Paper Menu >>
Journal Menu >>
 J. Biomedical Science and Engineering, 2008, 1, 178-181 Published Online November 2008 in SciRes. http://www.srpublishing.org/journal/jbise JBiSE Enhanced apoptosis and electrostatic acetylcholi- nesterase activity of abnormally hydrophobic envi- ronment in alzheimer’s plaques Rakesh Sharma1 and Soonjo Kwon2 1Department of Radiology and Molecular Biology, Columbia University, New York 10032. 2Department of Biological Engineering, Utah State University, Logan, Utah 84322. Correspondence should be addressed to Rakesh Sharma (rksz2004@yahoo.com). Received October 6, 2008; revised October 22, 2008; accepted October 22, 2008 ABSTRACT Alzheimer’s disease (AD) is considered a slow neuronal dysfunction process through hypoxia, ischemia and leads to apoptosis mediated senile plaques and neurofibrillary tangles (NFTs). Due to non-invasive approach of plaque characterization, computational techniques based on Brownian dynamics simulation are unique to speculate the electrostatic and kinetic properties of Acetylcho- linesterase (AChE). Typically the MRI spectros- copy high choline peak and enzyme specific to Alzheimer’s Disease (specificity constant (kcat/Km) of AChE) appeared associated with apoptosis and hypoxia. A simple display between synergy of cytokines, apoptosis, elevated AChE and choline is postulated as initial events. The events may be distributed heterogeneously within the senile plaques and neurofibrillary tangles (NFTs) of Alzheimer’s Disease (AD). The role of decreased brain AChE and synergy was associated with specific Magnetic Resonance Spectroscopic (MRS) pattern profiles in AD. These findings suggest that that the altered AChE and early apoptosis events in AD may be associated with specific MR spectral peak patterns. This study opens the possibility of reduced AChE levels causing high choline and reduced N-acetyl ace- tate (NAA) neurotransmitter by MRS after initial apoptosis and/or inflammation to make amyloid plaques in the cerebral tissue of Alzheimer’s disease (AD) patients. These results can be useful in clinical trials on AD lesions. Keywords: Alzheimer’s Disease, Acetylcholi- nesterase, Electrostatics, Dielectic effect, Ionic effect, Brownian dynamics, Apoptosis 1. INTRODUCTION 1.1. Alzheimer’s Disease Azheimer’s disease (AD) has manifestations of senile plaques and neurofibrillary tangles (NFTs) in the cerebral cortex involving hippocampus of Alzheimer’s brains. Many studies have shown that the levels of the reduced ACh neurotransmitter in AD brain had been curiosity in recent past. In this direction, promising success is claimed in drug therapeutic trials based on AChE inhibitors. In AD presumably five neurological cell groups are commonly seen around the cortex rich with AD lesions associated with the low AChE enzyme. Other important problem of AD therapeutics was solved by use of AChE inhibitor higher concentrations such as, tacrine, physostigmine, and BW284C51 to inhibit AChE within AD lesions. Simulta- neously, elevated levels of the ACh substrate also inhibit AChE activity. Serotonin, 5-hydroxytryptophan, car- boxypeptidase inhibitor, and bacitracin, had been good choice to effectively inhibit cholinesterase activity within plaques and tangles, but fail to alter the AChE activity in normal tissue at standard physiological conditions. The initial stages of amyloid plaque formation are not known