Journal of Cancer Therapy
Vol.4 No.3(2013), Article ID:31947,10 pages DOI:10.4236/jct.2013.43095
Lenalidomide and Arsenic Trioxide Have Independent Non-Interfering Effects When Used in Combination on Myeloma Cell Lines in Vitro
![]()
1Hematology Department, General Hospital, Tianjin Medical University, Tianjin, China; 2Immunology Program and Malignant Hematology Program, Moffitt Cancer Center and Research Institute, Tampa, USA.
Email: Sheng.Wei@moffitt.org
Copyright © 2013 Huaquan Wang et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received March 17th, 2013; revised April 18th, 2013; accepted April 26th, 2013
Keywords: Multiple Myeloma; Lenalidomide; Arsenic Trioxide; cdc25c
ABSTRACT
Multiple myeloma (MM) is a plasma cell neoplasm characterized for its fast evolution and for being practically incurable, presenting a strong need for the development of therapies to target it. Among those under study are lenalidomide and arsenic trioxide (ATO) which show individual clinical promise, although never tested together. However, the combination of ATO with thalidomide, another immunomodulatory drug and lenalidomide’s structural albeit less potent analog, have been tried clinically with some success. Therefore, we investigated the effect the combination of lenalidomide and ATO have on the MM-derived U266 and RPMI 8226 cell lines. We observed that both compounds have separate, non-interfering, anti-myeloma mechanisms with ATO demonstrating strong cytotoxic effects while lenalidomide’s role remains cytostatic and immunomodulatory. However, ATO decreases cdc25c, which helps sensitize cells to lenalidomide effects enhancing the efficacy of their interaction. Mechanistically the combination of these two agents decreased the expression of MDM2, without affecting p53 activation or its expression. Therefore, this short study provides the foundation to continue mechanistic studies of the combination of lenalidomide and ATO as a foundation for future clinical application.
1. Introduction
Multiple myeloma (MM) is a plasma cell neoplasm generally derived from one clone in the bone marrow and characterized by skeletal destruction, renal failure, anemia, and hypercalcemia [1,2]. It accounts for 1% of all malignancies, and it is the second most common hematological malignancy in the United States where the overall incidence rate is 4.4/100,000/year, remains largely incurable and has a decreased survival post-diagnosis demonstrating the dire need for therapies that can treat or eventually decimate the tumor [3,4]. To complicate matters, malignancy of the disease is not only enhanced by an increase in tumor burden but driven by the direct interaction of the malignant clones with the tumor microenvironment, which regulate immune activation and the secretion of cytokines involved in pro-tumor signaling pathways [4].
Among the novel agents introduced in the last decade are the immunomodulatory drugs (IMiD) thalidomide and its derivative lenalidomide because they target both the MM clone as well as its interaction with the tumor microenvironment [5-10]. Its antitumor activity seems mediated through modulation of both the cytokine and cellular tumor cell microenvironment, such as a decreases in the pro-survival cytokine IL-6, through a mechanisms requiring the downregulation of phosphatases like Cdc25C [8,11,12]. The cell division cycle (Cdc) 25 dual-specificity phosphatases are an important family member of proteins that regulate cell cycle progression [13-15]. Three members of this family exist in humans and play a crucial role in the activation of cyclin-dependent kinases (Cdk) through dephosphorylation. Cdc25C over-expression has been reported in various types of human malignancies including MM and, moreover, has been correlated with either poor prognosis or tumor aggressiveness [14]. Importantly, we have recently demonstrated that one of the main mechanisms of lenalidomide cytotoxicity is through the down-regulation of Cdc25c [11]. Early clinical investigations noted its efficacy in relapsed and/ or refractory MM patients and lenalidomide is now used successfully in combination with other agents such as dexamethasone [16-18]. However, these drugs also have several dose limiting side effects combined with a high rate of relapse. Therefore combinatorial modalities of lenalidomide provide a potential avenue to discover new therapies for refractory disease that prevents drug resistance, and hence the aim of continuing preclinical studies is to aid in the identification of novel targeted treatments to enhance tumor cytotoxicity and avoid drug resistance; a goal more attainable with combined therapy.
