Paper Menu >>
Journal Menu >>
 J. Biomedical Science and Engineering, 2008, 1, 200-203 Published Online November 2008 in SciRes. http://www.srpublishing.org/journal/jbise JBiSE Searching maximum quasi-bicliques from protein-protein interaction network Hong-Biao Liu1, Juan Liu1 & Lian Wang1 1School of Computer, Wuhan University, Wuhan 430079, China. Correspondence should be addressed to Juan Liu (liujuan@whu.edu.cn). Received September 10, 2008; revised September 28, 2008; accepted September 28, 2008 ABSTRACT Searching the maximum bicliques or bipartite subgraphs in a graph is a tough question. We proposed a new and efficient method, Searching Quasi-Bicliques (SQB) algorithm, to detect maximum quasi-bicliques from protein-protein interaction network. As a Divide-and-Conquer method, SQB consists of three steps: first, it divides the protein-protein interaction network into a number of Distance-2-Subgraphs; second, by combining top-down and branch-and-bound methods, SQB seeks quasi-bicliques from every Distance-2-Subgraph; third, all the redundant results are removed. We successfully applied our method on the Saccharomyces cerevisiae dataset and obtained 2754 distinct quasi-bicliques. Keywords: Searching Quasi-Bicliques algo- rithm; Quasi-biclique; Protein-Protein Interac- tion Network; Distance-2-Subgraph; Di- vide-and-Conquer method 1. INTRODUCTION As high-throughput technologies such as Yeast Two-Hybrid [1] and Affinity Purification/ Mass Spec- trometry [2] have made significant progress, human be- ings have collected a great number of protein-protein interaction datasets. It is meaningful to dig out substruc- tures from large-scale protein interaction data. Biclique, one kind of the substructures, is common in pro- tein-protein interaction network. Biclique often contains useful biologically meaningful units. For example, the biclique shown in Figure 1 indicates an “all-versus-all” predicted interaction subnetwork [3], Where most of the edges, each representing a protein-protein interac- tion,were approved by biological experiments. Further- more, six proteins on the left side all contain SH3 domain and four proteins on the right side are all with the SH3-binding motifs. Therefore mining biclique can help biologists unveil the cellular function at the molecular level. However, mining bicliques from graph (or pro- tein-protein interaction network in this study) is a com- putationally intensive work, and has been proven as NP complete [4, 5, 6]. Although many researchers [7, 8, 9, 10, 11] have developed some algorithms to solve the maxi- mum biclique problem, they often focused on some spe- cial characteristics of the graph, so the problem is still intractable. Therefore, in computational biology field, some researchers mined quasi-bicliques instead of exact bicliques. Li [12] used the “frequent pattern” developed by Agrawal [13] to find “all-versus-all subnetwork” (or quasi-biclique). The “existing closed itemset mining al- gorithms” (proposed by Agrawal [13]) only uses the size constraint on transaction sets to decrease search space, which brings a great number of small maximum bicliques and greatly influences the process speed. Here, we propose Searching Quasi-Bicliques (SQB) algorithm to detect maximum quasi-bicliques from pro- tein interaction network. By means of Di- vide-and-Conquer method, SQB partitions the pro- tein-protein interaction network into a mount of Dis tance-2-Subgraphs , each for one vertex, and only con- taining two kinds of nodes: those being connected with the vertex (we call them the direct neighbors), and those being reachable from the vertex by passing just one other node (we call them