if they are the result of metabolic defect leading to pathology. The mechanisms were reviewed by Lu et al. 2003 [12]. The process of inflammation in AD was described by Bamberger et al. 2002 [5]. There is continuous hunt of biomarkers useful in AD reported by Ankarcrona et al. 2002 [3]. However, the sequence of these events remains unknown. There are several reports showing that neurons die partly by apoptosis in the AD brain. Drugs blocking apoptosis could therefore be potentially useful for early prevention of neuronal cell death. Biomarkers for apop- tosis should be important tools in the evaluation of drug effects and in the diagnostics of AD. Future strategies are more likely to modify the course of the disease. The most widely accepted hypothesis on the etiopathogenesis of AD proposes that aggregates of beta amyloid (Abeta) form in the brain. Under normal conditions, the predominant amy- loid peptide secreted is Abeta(1-40) with about 10-15% being the longer 1-42 form. In AD, there appears to be an increase in the longer more toxic form which is proposed to trigger tau hyperphosphorylation and neural degenera- tion. Neurotoxicity is thought to be due to altered calcium regulation, mitochondrial damage and/or immune stimula- tion. One strategy for treating AD is the prevention of Abeta release or the blockade of it neurotoxic activity re- SciRes Copyright © 2008  R. Sharma / J. Biomedical Science and Engineering 1 (2008) 178-181 179 SciRes Copyright © 2008 JBiSE ported by Lu 2003 [12]. Present paper explains the electrostatics of lowered AChE catalytic activity in AD brain tissue over normal tissue with possibility of the protein-rich deposits associ- ated with the onset of AD. The paper further, illustrates the possibility of reduced AChE associated with high choline and reduced NAA peaks by MRS and initial events trig- gered by cytokines, apoptosis and inflammation to synthe- size amyloid protein as NFT plaques However, the high concentrations of protein-rich deposits in plaques and NFTs such as βAP, heparan and dermatan sulfate pro- teoglycans, serum amyloid P component, complement factors, and protein kinase C, had been active research over the increased the hydrophobicity of AChE in AD le- sions and abnormally reduced dielectric constant reported by Giambarella et al. 1997 [9]. The AChE catalysis has been explained as electrostatic steering mechanism where altered dielectric conditions seen by βAP deposition. Pos- sibly, dielectric constant shift in AD tissue also allows Coulombic interactions to permeate longer distances re- sulting with enhanced enzymatic activity and simultane- ously decreased ACh levels. MR spectral pattern of en- hanced choline also supports association with decreased ACh levels in AD. 1.2. Acetylcholinesterase The acetylcholinesterase enzyme (AChE) has 537 amino acid long polypeptide in the postsynaptic neural mem- branes of central nervous system and neuromuscular junc- tions by a glycosylphosphatidylinositol linkage. AChE catalyzes the hydrolysis of the acetylcholine (ACh) sub- strate neurotransmitter at cholinergic synapses. AChE hy- drolysis results in the termination of impulse transmission. The determination of the three-dimensional structure of AChE dimer enzyme comprises 12-stranded mixed β-sheet surrounded by 14 α-helices. These subunits assemble through disulfide linkage and hydrophobic interactions. The