Arsenic trioxide (ATO), a historically known treatment for many ailments, targets a wide variety of hematological and solid malignancies derived from several different tissue types [19,20]. It affects numerous intracellular signal transduction pathways and causes many alterations in cellular function, but it is primarily known for the induction of reduced viability, induction of apoptosis and growth inhibition of cancer cell lines at pharmacological concentrations [19,21-26]. Importantly, it has shown substantial efficacy in treating both newly diagnosed and relapsed patients with MM in particular as an option for the treatment of relapsing or refractory disease for which other therapies have failed [19,21,27-29]. Therefore, understanding the mechanisms of its action on myeloma cells will be critical to understand the potential for synergy between ATO and its combination with other chemotherapeutic agents to provide enhanced benefit in cancer therapy. Because of its ability to target the tumor, it has been postulated that a good combination would be the use of IMiDs in order to disrupt the tumor microenvironment while ATO targets the tumor directly. In particular, the combination of ATO with Thalidomide, from which lenalidomide derives, has produced favorable results in clinical trials for MM as well as other Malignancies such as Myelodysplastic syndrome (MDS) [30,31]. Therefore, in our current study we investigated the in vitro mechanism of the combination of lenalidomide and ATO with the hypothesis that lenalidomide is a more effective IMiD. We found that this drug combination has a greater effect on the induction MM cell susceptibility to apoptosis by ATO compared to either treatment alone. Additionally, ATO treatment does not impair the beneficial effects characteristic of lenalidomide.
2. Materials and Methods
2.1. Reagents
Lenalidomide (Celgene Corp., Summit, NJ) was dissolved in DMSO (final concentration, 0.1%). ATO was purchased from Sigma-Aldrich (St. Louis, MO) and the stock solution (10 mM) was prepared in 0.1 N NaOH. Cell Titer 96 Aqueous One Solution Cell Proliferation Assay was purchased from Promega (Madison, WI). FITC Annexin V Apoptosis Detection Kit was purchased from BD Pharmingen (Franklin Lakes, NJ). Antibodies for Western Blotting were purchased from the following suppliers: Cleaved PARP, Caspase-3, Bcl-2, P53, P53ser15, and mouse and rabbit horseradish peroxidase (HRP)- conjugated IgG (Cell Signaling Technology, Inc., Danvers, MA); MDM2 and PP2A (Abcam, Cambridge, MA); CDC25C (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) and ß-actin (Sigma-Aldrich, St. Louis, MO). Human IL-6 High Sensitivity ELISA kit was purchased from eBioscience (San Diego, CA). CycLex® Cdc25C Protein Phosphatase Fluorometric Assay Kit was purchased from MBL International (Woburn, MA).
2.2. Cell Culture
U266 and RPMI 8226 (8826) were purchased from the American Type Culture Collection (ATCC, Manassas, VA). U266 and 8226 cells were cultured in RPMI 1640 (Invitrogen, Carlsbad, CA) supplemented with penicillin (100 units/mL) and streptomycin (100 µg/mL) and 10% fetal bovine serum (v/v).
2.3. Methyl Tetrazolium Salt (MTS) Assay
MTS assays (Cell Titer 96 Aqueous One Solution Cell Proliferation Assay, Promega) were used to quantify the percentage of metabolically active cells that were either untreated or treated with lenalidomide, ATO or both drugs. Briefly, 2 × 104 cells per well were plated onto a 96-well plate. Lenalidomide, ATO, or both effects were tested at different ranges of times and concentrations and each specific time-point 20 μL of MTS reagent was added followed by incubation for an additional 4 hours at 37˚C. The plates were quantified by measuring the optical density at 490 nm (Benchmark Plus Microplate Spectrophotometer, Bio-Rad Laboratories (UK) Ltd.).
2.4. RNA Isolation and Reverse Transcription Reaction
Total RNA was purified from U266 and 8226 using TRIzol-Reagent according to the manufacturer’s instructions (Life Technologies). The purity and concentration were measured using a spectrophotometer. Reverse transcription (RT) reactions were performed using iScriptTM cDNA Synthesis kit (BIO-RAD). cDNA was synthesized by adding 1 μg of total RNA, 5× iScript Reaction Mix (4 μL), and 1 μL of iScript Reverse Transcriptase to a total volume of 20 μL. The reaction was incubated at 25˚C for 5 min, 42˚C for 30 min, and 85˚C for 5 min.
