on the basis of consensus criteria; however, the sensitivity of the criteria is relatively low [5]. Differentiating dementia with Lewy bodies from AD can be difficult because of the substantial overlap in clinical features. A classification of patients with AD and patients with other dementias accomplished by the combination of CSF Aβ42 and p-tau showed that CSF AD biomarker profile was seen in 47% of patients with DLB [2]. Therefore, a better biomarker system to distinguish DLB from AD is strongly desired.
Using the quantitative Aβ-immunoblot to analyze CSF samples of neuropathologically defined patients with AD and DLB, Bibl and colleagues found that absolute Aβ1- 42 and percentage term Aβ (42%) were significantly lowered in AD compared to non-demented controls (NDC). Oxidized absolute Aβ1-40ox and percentage Aβ (40ox%) were elevated in DLB as compared to NDC. Importantly it is reproductive on Aβ peptide patterns in neuropathologically characterized patients with AD and DLB. The results underline the usefulness of the CSF Aβ (42%) and Aβ (40ox%) as diagnostic biomarkers for AD and DLB, respectively [94,95].
It has been shown that significant between group differences were found for all CSF measures, and all except Aβ40, Aβ42 and Aβ42/p-tau differed between AD and DLB. The Aβ42/Aβ38 ratio was the measure that best discriminated between AD and DLB (accuracy, acc = 0.765; p < 0.005). Studies with independent and larger samples and priori cut-offs are needed to test this hypothesis [96]. However this study suggests that the level of Aβ38, another Aβ shorter form can potentially contribute in the diagnostic distinction between AD and DLB when combined with Aβ42. Single measures had low diagnostic accuracy, suggesting that developing a panel of biochemical markers is the most promising strategy.
To establish diagnostic accuracy and optimal cut-off levels of CSF tau phosphorylated at threonine 181 for discriminating AD from non-AD dementias in autopsy-confirmed dementia patients, CSF levels of Aβ42, t-tau and p-tau from patients with definite AD (n = 95) and non-AD dementias (n = 50) were determined with single-parameter ELISA kits. Optimal p-tau cut-off levels for differentiating AD from pooled non-AD dementias, DLB and frontotemporal dementia (FTD) were 50.4 pg/mL (acc = 0.73), 52.8 pg/mL (acc = 0.73) and 35.3 pg/mL (acc = 0.90), respectively. The highly significant difference in CSF p-tau levels comparing AD and non-AD dementias pointed to the potential value of CSF p-tau for discriminating AD from non-AD dementias [97]. The determination of the optimal cut-off levels of CSF biomarkers is a key issue for further validation and utility.
In another report, the utility of quantification of α-synuclein, Aβ and tau in the CSF of patients with DLB (n = 34), AD (n = 31) and other dementias (n = 21) was examined. Total and phosphorylated tau levels as well as Aβ40/42 and tau/Aβ42 ratios were significantly higher in AD patients than in patients with DLB and other dementias (both p < 0.01). CSF aSyn levels in DLB patients were significantly lower than those in patients with AD (p < 0.05) and other dementias (p < 0.01). CSF aSyn level correlated with the Aβ42 level in DLB patients (p = 0.01, r = 0.43). The study suggests that reduced levels of CSF aSyn in DLB may reflect the accumulation of aSyn with Lewy pathology in the brain and that quantification of CSF α-synuclein helps in the differentiation of DLB from AD and other dementias in the combination with Aβ42 and tau analysis [98].
However, other report suggested that CSF aSyn was not a useful diagnostic biomarker to differentiate DLB and/or PD (α-synucleinopathies) from AD. It only showed a trend of low CSF aSyn in DLB, but CSF aSyn levels did not differentiate between diagnostic groups (p = 0.16). However, higher ptau-181 and higher tau levels differentiated AD from DLB patients (p < 0.05). In addition, in DLB patients maybe lower CSF aSyn levels are related to worse cognitive performance [99]. This different between the two reports may be in part due to different kits they used, since in the early paper, they emphasized that a modified sandwich ELISA was developed which enables the sensitive quantification of CSF aSyn [98].
Since deficits in serotonergic and dopaminergic pathways seem more pronounced in DLB patients, CSF analysis of neurotransmitter metabolites, in addition to brain-specific proteins, may improve the differentiation between DLB and AD. Aerts et al retrospectively compared CSF concentrations of the neurotransmitter metabolites homovanillic acid (HVA), 5-hydroxyindolacetic acid (5-HIAA), and 3-methoxy-4-hydroxyphenylethyleneglycol (MHPG) and the brain-specific proteins t-tau, p-tau, and Aβ42 in patients with AD (n = 45) and with DLB (n = 23). The concentrations of all neurotransmitter metabolites, as well as those for t-tau and p-tau protein, were significantly lower in DLB compared to AD, irrespective of the diagnostic certainty (i.e., possible or probable). The currently used combination of Aβ42, p-tau, and t-tau yielded a sensitivity of 92.9% and a specificity of 90%. The addition of MHPG resulted in an increased sensitivity of 97.6% and a specificity of 95% for the discrimination between DLB and AD [100]. However, this exciting change of MHPG was not the case of another report, in which no significant correlations were found between AD and DLB groups [101].
Overall, it has been widely accepted that having a biochemical test, based on quantification of useful DLB biomarkers within CSF could be an effective diagnostic approach to improve the differential diagnosis. Although some of the investigated DLB CSF biomarkers are well within the clinical criteria for sensitivity and specificity, they all seem to be confounded by the overlap or contradictory data for each of the major groups of biomarkers (Aβ peptides, tau and aSyn). However, from increasing relevant reports, to better differentiate DLB from AD and other dementias, a conclusive proposal appears to emerge (Table 2). This is a combination of CSF measures: de

Table 2. Main changes of CSF biomarkers in DLB and AD.
creased aSyn, a correlation between CSF aSyn and Aβ42measures in DLB (characteristic for DLB only), and an elevated t-tau and p-tau profile in AD (differentiating AD from DLB) [5]. Progress in neurotransmitter CART discussed below may further increase the utility of biomarkers in diagnosis of DLB.
4. NEUROPEPTIDE CART AS A NEW BIOMARKER CANDIDATE FOR DEMENTIA WITH LEWY BODY
4.1. CART Has a Neurotrophic Role via Mitochondria and Antioxidant Mechanisms
CART peptide is broadly expressed in hypothalamus, cortex, hippocampus, amygdala, and pituitary and some endocrine glands. CART receptor or interaction partner has not been reported [102,103]. Using molecular genetics approaches we identified the first interaction protein for CART it is the mitochondrial protein SDH subunit B [11]. CART also increases SDH and mitochondrial complex II activities and ATP generation in both basal and ischemic conditions [11], suggesting the new mitochondrial mechanisms of CART’s neurotrophic or neuroprotective role.