enzyme structure shows structural characteristic of AChE as a deep (~20Å), narrow active site making en- zyme’s catalytic site Ser200, His440, and Glu327 at its base. The walls of this entity are lined with 14 highly conserved aromatic amino acids of active site. Positively charged ACh substrate toward the active site caused low-affinity cation-π interactions. Further, amino acid charge distribu- tion over AChE creates an electric field around the enzyme contributing to its enzymatic activity (electrostatic steering mechanism) involving its substrate, ACh [12]. Authors determined that the negative field drives the posi- tively-charged ACh substrate molecule toward the en- trance of its active site moiety and increases the catalytic rate of AChE. 1.3. Cytokines, Inflammation, Apoptosis, and Serum AChE Relationship in AD Inflammatory processes play a role in disease progression and pathology of AD, which involves the deposition of amyloid in the brain and the extensive loss of neurons. Amyloid plaque deposition is accompanied by the associa- tion of microglia with the senile plaque, and this interac- tion stimulates these cells to undergo phenotypic activa- tion and the subsequent expression of proinflammatory cytokines and neurotoxic products [5]. Inflammation has been reported in numerous neurodegenerative disorders such as Parkinson's disease, stroke and Alzheimer's disease (AD). In AD, the inflammatory response is mainly located to the vicinity of amyloid plaques. Cytokines, such as In- terleukin-1 (IL-1), Interleukin-6 (IL-6), Tumor Necrosis Factor alpha (TNF-α) and Transforming Growth Factor beta (TGF-β) have been clearly involved in this inflam- matory process. Although their expression is induced by the presence of amyloid-beta peptide, these cytokines are also able to promote the accumulation of amyloid beta peptide. Altogether, IL-1, IL-6, TNF-α and TGF-β should be considered as key players of a vicious circle leading to the progression of the disease reported by Cacquevel et al., 2004 [6]. Inflammatory stimuli also induce nitric oxide production, resulting in oxygen deficiency (hypoxia) and stimulating adenylate cyclase activity. Under these condi- tions, the rarte of apoptosis increases. Neuron dysfunction is partly due to apoptosis in the AD brain (Figure 1). 2. RESULTS AND DISCUSSION 2.1. Ionic Strength Computed rate constants of Torpedo californica AChE as model enzyme at various ionic strengths are given in Fig- ure 2. These values are compared with experimental bi- molecular association constants (kcat/Km) and enzymatic specificity constants (kcat/Km) of a related Electrophorus electricus AChE enzyme as reported by Nolte et al., 1980 [14]. Since the association constant considers the binding event of the reaction and the specificity constant describes both binding and subsequent catalytic turnover, k1 is the theoretical maximum value for the calculated diffu- sion-controlled rate constant, while kcat/Km sets the lower limit on these second-order reactions. As seen in Figure 2, the calculated rate constants found in this work lie be- tween these two extremes throughout the range of ionic strengths tested. This provides encouraging support for the ionic screening approximations used in this work. Fur- thermore, the decrease in the rate of AChE catalysis with increasing ionic strength provides strong evidence that an electrostatic steering mechanism plays a role in AChE kinetics. The similarity in the negative slope observed for both association and specificity constants indicates that the ligand binding step of the reaction is dependent upon sol- vent salt concentration. 