2.5. Analysis of mRNA Expression by Real-Time Quantitative-PCR (Q-PCR)
Q-PCR reactions were performed by means of iQ SYBR Green Supermix of Bio-Rad. Each reaction (25 μL) contained 12.5 μL of iQ SYBR green supermix, 0.25 μL of forward primer (20 μM), 0.25 μL of reverse primer (20 μM), 11 μL of RNase-free water, and 1.0 μL of cDNA. The following cycles were performed 1 × 3 min at 95˚C, 40 amplification cycles (15 s 95˚C, 60 s 56˚C), 1 × 1 min 95˚C, 1 × 1 min 55˚C and a melting curve (80 × 10 s 55˚C with an increase of 0.5˚C per 10 s). A negative control without cDNA template was run with every assay. The optimal melting point of dsDNA I and the efficiency of the reaction were optimized beforehand. Transcript copy number per individual was calculated by normalization to GAPDH expression. Oligonucleotide primers for amplifying Cdc25C (NC_000005) forward primer: 5’-AGA GCA AGA CCC TGT CTC AA-3’, Cdc25C reverse primer: 5’-TCT CAT CCT TCC TTC ACA GC-3’. IL-6 forward primer was (5’-AGA CAG CCA CTC ACC TCT TC-3’), and IL-6 reverse primer was (5’-TCT TTG CTG CTT TCA CAC AT-3’). P53 forward primer was (5’-GTA CAT CTG GCC TTG AAA CC-3’), and p53 reverse primer was (5’-AGC TGC CCA ACT GTA GAA AC-3’). MDM2 forward primer was (5’- GTC AAT CAG CAG GAA TCA TCG-3'), and MDM2 reverse primer was (5'- CCT TTT GAT CAC TCC CAC CTT -3’). The GAPDH forward primer was (5’-CAA AAG GGT CAT CAT CTC TGC-3’), and the GAPDH reverse primer was (5’-GAG GGG CCA TCC ACA GTC TTC-3’).
2.6. Cdc25C Phosphatase Assay
The phosphatase assay was performed as per manufacturer’s instructions. Briefly, 2.5 µL of rh-Cdc25C (0.1 µg/µL) was incubated for 30 min at 37˚C with the indicated concentrations of either Cdc25˚C Phosphatase inhibitor (CDC25 Phosphatase Inhibitor II, NSC 663284 (Calbiochem) or treatment with lenalidomide, ATO or both in 1× assay buffer. After incubation, samples were aliquoted into 96-well plates and 5 µL of the 10× fluorophospho substrate, OMFP, was added. After 15 min of incubation at room temperature, 25 μL of stop solution was added to each well. The sample was measured on a synergy fluorescent plate reader with excitation at 485 nm and emission at 528 nm. Percentage activity was calculated according to the equation (OD of test sample/OD of control) × 100.
2.7. Apoptosis Assays and Cell-Cycle Analysis
U266 and 8226 at a concentration of 2.0 × 105 cells per well were seeded in a 12-well plate. The cells were either untreated or treated with 20 μM of lenalidomide, 1 μM ATO or both for an additional 48 h. The plates were incubated for 4 d and analyzed for apoptosis using the Annexin V apoptosis kit (BD Pharmingen). Each well was resuspended in 1× binding buffer at a concentration of 1.0 × 106 cells per milliliter and stained with AnnexinFITC and propidium iodide. All samples were analyzed by flow cytometry using a FACScalibur within 30 min of staining. For cell-cycle analysis, cells were treated with lenalidomide, ATO, or both for 48 h at 37˚C, then washed and resuspended in BD Stain Buffer (106/ml). Cold ethanol (3 ml) was added while vortexing, and the cells were then incubated overnight at 4˚C and washed 2 times in stain buffer. One mL of PI staining solution (3.8 mM Na citrate, 50 μg/ml PI in PBS) was added to each cell pellet along with 50 μL of RNase A solution (10 μg/ml). Cell suspensions were incubated for 4 h at 4˚C before analysis on BD FACScalibur (BD BioSciences).
2.8. Western Blot Analysis
The cells were solubilized with lysis buffer (50mM TrisHCl (pH 7.4), 0.5% (v/v) Nonidet P-40 (Nonidet P-40), 150mM NaCl, 5mM EDTA, 50mM NaF, 1mM Na3VO4, 1mM phenylmethylsulfonyl fluoride, 10μg/ml leupeptin, and 10μg/ml aprotinin), and whole cell lysates (WCLs) were prepared by centrifugation at 12,000 g for 15 min at 4˚C. Proteins from WCLs were separated by SDS-PAGE (10% PAGE) and transferred onto PVDF membrane filters (Immobilon; Millipore). The membranes were immunoblotted with the respective antibodies, and bound antibodies were visualized with horseradish peroxidaseconjugated antibodies against mouse or rabbit IgGs (BioRad) using chemiluminescence reagents (Western Lightning; PerkinElmer Life Sciences).