Interestingly, in vivo study has shown that intracerebroventricularly injection of CART increases spatial learning and memory in rats subjected to the acquisition protocol in Morris water maze [103]. On the other hand, endogenous CART was upregulated in the frontal cortex, hypothalamus, amygdala, medulla oblongata and adrenal gland after the forced swim stress [104].
Recently, we reported that CART has a general cytoprotection in mammalian and human cells [12]. Using cis-parinaric acid as a sensitive reporting probe for peroxidation in membranes, and a lipid-soluble azo initiator of peroxyl radicals, we found that CART has an antioxidant property. Interestingly CART is preferentially localized in mitochondria, which are the main source of free radicals. In addition, this antioxidant and protective role is also seen in a mouse Parkinson’s disease model [12]. Therefore we proposed that CART is an endogenous antioxidant, a powerful antioxidant hormone. These new functions for this unique peptide combined with its transcriptional activity on some neurotrophic genes such as BDNF [105,106], enable mitochondria within both neurons and non-neuronal cells to cope with pathological processes and provide stabile local energy homeostasis in the brain which, in turn, is a prerequisite for neuronal survival. These mitochondria associated cytoprotection and trophic roles are particularly important for neurodegenerative diseases since most of them have a mitochondrial dysfunction or an energy failure condition.
4.2. CART as a New Biomarker for DLB
It has been shown that DLB patients displayed hypothalamic atrophy, whereas this region was not affected in AD patients [107]. CART is expressed highly in neurons in the hypothalamus. Although Lewy bodies have been described in the hypothalamus of DLB patients [108], extensive neuropathological studies of this region in DLB have not yet been reported. Using sensitive and a commercially available radioimmunoassay (RIA), Schultz K, et al. [90] found that CSF CART levels were significantly reduced by 30% in DLB patients compared to controls as well as to AD patients. Even though this was not a large scale study, proper design including RIA assay controls and statistical analyses made the data convincing. The results suggest that reduced CSF CART is a sign of hypothalamic dysfunction in DLB and that it may serve as a new biomarker for this DLB patient group. A large scale study in a clinic center or within multiple clinic centers in the future may further verify these findings. CART as a new promising biomarker together with other key markers is summarized in Table 2.
Interestingly, CSF dopamine metabolite (DOPAC) level is correlated with behavioral and psychological signs and symptoms of dementia (BPSD) in general [101]; intracerebroventricular injections of CART in rodents lead to increased dopaminergic activity in nucleus accumbens, striatum and hypothalamus [109]; and systemic injections of CART in a Parkinson’s disease mouse model preserve dopaminergic neurons in SN area [12]. Reduced CART levels may therefore be involved in causing or augmenting the dopaminergic hypofunction resulting in Parkinsonism, as well as other symptoms, including mood/psychiatric symptoms [110]. CART level is also reduced in AD patients, the precise mechanisms of CART underline these changes and the significance of CART in both DLB and AD diseases need further investigation in the future.
In addition, although the involvement of CART in mitochondria and neurotransmitter-neutrophin signal transduction in healthy and injured neurons is increasingly recognized, the impact of CART on the cellular processing of amyloid precursor protein, tau and aSyn, the key genes/proteins associated AD and DLB is currently unexplored.
5. PROBLEMS, CONCERNS AND CHARLENGES IN CSF BIOMARKERS
Recent decades study in neurodegenerative diseases has made significant progress. However, the cure treatment is still lacking. This is the greatest challenge in the related fields. Disease-modifying approaches for Alzheimer’s disease and dementia with LB might be most effective when initiated very early in the course, before the pathologic burden and synaptic and neuronal degeneration make it unlikely to recover. It has been widely accepted that biomarkers of disease may provide important avenues and clues of research to enhance the diagnosis of individuals with early AD and could assist in the early identification of those individuals at risk for developing AD. It has also been recognized that the core biomarkers reflect AD pathology, and are candidate markers for predicting future cognitive decline in healthy individuals and progression to dementia in patients who are cognitively impaired [73]. However, for such biomarkers to become clinically useful, long-term follow-up studies are necessary to evaluate the relevance of cross-sectional biomarker changes to the longitudinal course of the disease [111].
Even the CSF biomarkers Aβ42, t-tau, and p-tau demonstrate good diagnostic accuracy for AD, there are large variations in biomarker measurements between studies, and between and within laboratories. Importantly the Alzheimer’s association has initiated a global quality control program to estimate and monitor variability of measurements, quantify batch-to-batch assay variations, and identify sources of variability. In the report from the first two rounds of the program indicates that measurements of CSF AD biomarkers show large between-laboratory variability, likely caused by factors related to analytical procedures and the analytical kits. Standardization of laboratory procedures and efforts by kit vendors to increase kit performance might lower variability and will likely increase the usefulness of CSF AD biomarkers [112]. To evaluate and monitor the biomarker tests, except the standard (normal) values or value range should be listed, assay inner controls, kit internal as well as external controls should be included in each assay kit and the information may be necessary to be listed in the report. The assessment kit-associated study in the endocrinology, including the recent article regarding CSF CART expression determined by RIA or other methods may be good example [90,113-115].
In addition, overlap of current CSF core biomarkers with both controls, DLB and AD results in insufficient diagnostic accuracy, therefore the development of more specific and sensitive biomarkers for these disorders is needed.
To increase agents/kits specificity and avoid cross reaction, highly specific mouse monoclonal antibodies are preferred, since in the principle, small Aβ peptides (such as dominant Aβ40 and pathological Aβ42) are different in 2 or 3 amino acids only, monoclonal antibody can target the unique region/epitope to produce highly specific antibody, but polyclonal antibody or multiple colonial antibody recognizes multiple epitopes of the antigen (Aβ), including some common regions of Aβ40 and Aβ42. Notably, it has been reported that using such monoclonal antibody/kit already sensitively detected that plasma Aβ42 is significantly elevated in late onset AD (Aβ42 levels than controls, p < 0.0001, but Aβ40 p = 0.2) [8,116]. We recently verified that this Aβ42 highly specific antibody also works well in immunohistochemistry [117]. However, it seems that the polyclonal antibodies and related assay kits are more than monoclonal ones in the current market. This factor somewhat affects present antibody-based biomarker research and clinical utility.
These concerns and questions above may be resolved through a great collaboration from scientists/researchers, production companies, and governor/organization/association in the world. Certainly it is a real challenge in the related fields. I hope that this scientific review will further facilitate the task to move forward efficiently.