2.2. Substrate Radius To demonstrate the limited accessibility of this enzyme’s active site structure, simulations were performed using various substrate radii (Figure 2). It is reasonable to pre- dict increased rate constant values with a reduced substrate radius since the probability of a smaller substrate pene- trating the active site gorge and reacting with AChE is higher. The results shown in Figure 3 show this expected 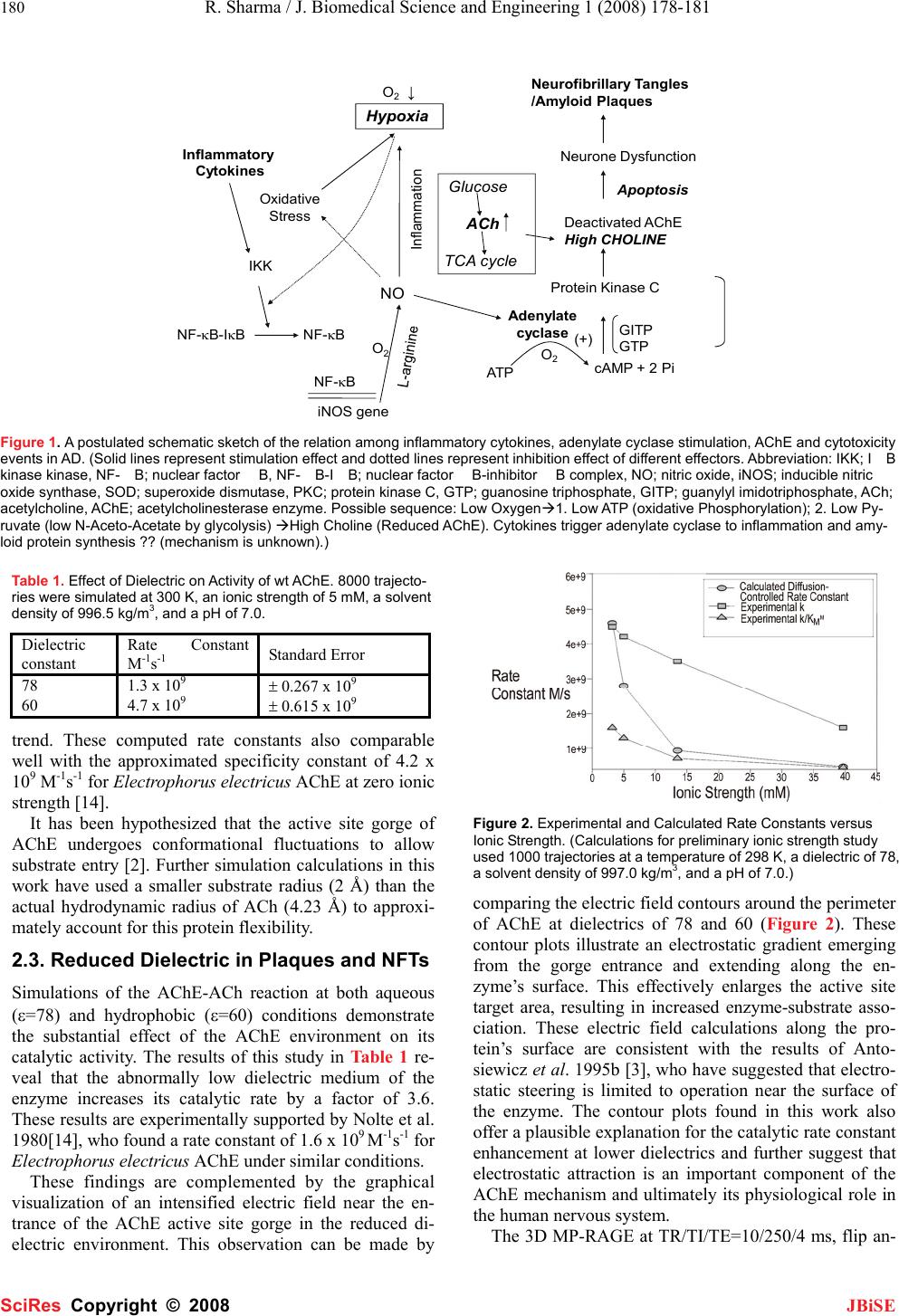 180 R. Sharma / J. Biomedical Science and Engineering 1 (2008) 178-181 SciRes Copyright © 2008 JBiSE Hypoxia O 2 ↓ NO Adenylate cyclase Oxidative Stress Inflammatory Cytokines IKK NF-κB-IκBNF-κB iNOS gene NF-κBATP cAMP + 2 Pi Protein Kinase C Deactivated AChE High CHOLINE O 2 GITP GTP Inflammation Neurone Dysfunction Apoptosis (+) Neurofibrillar y Tangles /Amyloid Plaques Glucose ACh TCA cycle O 2 Figure 1. A postulated schematic sketch of the relation among inflammatory cytokines, adenylate cyclase stimulation, AChE and cytotoxicity events in AD. (Solid lines represent stimulation effect and dotted lines represent inhibition effect of different effectors. Abbreviation: IKK; IB kinase kinase, NF-B; nuclear factor B, NF -B -IB; nuclear factor B -inhibitor B complex, NO; nitric oxide, iNOS; inducible nitric oxide synthase, SOD; superoxide dismutase, PKC; protein kinase C, GTP; guanosine triphosphate, GITP; guanylyl imidotriphosphate, ACh; acetylcholine, AChE; acetylcholinesterase enzyme. Possible sequence: Low OxygenÆ1. Low ATP (oxidative Phosphorylation); 2. Low Py- ruvate (low N-Aceto-Acetate by glycolysis) ÆHigh Choline (Reduced AChE). Cytokines trigger adenylate cyclase to inflammation and amy- loid protein synthesis ?? (mechanism is unknown).) Table 1. Effect of Dielectric on Activity of wt AChE. 8000 trajecto- ries were simulated at 300 K, an ionic strength of 5 mM, a solvent density of 996.5 kg/m3, and a pH of 7.0. Dielectric constant Rate Constant M-1s-1 Standard Error 78 1.3 x 109 ± 0.267 x 109 60 4.7 x 109 ± 0.615 x 109 trend. These computed rate constants also comparable well with the approximated specificity constant of 4.2 x 109 M-1s-1 for Electrophorus electricus AChE at zero ionic strength [14]. It has been hypothesized that the active site gorge of AChE undergoes conformational fluctuations to allow substrate entry [2]. Further simulation calculations in this work have used a smaller substrate radius (2 Å) than the actual hydrodynamic radius of ACh (4.23 Å) to approxi- mately account for this protein flexibility. 2.3. Reduced Dielectric in Plaques and NFTs Simulations of the AChE-ACh reaction at both aqueous (ε=78) and hydrophobic (ε=60) conditions demonstrate the substantial effect of the AChE environment on its catalytic activity. The results of this study in Table 1 re- veal that the abnormally low dielectric medium of the enzyme increases its catalytic rate by a factor of 3.6. These results are experimentally supported by Nolte et al. 1980[14], who found a rate constant of 1.6 x 109 M-1s-1 for Electrophorus electricus AChE under similar conditions. These findings are complemented by the graphical visualization of an