distance-2-neighbors). Next, through top-down and branch-and-bound methods, SQB tries to find the quasi-bicliques from all the Distance-2- Subgraphs. At last, SQB merges the redundant ones in the quasi-biclique clusters. We applied our algorithm on the Saccharomyces cerevisiae dataset and obtained 2754 distinct quasi-bicliques. Yfr024c Rvs167 Ysc84 Ypr154w Bbc1 Las17 Ypr171w Acf2 Ynl094w Ygr136w SH3 proteinsSH3-binding proteins Figure1. An all-versus-all predicted interaction subnetwork. SciRes Copyright © 2008  H. B. Liu / J. Biomedical Science and Engineering 1 (2008) 200-203 201 SciRes Copyright © 2008 JBiSE The organization of this paper is as follows. Section 2 states the maximum Quasi-biclique problem. Section 3 describes the SQB algorithm for finding the maximum quasi bicliques. Section 4 reports the results of the ap- plication of SQB algorithm on in a real proteomic data. The paper ends with conclusions and the future work. 2. MAXIMUM QUASI-BICLIQUE PROBLEM We use a simple graph like [14] to describe a pro- tein-protein interaction network. A vertex represents a kind of protein and an edge means there is an interaction between two kinds of proteins. Quasi-biclique is a graph G= (V, E), in which V can be divided into two non empty sets {V1, U1} and every vertex in V1 directly links to nearly every vertex in U1. The question of finding maximum quasi-biclique in a graph G= (V, E) can be formalized as following function. ,)),(( )),((max nmEVGf EVGf = subject to VUVVUV VmnUmVn ⊂⊂=∩ ≤+== 1,1,11 |,||,1||,1| φ where |V| denotes cardinality of the vertex set of the input graph, n and m should be greater than 1 and lower than |V|-2. A quasi-biclique is measured by the value of nm which actually is the number of interacting edges between two sets. In the following, we denote a quasi-bicluque as QB (V1, U1). 3. SQB ALGORITHM The main method of SQB is Divide-and-Conquer, which includes three parts. The first one is to seek every ver- tex’s Distance-2-Subgraph from a graph. The second one is to find every vertex’s quasi-biclique from its Dis- tance-2-Subgraph. The third one is to merge solutions: after finding every vertex’s quasi-bicliques, SQB puts all the quasi-bicliques together, removes the similar ones, prunes the smaller ones, and obtains the quasi-bicliques of the whole graph. The three parts of SQB are detailed in the following. 3.1. Finding Distance-2-Subgraph As some graph, especially the biological protein-protein interaction network, is very large, the process on the graph will need a very large memory space so it is not feasible in common applications. But it is obvious that the distance between any two vertexes in a quasi-biclique is not greater than 2. So if we want to find a quasi-biclique which in- cludes a specific vertex, we only need to consider the vertex and its related neighbors. The related neighbors are vertexes which are less than 3 in distance to the specific vertex. The induced subgraph, which consists of the ver- tex and its related neighbors, is denoted as Dis- tance-2-Subgraph. The edge status between any two ver- texes in an induced subgraph is the same as that in the original graph. SQB needs to find every vertex’s Dis- tance-2-Subgraph in order to obtain its maximum quasi-biclique. 