2.9. Statistical analysis
Data are presented as mean ± SD. Differences between two groups were evaluated by Student’s t-test; p < 0.05 was considered to be statistically significant.
3. Results
3.1. ATO’s Robust Effects on Cell Viability Slightly Increase after Combination with Lenalidomide
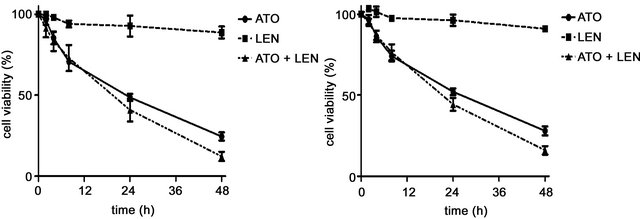
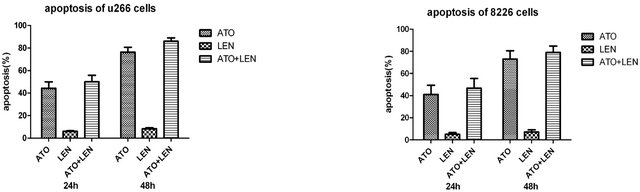
In order to compare in vitro the individual or combined effect of these drugs’ on MM cells we initially investigated the effects of ATO, lenalidomide of their combination on the viability the MM cell lines U266 and 8226. We treated cultured cells with either 1 μM ATO, 20 μM lenalidomide or both for 48 hours and tested the cellular viability in an MTS assay (Figures 1(a) and (b)). We observed that while lenalidomide only modestly inhibited cell growth ATO, with or without lenalidomide, had a profound effect on the viability of both cell lines. Interestingly, while together they increased the apoptotic effect it was in an additive manner, slightly increasing ATO’s apoptotic effects on these cells. Similarly, different concentrations of lenalidomide alone did not induce apoptosis of MM myeloma cells while ATO demonstrated a clear threshold for cytotoxicity of these cell lines (Figures 1(c) and (d)). To corroborate these results and also to understand the contribution of lenalidomide to apoptotic effects on these MM cells we repeated our experiments with ATO, lenalidomide or both at the same concentrations mentioned above and analyzed cells at either 24 or 48 hours by AnnexinV/PI expression in flow cytometry. Indeed, just as we observed for viability, the highest increases in apoptotic cells were in ATO treated cells either as a single agent or in combination (Figures 2(a) and (b)). In order to understand the apoptotic mechanisms of each agent or their combination we performed western blot on the lysates of MM cells treated with each one of them and measured for PARP, caspase 3 and the antiapoptotic molecule Bcl-2. Strong PARP cleavage was induced by the presence of ATO in the culture on both cell lines but lenalidomide had differential effects on both cell lines. Lenalidomide induced a slight increase in PARP cleavage in U266 cell but no caspase 3 cleavage while the reverse was seen in 8226 cells, no PARP cleavage but caspase 3 instead. In both lenalidomide had no effect on the expression of Bcl2. Therefore, the slight decrease in cellular viability induced by lenalidomide can be due to these apoptotic effects although it is clear that they do not add much to ATO as a cytotoxic agent.
 (a)
(a)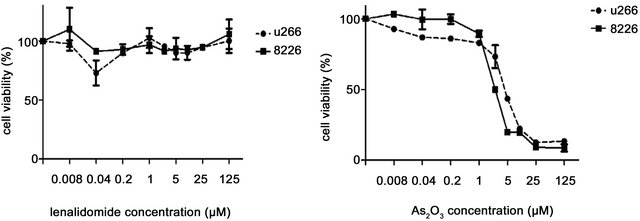 (b)
(b)
Figure 1. Effect of ATO, lenalidomide (LEN), or both on the viability of myeloma cell lines (a) U266 cells and (b) 8266 cells were treated with ATO (1 μM), LEN (20uM), or both at different time points and cell viability was measured in an MTS assay as described in the methods section. (c) Cell viability of U266 and 8226 myeloma cells was measured by MTS assay after treatment with increasing concentrations of ATO or (d) lenalidomide. For all the figures data are mean ± SD of three independent experiments.