6. CONCLUSIONS AND FUTURE DIRECTIONS
Intense multidisciplinary research has provided detailed knowledge of the molecular pathogenesis of neurodegenerative diseases, including Alzheimer’s dementia and dementia with Lewy bodies. Among these, pathologyassociated biomarkers in the cerebrospinal fluid, ideally amyloid-β (Aβ42), total-tau (t-tau), phosphorylated tau (p-tau) and α-synuclein (aSyn) have been extensively studied. These markers especially their combination have demonstrated good diagnostic accuracy for AD and DLB. These biomarkers reflect disease pathology, and are candidate markers for predicting future cognitive decline in healthy individuals and progression to dementia in patients who are cognitively impaired. It is very important for diagnosis and treatment of these progressive diseases in its early stages prior to the occurrence of irreversible neuronal damage; hence these CSF biomarkers are crucial in a routine clinical setting in the future. Further improving and standardizing the present assay systems, and identifying new, specific biomarkers through mass spectrometry (MS)-based different quantitative proteomics and other approaches are the exciting challenges in the field.
Importantly, not only in traditional AD patients, but also in people with mild cognitive impairment, CSF Aβ42 levels are significantly decreased comparing to healthy adults. This reflects abnormal amyloid metabolism, one of the most important changes in the brain, suggesting the formation of some amyloid plaques (even tiny or diffused plaque deposits) in the hippocampus and cortex. At the same time or later, total tau levels, especially p-tau levels are significantly increased in AD patients comparing to healthy controls. Specific and sensitive assay or kit-based optimal cut-off levels of the CSF biomarkers may be determined in the future.
Patients with DLB show either normal levels of these core CSF biomarkers or slightly elevated t-tau and slightly decreased Aβ42, while p-tau levels are around normal, or only slightly changed. This feature is significantly different from AD and can be the first key point of the differential diagnosis between DLB and AD. Furthermore, aSyn is decreased in most DLB cases comparing to healthy controls as well as AD patients. This is the secondary point for the differential diagnosis. In addition, among other biomarker candidates, endogenous antioxidant CART is a highly promising one since deficits in serotonergic and dopaminergic pathways seem more pronounced in DLB patients, and CART can modulate these pathways [110] especially clinic practice shows that CART level in CSF is significantly decreased in DLB patients comparing to AD as well as healthy controls [90]. Combination of CART with the core markers would further increase the diagnostic accuracy.
Therefore, in conclusion, the combination of CART and the brain specific proteins Aβ42, t-tau, p-tau and aSyn in CSF will be associated with the clinical diagnosis of DLB and discriminated between AD and DLB with highly diagnostic accuracy, suggesting this combination as a new, potential biomarker panel for DLB. If this diagnosis panel is to be confirmed by new investigations, it should be a great advance in neurodegenerative disease field, and it should bring big benefits to patients.
7. ACKNOWLEDGEMENTS
This project was supported by the American Heart Association (AHA award, Beginning Grant-in-Aid no. 0565527Z) and the National Center for Research Resources and the Office of Research Infrastructure Programs (ORIP) of the National Institutes of Health through Grant Number RR000163.
![]()
![]()
REFERENCES
- Knopman, D.S., DeKosky, S.T., Cummings, J.L., Chui, H., Corey-Bloom, J., Relkin, N., Small, G.W., Miller, B. and Stevens, J.C. (2001) Practice parameter: Diagnosis of dementia (an evidence-based review). Neurology, 56, 1143-1153.
- Schoonenboom, N.S., Reesink, F.E., Verwey, N.A., Kester, M.I., Teunissen, C.E., van de Ven, P.M., Pijnenburg, Y.A., Blankenstein, M.A., Rozemuller, A.J., Scheltens, P., et al. (2012) Cerebrospinal fluid markers for differential dementia diagnosis in a large memory clinic cohort. Neurology, 78, 47-54. doi:10.1212/WNL.0b013e31823ed0f0
- McKeith, I.G., Burn, D.J., Ballard, C.G., Collerton, D., Jaros, E., Morris, C.M., McLaren, A., Perry, E.K., Perry, R., Piggott, M.A., et al. (2003) Dementia with lewy bodies. Seminars in Clinical Neuropsychiatry, 8, 46-57. doi:10.1053/scnp.2003.50006
- Jellinger, K.A. (2009) Formation and development of lewy pathology: A critical update. Journal of Neurology, 256, 270-279. doi:10.1007/s00415-009-5243-y
- Mukaetova-Ladinska, E.B., Monteith, R. and Perry, E.K. (2010) Cerebrospinal fluid biomarkers for dementia with lewy bodies. International Journal of Alzheimers’s Disease, 2010, Article ID 536538. doi:10.4061/2010/536538
- Lin, M.T. and Beal, M.F. (2006) Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature, 443, 787-795. doi:10.1038/nature05292
- Martin, L.J. (2010) Mitochondrial and cell death mechanisms in neurodegenerative diseases. Pharmaceuticals (Basel), 3, 839-915.
- Mao, P. and Reddy, P.H. (2011) Aging and amyloid beta-induced oxidative DNA damage and mitochondrial dysfunction in alzheimer's disease: Implications for early intervention and therapeutics. Biochimica et Biophysica Acta, 1812, 1359-1370.
- Hasan, M.K. and Mooney, R.P. (2011) The predisposing factors, biological markers, neuroimaging techniques and medical complications associated with alzheimer’s disease. The West Virginia Medical Journal, 107, 26-29.
- Cheung, W.W. and Mao, P. (2012) Recent advances in obesity: Genetics and beyond. ISRN Endocrinology, 2012, Article ID 536905. doi:10.5402/2012/536905
- Mao, P., Ardeshiri, A., Jacks, R., Yang, S., Hurn, P.D. and Alkayed, N.J. (2007) Mitochondrial mechanism of neuroprotection by CART. European Journal of Neuroscience, 26, 624-632. doi:10.1111/j.1460-9568.2007.05691.x
- Mao, P., Meshul, C.K., Thuillier, P., Goldberg, N.R. and Reddy, P.H. (2012) CART peptide is a potential endogenous antioxidant and preferentially localized in mitochondria. PLoS One, 7, Article ID e29343. doi:10.1371/journal.pone.0029343
- Hyman, B.T., Phelps, C.H., Beach, T.G., Bigio, E.H., Cairns, N.J., Carrillo, M.C., Dickson, D.W., Duyckaerts, C., Frosch, M.P., Masliah, E., et al. (2012) National institute on aging-Alzheimer’S association guidelines for the neuropathologic assessment of alzheimer’s disease. Alzheimers Dement, 8, 1-13. doi:10.1016/j.jalz.2011.10.007
- Hebert, L.E., Scherr, P.A., Bienias, J.L., Bennett, D.A. and Evans, D.A. (2003) Alzheimer disease in the US population: Prevalence estimates using the 2000 census. Archives of Neurology, 60, 1119-1122. doi:10.1001/archneur.60.8.1119
- McKeith, I.G., Dickson, D.W., Lowe, J., Emre, M., O’Brien, J.T., Feldman, H., Cummings, J., Duda, J.E., Lippa, C., Perry, E.K., et al. (2005) Diagnosis and management of dementia with lewy bodies: Third report of the DLB consortium. Neurology, 65, 1863-1872. doi:10.1212/01.wnl.0000187889.17253.b1
- Kobayashi, M., Sato, T., Sato, A. and Imamura, T. (2009) Oldest-old dementia in a japanese memory clinic. Brain Nerve, 61, 972-978.