3.2. Detecting Maximum Quasi-bicliques After finding every vertex’s Distance-2-Subgraph, SQB begins to find the quasi-biclique. This process, detecting quasi-bicliques, is the essential part of SQB. SQB uses the size (nm) to measure a quasi-biclique and it is crucial to know the specific value of n and m of a maximum quasi-biclique. As n and m are in a limited range, SQB tests the values of n and m from the upper limit to the lower one. If a graph has a quasi-biclique QB(|V|=n, |U|=m), the vertexes in the graph with degree lower than n and m should not be in the QB, so SQB removes these smaller vertexes during the process. Furthermore, if the test value of n and m are greater, SQB can remove more vertexes and increase the speed of the process. Before explaining our program, we introduce how to split a graph. We use a complex data structure CD to store the V1 set, U1 set and the induced subgraph G of V1 set and U1 set. The program splits the graph at the V1 set, and U1 set in turn. The program chooses the vertex in V1 or U1 with largest degree and labels it so that next time, the program avoids splitting at the same vertex again. For example, if the program chooses v15 as the candidate vertex, it then produces four sets V150, V151, U150, and U151. The first set V150 includes v15 and vertexes in V1 which has a distance of 2 to vertex v15. The second set V151 consists of elements in V1 except v15. The third set U150 contains vertexes in U1 which is the direct neighbor of vertex v15. The fourth set U151 is the same as U1. Next, the program produces induced graph G150 which con- tains vertexes V150 and U150, then puts V150, U150, G150 into data structure CD150(V150, U150, G150). In the same way, it gets another data structure CD151(V151, U151, G151). The algorithm of detecting quasi-bicliques is listed in the end of this subsection. The algorithm consists of a FOR loop that begins from 20 to 2. (20 is an experimental value which should be increased with the growing of nodes of the input graph). At first SQB uses the sub-function Search_k_Quasi_Bicliques(G, k) to test whether the graph contains quasi-bicliques in which the |V|>20 and |U|>20. If the sub-function finds it’s true, the FOR loop terminates, otherwise the FOR loop decreases the test value by one and continues to test, until the sub-function finds quasi-bicliques or the test value lower than 2. The sub-function func Search_k_Quasi_Bicliques(G, k) is the key component of SQB. At first, the input graph G’s vertex set is divided into two parts, V1 and U1. Next, V1 and U1, and G are put into complex data structure CD(V1, U1, G). CD is put into a buffer BUFFER. Next, the pro- gram go into a WHILE loop. This loop’s terminate con- dition is that the buffer is empty. During the loop, first, the program removes one element from BUFFER and puts it into CD0(V01, U01, G0), then deletes vertex in V01 with  202 H. B. Liu / J. Biomedical Science and Engineering 1 (2008) 200-203 SciRes Copyright © 2008 JBiSE degree lower than k and deletes vertex in U01 with degree lower than k. Next, the program judges whether CD0(V01, U01, G0) is a quasi-biclique. If the CD0 is a quasi-biclique and the size is greater than the current maximum value, the program outputs CD0 and puts the current maximum value as CD0’s size. If it is not a quasi-biclique and its size greater than current maximum value, the program splits the CD0 into two units (CD01, CD02) and puts them into the BUFFER. Otherwise, the program discards the left smaller ones. Part of SQB Algorithm Input. Graph G(V, E) Output. The maximum quasi-bicliques in G(V, E) func Search_Maximum_Quasi_Bicliques() = for tt from 20 to 2 step-1 do OUT= Search_k_Quasi_Bicliques(G, tt) If OUT is not empty Output OUT and Stop the program end for end func Search_Maximum_Quasi_Bicliques() func Search_k_Quasi_Bicliques(G, k)= Divide vertex set V into two sets, V1 and U1 Put V1, V2, G into Complex Data Structure CD(V1, U1, G) Put CD into BufferBUFFER While(BUFFER is not empty) Remove first element in BUFFER to CD0(V01, U01, G0) Delete vertex in V01 with