intensified electric field near the en- trance of the AChE active site gorge in the reduced di- electric environment. This observation can be made by Figure 2. Experimental and Calculated Rate Constants versus Ionic Strength. (Calculations for preliminary ionic strength study used 1000 trajectories at a temperature of 298 K, a dielectric of 78, a solvent density of 997.0 kg/m3, and a pH of 7.0.) comparing the electric field contours around the perimeter of AChE at dielectrics of 78 and 60 (Figure 2). These contour plots illustrate an electrostatic gradient emerging from the gorge entrance and extending along the en- zyme’s surface. This effectively enlarges the active site target area, resulting in increased enzyme-substrate asso- ciation. These electric field calculations along the pro- tein’s surface are consistent with the results of Anto- siewicz et al. 1995b [3], who have suggested that electro- static steering is limited to operation near the surface of the enzyme. The contour plots found in this work also offer a plausible explanation for the catalytic rate constant enhancement at lower dielectrics and further suggest that electrostatic attraction is an important component of the AChE mechanism and ultimately its physiological role in the human nervous system. The 3D MP-RAGE at TR/TI/TE=10/250/4 ms, flip an-  R. Sharma / J. Biomedical Science and Engineering 1 (2008) 178-181 181 SciRes Copyright © 2008 JBiSE A B C D Figure 3. A representative 1.5 Tesla MRI T1 axial image of AD brain (panel A) with post-segmentation (panel B) showin neurofi- brillary tangle with arrow.(The scan setting were: 3D MP-RAGE were TR/TI/TE=10/250/4 ms, flip angle 15,1.0 x 1.0 mm2 reso- lution, and 1.4 mm thick partitions showing VOI and spectroscopic voxels and selective Choline and NAA spectral peaks from right and left sides to compare metabolites in cortical and ventricular regions (Choline peak on right panel D). For details see reference (Sharma 2005).) gle 15, 1.0 x 1.0 mm2 resolution, and 1.4 mm thick parti- tions in our previous report showed NAA/Cr, NAA/Cho were significantly reduced (p < 0.02 and p value < 0.03 respectively) in AD compared with elderly controls due to reductions of NAA after NAA correction in AD. Fur- thermore, the difference of hippocampal NAA between the groups without atrophy correction (which reflects both NAA and volume changes) was about 40% larger than with correction of atrophy as shown in Figure 3. The major elevated peak was choline at 3.00 ppm. These finding suggest a possibility of reduced glycolysis leading to low N-acetyl acetate formation and choline accumula- tion indirectly reducing TCA cycle to generate enough ATP in localized tissue. Low ATP and oxygen are well understood to lead inflammation and amyloid plaque formation. 3. CONCLUSIONS Abnormal Magnetic Resonance spectral NAA, choline peak patterns are associated with low AChE enzyme ac- tivity in AD with possible enhanced ACh breakdown and surrounding electrostatic field of this enzyme. However, the association of cytokines, apoptosis to lead hypoxia and inflammation in amyloid plaque in AD formation is unclear. REFERENCES [1] S. Allison, R. Bacquet, and J. McCammon. (1988) Simulation of the Diffusion-Controlled Reaction between Superoxide and Super- oxide Dismutase. II. Detailed Models. Biopolymers, Vol. 27, 251-269. [2] J. Antosiewicz, J. McCammon, S. Wlodek, and