- Gravina, S.A., Ho, L., Eckman, C.B., Long, K.E., Otvos, L. Jr., Younkin, L.H., Suzuki, N. and Younkin, S.G. (1995) Amyloid beta protein (A beta) in alzheimer’s disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43). The Journal of Biological Chemistry, 270, 7013-7016.
- Martorana, A., Esposito, Z. and Koch, G. (2010) Beyond the cholinergic hypothesis: Do current drugs work in alzheimer's disease? CNS Neuroscience & Therapeutics, 16, 235-245.
- Hut, R.A. and Van der Zee, E.A. (2011) The cholinergic system, circadian rhythmicity, and time memory. Behavioural Brain Research, 221, 466-480. doi:10.1016/j.bbr.2010.11.039
- Robinson, L., Platt, B. and Riedel, G. (2011) Involvement of the cholinergic system in conditioning and perceptual memory. Behavioural Brain Research, 221, 443-465. doi:10.1016/j.bbr.2011.01.055
- Kar, S., Slowikowski, S.P., Westaway, D. and Mount, H.T. (2004) Interactions between beta-amyloid and central cholinergic neurons: Implications for Alzheimer’s disease. Journal of Psychiatry and Neuroscience, 29, 427-441.
- Pakaski, M. and Kalman, J. (2008) Interactions between the amyloid and cholinergic mechanisms in Alzheimer’s disease. Neurochemistry Intnational, 53, 103-111. doi:10.1016/j.neuint.2008.06.005
- Schliebs, R. and Arendt, T. (2011) The cholinergic system in aging and neuronal degeneration. Behavioural Brain Research, 221, 555-563. doi:10.1016/j.bbr.2010.11.058
- Mao, P. and Reddy, P.H. (2010) Is multiple sclerosis a mitochondrial disease? Biochimica et Biophysica Acta, 1802, 66-79.
- Beal, M.F. (2005) Mitochondria take center stage in aging and neurodegeneration. Annals of Neurology, 58, 495-505. doi:10.1002/ana.20624
- Swerdlow, R.H. (2011) Alzheimer’s disease pathologic cascades: Who comes first, what drives what. Neurotoxicity Research, in press.
- Ittner, L.M. and Gotz, J. (2011) Amyloid-beta and tau—A toxic pas de deux in Alzheimer’s disease. Nature Reviews Neuroscience, 12, 65-72. doi:10.1038/nrn2967
- Reddy, P.H. (2011) Abnormal tau, mitochondrial dysfunction, impaired axonal transport of mitochondria, and synaptic deprivation in Alzheimer’s disease. Brain Research, 1415, 136-148. doi:10.1016/j.brainres.2011.07.052
- Nunomura, A., Perry, G., Aliev, G., Hirai, K., Takeda, A., Balraj, E.K., Jones, P.K., Ghanbari, H., Wataya, T., Shimohama, S., et al. (2001) Oxidative damage is the earliest event in alzheimer disease. Journal of Neuropathology & Experimental Neurology, 60, 759-767.
- Lauren, J., Gimbel, D.A., Nygaard, H.B., Gilbert, J.W. and Strittmatter, S.M. (2009) Cellular prion protein mediates impairment of synaptic plasticity by amyloid-beta oligomers. Nature, 457, 1128-1132. doi:10.1038/nature07761
- Leuner, K., Schutt, T., Kurz, C., Eckert, S.H., Schiller, C., Occhipinti, A., Mai, S., Jendrach, M., Eckert, G.P., Kruse, S.E., et al. (2012) Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxidants & Redox Signaling, 16, 1421-1433. doi:10.1089/ars.2011.4173
- Kroner, Z. (2009) The relationship between Alzheimer’s disease and diabetes: Type 3 diabetes? Alternative Medicine Review, 14, 373-379.
- Gotz, J., Ittner, L.M. and Lim, Y.A. (2009) Common features between diabetes mellitus and Alzheimer’s disease. Cellular and Molecular Life Sciences, 66, 1321-1325.
- Akter, K., Lanza, E.A., Martin, S.A., Myronyuk, N., Rua, M. and Raffa, R.B. (2011) Diabetes mellitus and Alzheimer’s disease: Shared pathology and treatment? British Journal of Clinical Pharmacology, 71, 365-376. doi:10.1111/j.1365-2125.2010.03830.x
- Lim, Y.A., Rhein, V., Baysang, G., Meier, F., Poljak, A., Raftery, M.J., Guilhaus, M., Ittner, L.M., Eckert, A. and Gotz, J. (2010) Abeta and human amylin share a common toxicity pathway via mitochondrial dysfunction. Proteomics, 10, 1621-1633. doi:10.1002/pmic.200900651
- Kalaria, R.N. (2000) The role of cerebral ischemia in Alzheimer’s disease. Neurobiology of Aging, 21, 321-330. doi:10.1016/S0197-4580(00)00125-1
- Quinn, J. (2003) Vascular dementia. Journal of the American Medical Directors Association, 4, S155-S161. doi:10.1016/S1525-8610(04)70407-7
- Auld, D.S., Kornecook, T.J., Bastianetto, S. and Quirion, R. (2002) Alzheimer’s disease and the basal forebrain cholinergic system: Relations to beta-amyloid peptides, cognition, and treatment strategies. Progress in Neurobiology, 68, 209-245. doi:10.1016/S0301-0082(02)00079-5
- Schliebs, R. and Arendt, T. (2006) The significance of the cholinergic system in the brain during aging and in Alzheimer’s disease. Journal of Neural Transmission, 113, 1625-1644. doi:10.1007/s00702-006-0579-2
- Daulatzai, M.A. (2010) Early stages of pathogenesis in memory impairment during normal senescence and Alzheimer’s disease. Journal of Alzheimer’s Disease, 20, 355- 367.