degree lower than k Delete vertex in U01 with degree lower than k If CD0(V01, U01, G0) is Quasi_Bicliques then output CD0 Else if CD0(V01, U01, G0) is not empty then Split CD0 into CD01, CD02 at one unlabeled vertex put CD01, CD02 into BUFFER end while end func Search_k_Quasi_Bicliques() 3.3. Pruning Redundant Quasi-bicliques Through the above steps, SQB obtains every vertex’s quasi-bicliques. As some vertexes might have similar quasi-bicliques, SQB needs to remove redundant ones and obtains the overall distinct quasi-bicliques of the whole graph. SQB deletes one between any two quasi-bicliques QB1(V1, U1) and QB2(V2, U2) if they meet the follow- ing rules: If V2))(U1U2)((V1||2))(U1 )21((⊆∩⊆⊆ ∩ ⊆UVV is true, then delete QB1. If ))12()12((||))1(U2 )12((VUUVUVV ⊆∩⊆⊆ ∩ ⊆ is true, then delete QB2. The first rule means that if V1 is V2’s subset and U1 is U2’s subset, or if V1 is U2’s subset and U1 is V2’s subset, then QB1 is a part of QB2, QB1 can be deleted. The second rule is opposite to the first one. Otherwise, if two quasi-bicliques match neither of the above two rules, SQB keeps both of them. After the pruning operation, SQB obtains the distinct quasi-bicliques of the whole graph and the biggest one is the optimum one of the whole graph. 4. APPLICATION OF SQB The experiment was done on our web server which con- sisted of two Pentium 2 PCs with 4.8 GHZ CPU and 2G RAM. The Saccharomyces cerevisiae dataset Y78, de- rived from [15], consists of 78,390 protein-protein inter- actions, including 5321 proteins. During our experiments, we removed vertexes with degree 1 because they could not produce a biclique. The input graph of our program is with node number 4546. At first, we produced 4546 dis- tinct Distance-2-Subgraphs according to every vertex’s neighbors and their neighbors. The maximum subgraph is with 3164 nodes and the average value is 746.1. So the questions are very tough. About eighty percent of the vertexes have a process time less than 20 seconds and half of them are processed within 1 second. The average time is about 13.4 seconds. Giving the large input graphs, the performance is very remarkable. During our experiments we predicted 5616 quasi-bi- cliques which include empty or redundant ones. A small number of vertexes have more than one maximum quasi-bicliques, so the number of quasi-bicliques is greater than 4546, the number of Distance-2-Subgraphs. Table 1. The Maximum Quasi-Bicliques (YPL212C). |V|=26 YBL038W YBR251W YBR283C YDL136W YDL191W YEL050C YGL103W YGL123W YGR034W YGR220C YIL018W YIL021W YJL063C YLR075W YLR344W YLR378C YMR260C YNL178W YNL284C YNR037C YOL040C YOL127W YOR063W YPL131W YPL183W-A YPR110C |U|=62 YBL087C YBL091C YBR031W YBR048W YBR146W YCR031C YDL061C YDL083C YDL140C YDL202W YDR012W YDR025W YDR101C YDR116C YDR226W YDR237W YDR418W YDR450W YEL054C YER117W YER170W YFL001W YGL063W YGL068W YGL135W YGL147C YGR085C YHR147C YIL133C YJL177W YJL190C YJL191W YKL009W YKL024C YKL170W YKL180W YLR244C YLR340W YLR367W YLR388W YML010W YML025C YML026C YMR143W YMR158W YMR188C YNL067W YNL069C YNL081C YNL177C YNL185C YNL306W YOR116C YOR150W YOR151C YOR207C YOR341W YPL212C YPL220W YPR010C YPR102C YPR166C 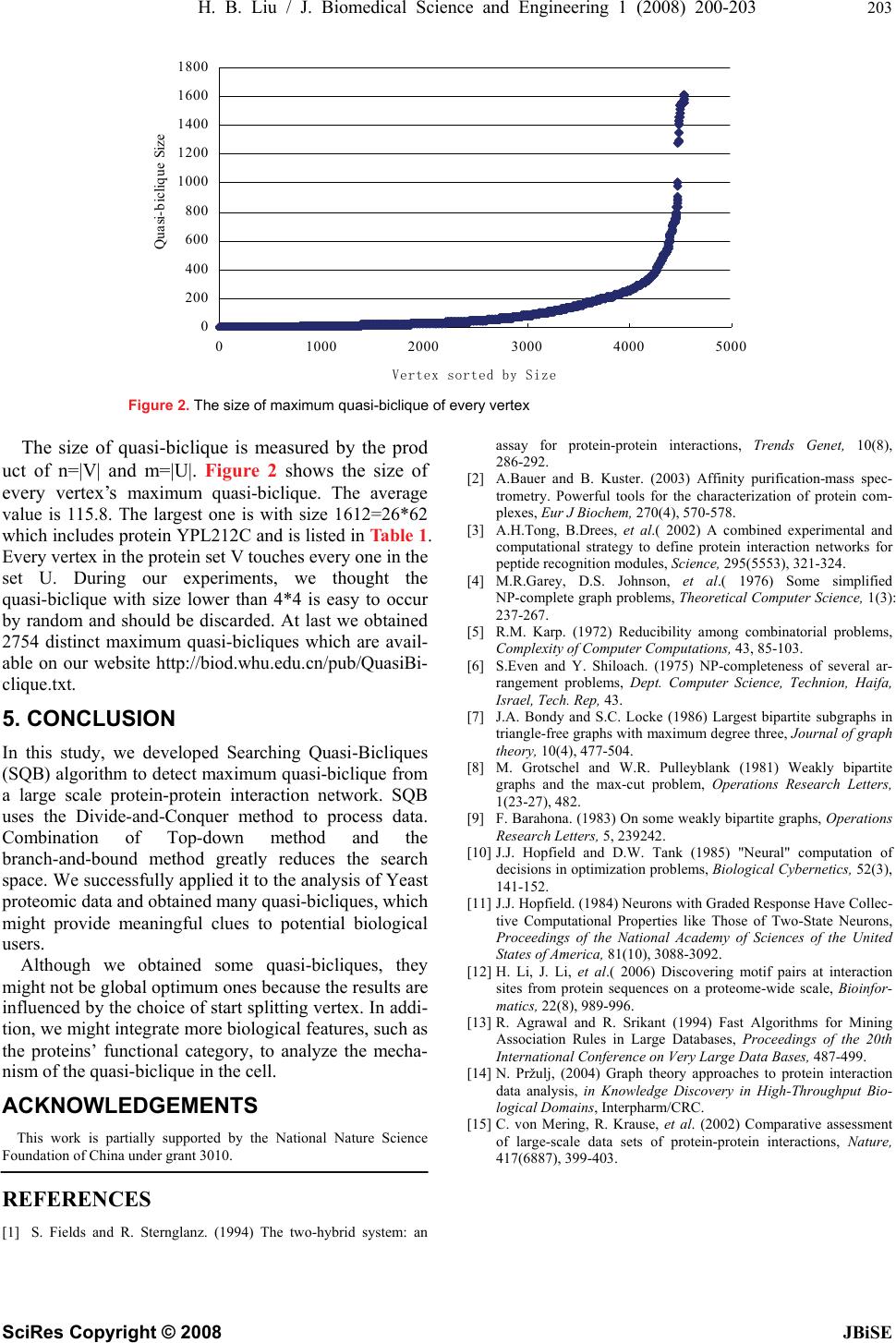 H. B. Liu / J. Biomedical Science and Engineering 1 (2008) 200-203 203 SciRes Copyright © 2008 JBiSE 0 200 400 600 800 1000 1200 1400 1600 1800 01000 20003000 4000 5000 Vertex sorted by Size Quasi-biclique Size Figure 2. The size of maximum quasi-biclique of every vertex The size of quasi-biclique is measured by the prod uct of n=|V| and m=|U|. Figure 2 shows the size of every vertex’s maximum quasi-biclique. The average value is 115.8. The largest one is with size 