M. Gilson. (1995) Simulation of Charge-Mutant Acetylcholinesterases. Biochemistry, Vol. 34, pg. 4211-4219. [3] M. Ankarcrona, B. Winblad. (2005) Biomarkers for apoptosis in Alzheimer's disease. Int J Geriatr Psychiatry, 20, 101–105. [4] J. Antosiewicz, S. Wlodek, J. McCammon. (1996) Acetylcholi- nesterase: Role of the Enzyme’s Charge Distribution in Steering Charged Ligands Toward the Active Site. Biopolymers, Vol. 39, 85-94. [5] M.E. Bamberger and G. E. Landreth. (2002) Inflammation, Apop- tosis, and Alzheimer's Disease. The Neuroscientist, Vol. 8, 276-283. [6] M. Cacquevel, N. Lebeurrier, S. Cheenne, and D. Vivien. (2004) Cytokines in Neuroinflammation and Alzheimer's Disease. Current Drug Targets, Vol. 5, 529-534. [7] K. Davis and P. Powchik. (1995) The Lancet, Vol. 345, 625-630. [8] D. Ermak and J. McCammon. (1978) Brownian Dynamics with Hydrodynamic Interactions.J. Chem. Phys., Vol. 69, 1352-1360. [9] U. Giambarella, T. Yamatsuji, T. Okamoto, T. Matsui, T. Ikezu. Y. Murayama, M.A. Levine, A. Katz, N. Gautam, and I. Nishimoto. (1997) G protein complex -mediated apoptosis by familial Alzheimer's disease mutant of APP. The EMBO Journal, Vol. 14, 4897–4907. [10] T. Golde, S Estus, L Younkin, D Selkoe, and S Younkin. (1992) Processing of the Amyloid Protein Precursor to Potentially Amy- loidogenic Derivatives. Science, Vol. 255, 728-730. [11] J. Hardy and D. Allsop. (1991) Amyloid Deposition as the Central Event in the Aetiology of Alzheimer’s Disease.Trends in Pharm. Sci., Vol. 12, pg. 383-388. [12] D. Lu. (2003) Mechanisms of apoptosis in Alzheimers disease. Journal of Neurochemistry,Vol. 87, 733-41. [13] R. Nitsch, B. Slack, R. Wurtman and J. Growdon. (1992) Release of Alzheimer Amyloid Precursor Derivatives Stimulated by Activa- tion of Muscarinic Acetylcholine Receptors. Science, Vol. 258, pg. 304-307. [14] H. Nolte, T. Rosenberry and E. Neumann. (1980) Effective Charge on Acetylcholinesterase Active Sites Determined from the Ionic Strength Dependence of Association Rate Constants with Cationic Ligands. Biochemistry, Vol. 19, 3705-3711. [15] S. Northrup, S. Allison, and J. McCammon. (1984) Brownian Dy- namics Simulation of Diffusion-Influenced Bimolecular Reactions. J. Chem. Phys., Vol. 80, 1517-1524. [16] F. Rose. (1981) Metabolic Disorders of the Nervous System, Pit- man, London, pg. 411. [17] C. Schätz., C. Geula, and M. Mesulam. (1990) Competitive Sub- strate Inhibition in the Histochemistry of Cholinesterase Activity in AD. Neurosci. Lttrs., Vol. 117, 56-61. [18] A. Shafferman, A. Ordentlich, D. Barak, C. Kronman, R. Ber, T. Bino, N. Ariel, R. Osman, and B. Velan. (1994) Electrostatic attrac- tion by surface charge does not contribute to the catalytic efficiency of acetylcholinesterase. EMBO, Vol. 13, 3448-3455. [19] R. Sharma. (2005) Molecular Imaging by Proton Magentic Reso- nance Imaging (MRI) and MR Spectroscopic Imaging (MRSI) in Neurodegeneration. Informatica Medica Slovenica, Vol. 10, 35-55. [20] J. Sussman, M. Harel, F. Frolow, C. Oefner, A. Goldman, L. Toker, and I. Silman. (1991) Atomic Structure of Acetylcholinesterase from Torpedo californica: A Prototypic Acetylcholine-Binding Protein. Science, Vol. 253, 872-878. [21] D. Voet and J. Voet. (1995) Biochemistry, 2nd Edition, John Wiley & Sons, Inc., New York, 317. [22] D. Voet and J. Voet. (1995) Biochemistry, 2nd Edition, John Wiley & Sons, Inc., New York, 317. Choline NAA |