 (a)
(a)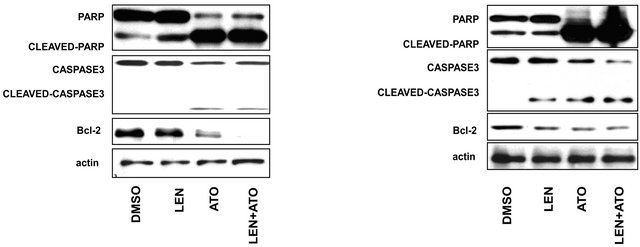 (b)
(b)
Figure 2. Apoptosis analysis of myeloma cells after treatment by ATO, LEN, or both (a) U266 cells and (b) 8266 cells were treated with ATO (1uM), LEN (20 μM), or their combination for either 24 or 48 hours after which apoptosis was assessed by Annexin-V/PI staining by flow cytometry in a FACScalibur. Data represents the mean ± SD of three independent experiments. (c) U266 and (d) 8266 cells were treated with ATO (1 μM), LEN (20 μM), or both for 48 hours before cell lysis. Lysates where then analyzed by western blot for the cleavage of PARP and caspase-3 as well as the presence of Bcl-2. Actin was used as a loading control.
3.2. Treatment with ATO Does Not Affect Lenalidomide’s IMiD Activity
Use One of the main cytokines affected by lenalidomide is IL-6. We therefore wanted to understand if ATO can provide the same effects or if the combination of ATO and lenalidomide could maintain the beneficial effects seen with lenalidomide. Cells were cultured with ATO (1 μM), lenalidomide (20 μM) or both and the total RNA and cell culture supernatants were collected. IL-6 mRNA expression, as measured by real time RT-PCR, demonstrates that while ATO significantly downregulates IL-6 in myeloma cells, lenalidomide’s effect on IL-6 inhibition is much stronger. Furthermore, the combination of lenalidomide and ATO has an additive effect, demonstrating the highest downregulation of IL-6 expression on both cell lines (Figures 3(a) and (b)). Conversely, while U266 IL-6 secretion levels were downregulated throughout, we did not observe a significant difference with either compound on the secretion of IL-6 by 8226 cells indicating that there is still a difference between those cell lines that prevents the downregulation of IL-6 secretion even if its production is reduced (Figures 3(c) and (d)).
3.3. Treatment with Lenalidomide and ATO Decreases the Expression of MDM2, but Had No Affect p53 Levels or Activity
Next we tested the effect these compounds had on P53 and MDM2. Western blot analysis revealed that total protein levels of P53 were unaffected. Additionally, P53 activity was also unaltered by treatment as measured by the phosphorylation of P53 at the serine 15 residue. Both
 (a)
(a)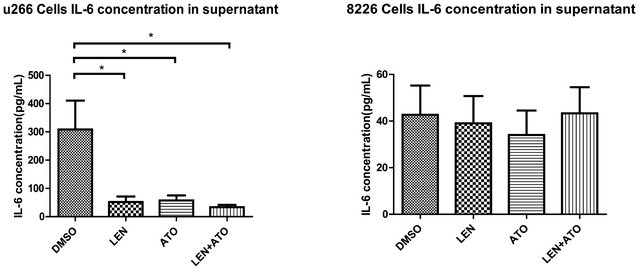 (b)
(b)
Figure 3. Gene and protein expression of IL-6 in myeloma cells after treatment with ATO, LEN, or both (a) U266 cells and (b) 8266 cells were treated with ATO (1 μM), LEN (20 μM), or both for 48 hours before lysing the cells with TRIzol reagent for total RNA isolation. Messenger RNA expression was assessed by real-time PCR and results were analyzed by the DDCt method. The supernatants of (c) U266 cells and (d) 8266 cells treated with ATO (1 μM), LEN (20 μM), or both for 48 hours where saved at –80˚C until use in an ELISA assay. Data are means ± SD of three independent experiments.
lenalidomide and ATO were able to downregulate the expression of MDM2. Not surprisingly, MDM2 was also downregulated when the drugs were combined (Figure 4(a)). Gene expression experiments confirmed our protein results where there were no apparent changes in P53 but downregulation of MDM2 was observed after any of the treatments (Figures 4(b) and (c)). These effects were observed in both cell lines tested, but in the case of 8226 cells, the combination of lenalidomide and ATO resulted in undetectable MDM2 protein levels.