- Tamagno, E., Guglielmotto, M., Aragno, M., Borghi, R., Autelli, R., Giliberto, L., Muraca, G., Danni, O., Zhu, X., Smith, M.A., et al. (2008) Oxidative stress activates a positive feedback between the gammaand beta-secretase cleavages of the beta-amyloid precursor protein. Journal of Neurochemistry, 104, 683-695.
- Shen, C., Chen, Y., Liu, H., Zhang, K., Zhang, T., Lin, A. and Jing, N. (2008) Hydrogen peroxide promotes abeta production through JNK-dependent activation of gamma-secretase. The Journal of Biological Chemistry, 283, 17721-17730. doi:10.1074/jbc.M800013200
- De Leon, M.J., DeSanti, S., Zinkowski, R., Mehta, P.D., Pratico, D., Segal, S., Clark, C., Kerkman, D., DeBernardis, J., Li, J., et al. (2004) MRI and CSF studies in the early diagnosis of Alzheimer’s disease. Journal of Internal Medicine, 256, 205-223. doi:10.1111/j.1365-2796.2004.01381.x
- Brys, M., Glodzik, L., Mosconi, L., Switalski, R., De Santi, S., Pirraglia, E., Rich, K., Kim, B.C., Mehta, P., Zinkowski, R., et al. (2009) Magnetic resonance imaging improves cerebrospinal fluid biomarkers in the early detection of Alzheimer’s disease. Journal of Alzheimer’s Disease, 16, 351-362.
- Blennow, K. and Zetterberg, H. (2010) Is it time for biomarker-based diagnostic criteria for prodromal Alzheimer’s disease? Alzheimer’s Research & Therapy, 2, 8. doi:10.1186/alzrt31
- McKhann, G.M., Knopman, D.S., Chertkow, H., Hyman, B.T., Jack, C.R. Jr., Kawas, C.H., Klunk, W.E., Koroshetz, W.J., Manly, J.J., Mayeux, R., et al. (2011) The diagnosis of dementia due to Alzheimer’s disease: Recommendations from the national institute on aging-Alzheimer’s association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s and Dementia, 7, 263-269. doi:10.1016/j.jalz.2011.03.005
- Prvulovic, D. and Hampel, H. (2011) Amyloid beta (abeta) and phospho-tau (p-tau) as diagnostic biomarkers in alzheimer’s disease. Clinical Chemistry and Laboratory Medicine, 49, 367-374. doi:10.1515/cclm.2011.087
- Vos, S., van Rossum, I., Burns, L., Knol, D., Scheltens, P., Soininen, H., Wahlund, L.O., Hampel, H., Tsolaki, M., Minthon, L., et al. (2012) Test sequence of CSF and MRI biomarkers for prediction of AD in subjects with MCI. Neurobiology of Aging, in press. doi:10.1016/j.neurobiolaging.2011.12.017
- McKeith, I.G., Galasko, D., Kosaka, K., Perry, E.K., Dickson, D.W., Hansen, L.A., Salmon, D.P., Lowe, J., Mirra, S.S., Byrne, E.J., et al. (1996) Consensus guidelines for the clinical and pathologic diagnosis of dementia with lewy bodies (DLB): Report of the consortium on DLB international workshop. Neurology, 47, 1113-1124.
- Leverenz, J.B., Umar, I., Wang, Q., Montine, T.J., McMillan, P.J., Tsuang, D.W., Jin, J., Pan, C., Shin, J., Zhu, D., et al. (2007) Proteomic identification of novel proteins in cortical lewy bodies. Brain Pathology, 17, 139- 145. doi:10.1111/j.1750-3639.2007.00048.x
- Tofaris, G.K. and Spillantini, M.G. (2005) Alpha-synuclein dysfunction in lewy body diseases. Movement Disordersrs, 20, S37-S44. doi:10.1002/mds.20538
- Volles, M.J. and Lansbury, P.T. Jr. (2003) Zeroing in on the pathogenic form of alpha-synuclein and its mechanism of neurotoxicity in Parkinson’s disease. Biochemistry, 42, 7871-7878. doi:10.1021/bi030086j
- Paleologou, K.E., Kragh, C.L., Mann, D.M., Salem, S.A., Al-Shami, R., Allsop, D., Hassan, A.H., Jensen, P.H. and El-Agnaf, O.M. (2009) Detection of elevated levels of soluble alpha-synuclein oligomers in post-mortem brain extracts from patients with dementia with lewy bodies. Brain, 132, 1093-1101. doi:10.1093/brain/awn349
- Tofaris, G.K., Kim, H.T., Hourez, R., Jung, J.W., Kim, K.P. and Goldberg, A.L. (2011) Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomallysosomal pathway. Proceedings of the National Academy of Sciences USA, 108, 17004-17009. doi:10.1073/pnas.1109356108
- MacInnes, N., Iravani, M.M., Perry, E., Piggott, M., Perry, R., Jenner, P. and Ballard, C. (2008) Proteasomal abnormalities in cortical lewy body disease and the impact of proteasomal inhibition within cortical and cholinergic systems. Journal of Neural Transmission, 115, 869-878. doi:10.1007/s00702-008-0027-6
- Uversky, V.N. (2007) Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. Journal of Neurochemistry, 103, 17-37.
- Sugeno, N., Takeda, A., Hasegawa, T., Kobayashi, M., Kikuchi, A., Mori, F., Wakabayashi, K. and Itoyama, Y. (2008) Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death. The Journal of Biological Chemistry, 283, 23179-23188. doi:10.1074/jbc.M802223200
- Breydo, L., Wu, J.W. and Uversky, V.N. (2012) Alphasynuclein misfolding and Parkinson’s disease. Biochimica et Biophysica Acta, 1822, 261-285.