1612=26*62 which includes protein YPL212C and is listed in Table 1. Every vertex in the protein set V touches every one in the set U. During our experiments, we thought the quasi-biclique with size lower than 4*4 is easy to occur by random and should be discarded. At last we obtained 2754 distinct maximum quasi-bicliques which are avail- able on our website http://biod.whu.edu.cn/pub/QuasiBi- clique.txt. 5. CONCLUSION In this study, we developed Searching Quasi-Bicliques (SQB) algorithm to detect maximum quasi-biclique from a large scale protein-protein interaction network. SQB uses the Divide-and-Conquer method to process data. Combination of Top-down method and the branch-and-bound method greatly reduces the search space. We successfully applied it to the analysis of Yeast proteomic data and obtained many quasi-bicliques, which might provide meaningful clues to potential biological users. Although we obtained some quasi-bicliques, they might not be global optimum ones because the results are influenced by the choice of start splitting vertex. In addi- tion, we might integrate more biological features, such as the proteins’ functional category, to analyze the mecha- nism of the quasi-biclique in the cell. ACKNOWLEDGEMENTS This work is partially supported by the National Nature Science Foundation of China under grant 3010. REFERENCES [1] S. Fields and R. Sternglanz. (1994) The two-hybrid system: an assay for protein-protein interactions, Trends Genet, 10(8), 286-292. [2] A.Bauer and B. Kuster. (2003) Affinity purification-mass spec- trometry. Powerful tools for the characterization of protein com- plexes, Eur J Biochem, 270(4), 570-578. [3] A.H.Tong, B.Drees, et al.( 2002) A combined experimental and computational strategy to define protein interaction networks for peptide recognition modules, Science, 295(5553), 321-324. [4] M.R.Garey, D.S. Johnson, et al.( 1976) Some simplified NP-complete graph problems, Theoretical Computer Science, 1(3): 237-267. [5] R.M. Karp. (1972) Reducibility among combinatorial problems, Complexity of Computer Computations, 43, 85-103. [6] S.Even and Y. Shiloach. (1975) NP-completeness of several ar- rangement problems, Dept. Computer Science, Technion, Haifa, Israel, Tech. Rep, 43. [7] J.A. Bondy and S.C. Locke (1986) Largest bipartite subgraphs in triangle-free graphs with maximum degree three, Journal of graph theory, 10(4), 477-504. [8] M. Grotschel and W.R. Pulleyblank (1981) Weakly bipartite graphs and the max-cut problem, Operations Research Letters, 1(23-27), 482. [9] F. Barahona. (1983) On some weakly bipartite graphs, Operations Research Letters, 5, 239242. [10] J.J. Hopfield and D.W. Tank (1985) "Neural" computation of decisions in optimization problems, Biological Cybernetics, 52(3), 141-152. [11] J.J. Hopfield. (1984) Neurons with Graded Response Have Collec- tive Computational Properties like Those of Two-State Neurons, Proceedings of the National Academy of Sciences of the United States of America, 81(10), 3088-3092. [12] H. Li, J. Li, et al.( 2006) Discovering motif pairs at interaction sites from protein sequences on a proteome-wide scale, Bioinfor- matics, 22(8), 989-996. [13] R. Agrawal and R. Srikant (1994) Fast Algorithms for Mining Association Rules in Large Databases, Proceedings of the 20th International Conference on Very Large Data Bases, 487-499. [14] N. Pržulj, (2004) Graph theory approaches to protein interaction data analysis, in Knowledge Discovery in High-Throughput Bio- logical Domains, Interpharm/CRC. [15] C. von Mering, R. Krause, et al. (2002) Comparative assessment of large-scale data sets of protein-protein interactions, Nature, 417(6887), 399-403. |