- Beach, T.G., Adler, C.H., Sue, L.I., Peirce, J.B., Bachalakuri, J., Dalsing-Hernandez, J.E., Lue, L.F., Caviness, J.N., Connor, D.J., Sabbagh, M.N., et al. (2008) Reduced striatal tyrosine hydroxylase in incidental lewy body disease. Acta Neuropathologica, 115, 445-451. doi:10.1007/s00401-007-0313-7
- Dalfo, E., Portero-Otin, M., Ayala, V., Martinez, A., Pamplona, R. and Ferrer, I. (2005) Evidence of oxidative stress in the neocortex in incidental lewy body disease. Journal of Neuropathology & Experimental Neurology, 64, 816-830. doi:10.1097/01.jnen.0000179050.54522.5a
- Dalfo, E. and Ferrer, I. (2008) Early alpha-synuclein lipoxidation in neocortex in lewy body diseases. Neurobiology of Aging, 29, 408-417. doi:10.1016/j.neurobiolaging.2006.10.022
- Navarro, A., Boveris, A., Bandez, M.J., Sanchez-Pino, M.J., Gomez, C., Muntane, G. and Ferrer, I. (2009) Human brain cortex: Mitochondrial oxidative damage and adaptive response in parkinson disease and in dementia with lewy bodies. Free Radical Biology & Medicine, 46, 1574-1580. doi:10.1016/j.freeradbiomed.2009.03.007
- Mao, P., Gallagher, P., Nedungadi, S., Manczak, M., Shirendeb, U.P., Kohama, S.G., Ferguson, B., Park, B.S. and Reddy, P.H. (2012) Mitochondrial DNA deletions and differential mitochondrial DNA content in rhesus monkeys: Implications for aging. Biochimica et Biophysica Acta, 1822, 111-119.
- Kume, K., Kikukawa, M., Hanyu, H., Takata, Y., Umahara, T., Sakurai, H., Kanetaka, H., Ohyashiki, K., Ohyashiki, J.H. and Iwamoto, T. (2012) Telomere length shortening in patients with dementia with lewy bodies. European Journal of Neurology. doi:10.1111/j.1468-1331.2011.03655.x
- Ahmad, A., Burns, C.S., Fink, A.L. and Uversky, V.N. (2012) Peculiarities of copper binding to alpha-synuclein. Journal of Biomolecular Structure & Dynamics, 29, 825- 842.
- Gomez, A. and Ferrer, I. (2009) Increased oxidation of certain glycolysis and energy metabolism enzymes in the frontal cortex in lewy body diseases. Journal of Neuroscience Research, 87, 1002-1013. doi:10.1002/jnr.21904
- Manczak, M., Mao, P., Calkins, M.J., Cornea, A., Reddy, A.P., Murphy, M.P., Szeto, H.H., Park, B. and Reddy, P.H. (2010) Mitochondria-targeted antioxidants protect against amyloid-beta toxicity in alzheimer’s disease neurons. Journal of Alzheimer’s Disease, 20, S609-631.
- Braak, H., Del Tredici, K., Rub, U., de Vos, R.A., Steur, E.N.J. and Braak, E. (2003) Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of Aging, 24, 197-211. doi:10.1016/S0197-4580(02)00065-9
- Leverenz, J.B., Hamilton, R., Tsuang, D.W., Schantz, A., Vavrek, D., Larson, E.B., Kukull, W.A., Lopez, O., Galasko, D., Masliah, E., et al., (2008) Empiric refinement of the pathologic assessment of lewy-related pathology in the dementia patient. Brain Pathology, 18, 220-224. doi:10.1111/j.1750-3639.2007.00117.x
- Beach, T.G., Adler, C.H., Lue, L., Sue, L.I., Bachalakuri, J., Henry-Watson, J., Sasse, J., Boyer, S., Shirohi, S., Brooks, R., et al. (2009) Unified staging system for lewy body disorders: Correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathologica, 117, 613-634. doi:10.1007/s00401-009-0538-8
- Montine, T.J., Phelps, C.H., Beach, T.G., Bigio, E.H., Cairns, N.J., Dickson, D.W., Duyckaerts, C., Frosch, M.P., Masliah, E., Mirra, S.S., et al. (2012) National institute on aging-Alzheimer’s association guidelines for the neuropathologic assessment of Alzheimer’s disease: A practical approach. Acta Neuropathologica, 123, 1-11. doi:10.1007/s00401-011-0910-3
- Archer, T., Kostrzewa, R.M., Beninger, R.J. and Palomo, T. (2011) Staging neurodegenerative disorders: Structural, regional, biomarker, and functional progressions. Neurotoxicity Research, 19, 211-234. doi:10.1007/s12640-010-9190-2
- Blennow, K., Hampel, H., Weiner, M. and Zetterberg, H. (2010) Cerebrospinal fluid and plasma biomarkers in alzheimer disease. Nature Reviews Neuroscience, 6, 131- 144. doi:10.1038/nrneurol.2010.4
- Galasko, D., Clark, C., Chang, L., Miller, B., Green, R.C., Motter, R. and Seubert, P. (1997) Assessment of CSF levels of tau protein in mildly demented patients with Alzheimer’s disease. Neurology, 48, 632-635.
- Andreasen, N., Minthon, L., Clarberg, A., Davidsson, P., Gottfries, J., Vanmechelen, E., Vanderstichele, H., Winblad, B. and Blennow, K. (1999) Sensitivity, specificity, and stability of CSF-tau in AD in a community-based patient sample. Neurology, 53, 1488-1494.
- Arai, H., Higuchi, S. and Sasaki, H. (1997) Apolipoprotein E genotyping and cerebrospinal fluid tau protein: Implications for the clinical diagnosis of Alzheimer’s disease. Gerontology, 43, 2-10. doi:10.1159/000213879
- Kurz, A., Riemenschneider, M., Buch, K., Willoch, F., Bartenstein, P., Muller, U. and Guder, W. (1998) Tau protein in cerebrospinal fluid is significantly increased at the earliest clinical stage of Alzheimer’s disease. Alzheimer Disease and Associated Disorders, 12, 372-377. doi:10.1097/00002093-199812000-00020
- Arai, H., Morikawa, Y., Higuchi, M., Matsui, T., Clark, C.M., Miura, M., Machida, N., Lee, V.M., Trojanowski, J.Q. and Sasaki, H. (1997) Cerebrospinal fluid tau levels in neurodegenerative diseases with distinct tau-related pathology. Biochemical and Biophysical Research Communications, 236, 262-264. doi:10.1006/bbrc.1997.6908
- Hampel, H., Teipel, S.J., Fuchsberger, T., Andreasen, N., Wiltfang, J., Otto, M., Shen, Y., Dodel, R., Du, Y., Farlow, M., et al. (2004) Value of CSF beta-amyloid1-42 and tau as predictors of Alzheimer’s disease in patients with mild cognitive impairment. Molecular Psychiatry, 9, 705-710.
- Hansson, O., Zetterberg, H., Buchhave, P., Londos, E., Blennow, K. and Minthon, L. (2006) Association between CSF biomarkers and incipient Alzheimer’s disease in patients with mild cognitive impairment: A follow-up study. The Lancet Neurology, 5, 228-234. doi:10.1016/S1474-4422(06)70355-6
- Fagan, A.M., Roe, C.M., Xiong, C., Mintun, M.A., Morris, J.C. and Holtzman, D.M. (2007) Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Archives of Neurology, 64, 343-349. doi:10.1001/archneur.64.3.noc60123
- Mattsson, N., Zetterberg, H., Hansson, O., Andreasen, N., Parnetti, L., Jonsson, M., Herukka, SK, van der Flier, W.M., Blankenstein, M.A., Ewers, M., Rich, K., Kaiser, E., Verbeek, M., Tsolaki, M., Mulugeta, E., Rosén, E., Aarsland, D., Visser, P.J., Schröder, J., Marcusson, J., de Leon, M., Hampel, H., Scheltens, P., Pirttilä, T., Wallin, A., Jönhagen, M.E., Minthon, L., Winblad, B. and Blennow, K. (2009) CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. The Journal of the American Medical Association, 302, 385- 393. doi:10.1001/jama.2009.1064
- Jahn, H., Wittke, S., Zurbig, P., Raedler, T.J., Arlt, S., Kellmann, M., Mullen, W., Eichenlaub, M., Mischak, H. and Wiedemann, K. (2011) Peptide fingerprinting of Alzheimer’s disease in cerebrospinal fluid: Identification and prospective evaluation of new synaptic biomarkers. PLoS One, 6, Article ID e26540. doi:10.1371/journal.pone.0026540
- Stomrud, E., Hansson, O., Zetterberg, H., Blennow, K., Minthon, L. and Londos, E. (2010) Correlation of longitudinal cerebrospinal fluid biomarkers with cognitive decline in healthy older adults. Archives of Neurology, 67, 217-223. doi:10.1001/archneurol.2009.316
- Buchhave, P., Minthon, L., Zetterberg, H., Wallin, A.K., Blennow, K. and Hansson, O. (2012) Cerebrospinal fluid levels of beta-amyloid 1-42, but not of tau, are fully changed already 5 to 10 years before the onset of alzheimer dementia. Archives of General Psychiatry, 69, 98- 106. doi:10.1001/archgenpsychiatry.2011.155
- Lanari, A. and Parnetti, L. (2009) Cerebrospinal fluid biomarkers and prediction of conversion in patients with mild cognitive impairment: 4-year follow-up in a routine clinical setting. Scientific World Journal, 9, 961-966. doi:10.1100/tsw.2009.106
- Santos, A.N., Ewers, M., Minthon, L., Simm, A., Silber, R.E., Blennow, K., Prvulovic, D., Hansson, O. and Hampel, H. (2012) Amyloid-beta oligomers in cerebrospinal fluid are associated with cognitive decline in patients with Alzheimer’S disease. Journal of Alzheimer’s Disease, in press.
- Gao, C.M., Yam, A.Y., Wang, X., Magdangal, E., Salisbury, C., Peretz, D., Zuckermann, R.N., Connolly, M.D., Hansson, O., Minthon, L., et al. (2010) Abeta40 oligomers identified as a potential biomarker for the diagnosis of Alzheimer’S disease. PLoS One, 5, Article ID e15725. doi:10.1371/journal.pone.0015725
- Straten, G., Eschweiler, G.W., Maetzler, W., Laske, C. and Leyhe, T. (2009) Glial cell-line derived neurotrophic factor (GDNF) concentrations in cerebrospinal fluid and serum of patients with early Alzheimer’s disease and normal controls. Journal of Alzheimer’s Disease, 18, 331- 337.
- Schultz, K., Wiehager, S., Nilsson, K., Nielsen, J.E., Lindquist, S.G., Hjermind, L.E., Andersen, B.B., Wallin, A., Nilsson, C. and Petersen, A. (2009) Reduced CSF CART in dementia with lewy bodies. Neuroscience Letters, 453, 104-106. doi:10.1016/j.neulet.2009.02.008
- Sha, D., Li, L., Ye, L., Liu, R. and Xu, Y. (2009) Icariin inhibits neurotoxicity of beta-amyloid by upregulating cocaine-regulated and amphetamine-regulated transcripts. Neuroreport, 20, 1564-1567. doi:10.1097/WNR.0b013e328332d345
- Jiang, H., Hampel, H., Prvulovic, D., Wallin, A., Blennow, K., Li, R. and Shen, Y. (2011) Elevated CSF levels of TACE activity and soluble TNF receptors in subjects with mild cognitive impairment and patients with Alzheimer’s disease. Molecular Neurodegeneration, 6, 69. doi:10.1186/1750-1326-6-69
- Laske, C., Stransky, E., Hoffmann, N., Maetzler, W., Straten, G., Eschweiler, G.W. and Leyhe, T. (2010) Macrophage colony-stimulating factor (M-CSF) in plasma and CSF of patients with mild cognitive impairment and Alzheimer’s disease. Current Alzheimer Research, 7, 409-414. doi:10.2174/156720510791383813
- Bibl, M., Mollenhauer, B., Lewczuk, P., Esselmann, H., Wolf, S., Trenkwalder, C., Otto, M., Stiens, G., Ruther, E., Kornhuber, J., et al. (2007) Validation of amyloid-beta peptides in CSF diagnosis of neurodegenerative dementias. Molecular Psychiatry, 12, 671-680. doi:10.1038/sj.mp.4001967
- Mollenhauer, B., Esselmann, H., Trenkwalder, C., SchulzSchaeffer, W., Kretzschmar, H., Otto, M., Wiltfang, J. and Bibl, M. (2011) CSF amyloid-beta peptides in neuropathologically diagnosed dementia with lewy bodies and alzheimer's disease. Journal of Alzheimer’s Disease, 24, 383-391.
- Mulugeta, E., Londos, E., Ballard, C., Alves, G., Zetterberg, H., Blennow, K., Skogseth, R., Minthon, L. and Aarsland, D. (2011) CSF amyloid beta38 as a novel diagnostic marker for dementia with lewy bodies. Journal of Neurology, Neurosurgery & Psychiatry, 82, 160-164. doi:10.1136/jnnp.2009.199398
- Koopman ,K., Le Bastard, N., Martin, J.J., Nagels, G., De Deyn, P.P. and Engelborghs, S. (2009) Improved discrimination of autopsy-confirmed Alzheimer’s disease (AD) from non-AD dementias using CSF P-tau(181P). Neurochemistry International, 55, 214-218. doi:10.1016/j.neuint.2009.02.017
- Kasuga, K., Tokutake, T., Ishikawa, A., Uchiyama, T., Tokuda, T., Onodera, O., Nishizawa, M. and Ikeuchi, T. (2010) Differential levels of alpha-synuclein, beta-amyloid42 and tau in CSF between patients with dementia with lewy bodies and Alzheimer’s disease. Journal of Neurology, Neurosurgery & Psychiatry, 81, 608-610. doi:10.1136/jnnp.2009.197483
- Reesink, F.E., Lemstra, A.W., van Dijk, K.D., Berendse, H.W., van de Berg, W.D., Klein, M., Blankenstein, M.A., Scheltens, P., Verbeek, M.M. and van der Flier, W.M. (2010) CSF alpha-synuclein does not discriminate dementia with lewy bodies from Alzheimer’s disease. Journal of Alzheimer’s Disease, 22, 87-95.
- Aerts, M.B., Esselink, R.A., Claassen, J.A., Abdo, W.F., Bloem, B.R. and Verbeek, M.M. (2011) CSF tau, Abeta42, and MHPG differentiate dementia with lewy bodies from Alzheimer’s disease. Journal of Alzheimer’s Disease, 27, 377-384.
- Engelborghs, S., Vloeberghs, E., Le Bastard, N., Van Buggenhout, M., Marien, P., Somers, N., Nagels, G., Pickut, B.A. and De Deyn, P.P. (2008) The dopaminergic neurotransmitter system is associated with aggression and agitation in frontotemporal dementia. Neurochemistry International, 52, 1052-1060. doi:10.1016/j.neuint.2007.10.018
- Rogge, G., Jones, D., Hubert, G.W., Lin, Y. and Kuhar, M.J. (2008) CART peptides: Regulators of body weight, reward and other functions. Nature Reviews Neuroscience, 9, 747-758. doi:10.1038/nrn2493
- Upadhya, M.A., Nakhate, K.T., Kokare, D.M., Singru, P.S. and Subhedar, N.K. (2011) Cocaineand amphetamineregulated transcript peptide increases spatial learning and memory in rats. Life Sciences, 88, 322-334. doi:10.1016/j.lfs.2010.12.008
- Balkan, B., Keser, A., Gozen, O., Koylu, E.O., Dagci, T., Kuhar, M.J. and Pogun, S. (2012) Forced swim stress elicits region-specific changes in CART expression in the stress axis and stress regulatory brain areas. Brain Research, 1432, 56-65. doi:10.1016/j.brainres.2011.11.006
- Mao, P. and Jacks, R. (2007) Transcriptional activity by cocaine-amphetamine-regulated transcript. Molecular Psychiatry, 12, 223-224. doi:10.1038/sj.mp.4001928
- Wu, B., Hu, S., Yang, M., Pan, H. and Zhu, S. (2006) CART peptide promotes the survival of hippocampal neurons by upregulating brain-derived neurotrophic factor. Biochemical and Biophysical Research Communications, 347, 656-661. doi:10.1016/j.bbrc.2006.06.117
- Whitwell, J.L., Weigand, S.D., Shiung, M.M., Boeve, B.F., Ferman, T.J., Smith, G.E., Knopman, D.S., Petersen, R.C., Benarroch, E.E., Josephs, K.A., et al. (2007) Focal atrophy in dementia with lewy bodies on MRI: A distinct pattern from Alzheimer’s disease. Brain, 130, 708-719. doi:10.1093/brain/awl388
- Piao, Y.S., Wakabayashi, K., Hayashi, S., Yoshimoto, M. and Takahashi, H. (2000) Aggregation of alpha-synuclein/NACP in the neuronal and glial cells in diffuse lewy body disease: A survey of six patients. Clinical Neuropathology, 19, 163-169.
- Shieh, K.R. (2003) Effects of the cocaineand amphetamine-regulated transcript peptide on the turnover of central dopaminergic neurons. Neuropharmacology, 44, 940- 948. doi:10.1016/S0028-3908(03)00095-9
- Mao, P. (2011) Potential antidepressant role of neurotransmitter CART: Implications for mental disorders. Depression Research and Treatment, 2011, Article ID 762139. doi:10.1155/2011/762139
- Biagioni, M.C. and Galvin, J.E. (2011) Using biomarkers to improve detection of Alzheimer’s disease. Neurodegener Disease Managment, 1, 127-139.
- Mattsson, N., Andreasson, U., Persson, S., Arai, H., Batish, S.D., Bernardini, S., Bocchio-Chiavetto, L., Blankenstein, M.A., Carrillo, M.C., Chalbot, S., et al. (2011) The Alzheimer’s association external quality control program for cerebrospinal fluid biomarkers. Alzheimer’s & Dementia, 7, 386-395.
- Bech, P., Winstanley, V., Murphy, K.G., Sam, A.H., Meeran, K., Ghatei, M.A. and Bloom, S.R. (2008) Elevated cocaineand amphetamine-regulated transcript immunoreactivity in the circulation of patients with neuroendocrine malignancy. The Journal of Clinical Endocrinology & Metabolism, 93, 1246-1253. doi:10.1210/jc.2007-1946
- Rosengren, L., Vanmechelen, E. and Blennow, K. (2005) Simultaneous measurement of beta-amyloid(1-42), total tau, and phosphorylated tau (Thr181) in cerebrospinal fluid by the xMAP technology. Clinical Chemistry, 51, 336-345. doi:10.1373/clinchem.2004.039347
- Shaw, L.M., Vanderstichele, H., Knapik-Czajka, M., Figurski, M., Coart, E., Blennow, K., Soares, H., Simon, A,J., Lewczuk, P., Dean, R.A., Siemers, E., Potter, W., Lee, V.M. and Trojanowski, J.Q. (2011) Qualification of the analytical and clinical performance of CSF biomarker analyses in ADNI. Acta Neuropathologica, 121, 597-609. doi:10.1007/s00401-011-0808-0
- Ertekin-Taner, N., Younkin, L.H., Yager, D.M., Parfitt, F., Baker, M.C., Asthana, S., Hutton, M.L., Younkin, S.G. and Graff-Radford, N.R. (2008) Plasma amyloid beta protein is elevated in late-onset Alzheimer’s disease families. Neurology, 70, 596-606. doi:10.1212/01.wnl.0000278386.00035.21
- Mao, P., Manczak, M., Calkins, M.J., Truong, Q., Reddy, T.P., Reddy, A.P., Shirendeb, U., Lo, H.H., Rabinovitch, P.S. and Reddy, P.H. (2012) Mitochondria-targeted catalase reduces abnormal APP processing, amyloid beta production and BACE1 in a mouse model of Alzheimer’s disease: Implications for neuroprotection and lifespan extension. Human Molecular Genetics, 21.
NOTES
*The author declares no conflict of interest.