3.4. Combined Treatment Strongly Downregulates the Expression of cdc25c at Both Protein and Gene Expression Levels
As discussed in the introduction, we recently reported that lenalidomide’s action is enhanced in MDS patients with 5(q) deletion, and this is proposed to be due to the haplodeficiency of PP2A and Cdc25c. Considering this, we propose that downregulation of these molecules by other methods could open the possibilities to use lenalidomide to treat other diseases. We next tested whether treatment with ATO could downregulate Cdc25C, PP2A, or both. We found that Cdc25C, but not PP2A was down-regulated in the presence of lenalidomide, ATO, or both compounds (Figures 5(a) and (d)). Although there only was a modest decrease in Cdc25C’ protein levels with treatment of lenalidomide or ATO alone, this effect was much more profound when the cells were treated with both drugs (Figures 5(a), (b), (d) and (e)). We also measured the phosphatase activity of Cdc25C, and similar to what we saw with protein expression, Cdc25C phosphatase activity was inhibited only slightly by lenalidomide, but when the cells were treated with both lenalidomide and ATO, inhibition was similar to what we
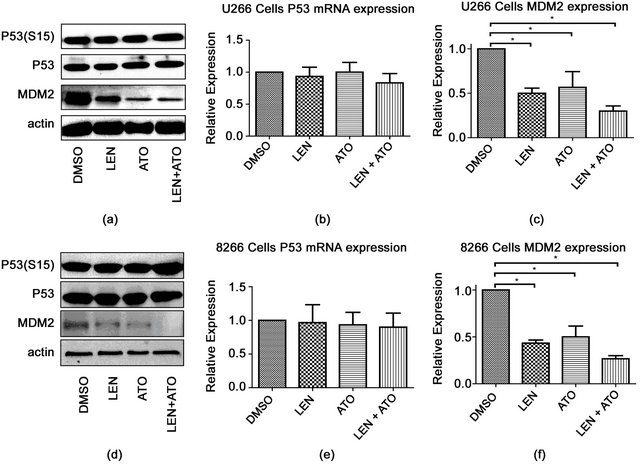
Figure 4. Effect of ATO, LEN, or their combination on p53 and mdm2 in myeloma cell lines after treatment (a) U266 cells were treated with either 0.2% DMSO, ATO (1uM), LEN (20 μM), or both for 48 hours after which protein lysates were analyzed by western blot for the presence of P53 or its phosphorylated form (S15) or MDM2. Actin served as a loading control. Total RNA was also collected from treated U266 as in (a) and assessed for changes in expression in (b) P53 and (c) MDM2 by the DDCt method using the primers described in the methods section. The same experiment was repeated for 8226 cells and (d) protein expression, as well as (e) P53 and (f) MDM2 gene expression were assessed as in (a). Data represents the mean ± SD of three independent experiments.
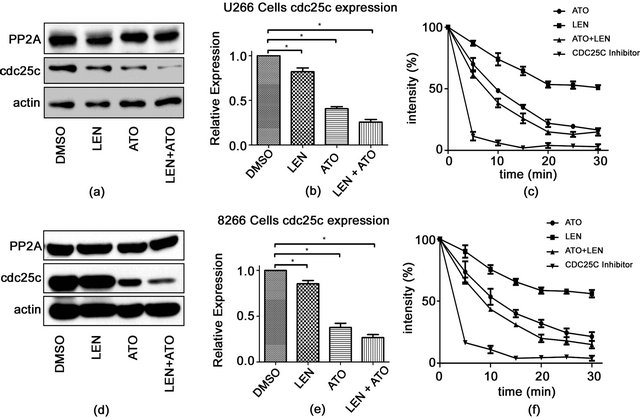
Figure 5. Changes in cdc25c expression and activity in myeloma cell lines after treatment with ATO, LEN, or both (a) U266 cells were treated with either 0.2% DMSO, ATO (1 μM), LEN (20 μM), or both for 48hours after which protein lysates were analyzed by western blot for the presence of either PP2A or cdc25c. Actin served as a loading control. (b) Total RNA was also collected from treated U266 as in (a) and assessed for changes in expression of cdc25c by the DDCt method using the primers described in the methods section. (c) Specific phosphatase activity of cdc25c was measured after treatment by kit as per the manufacturer’s protocol (see Materials and methods section) after collecting at various time points. The same experiment was repeated for 8226 cells with (d) protein expression, (e) cdc25c gene expression and (f) activity assessed as in (a). Data represents the mean ± SD of three independent experiments.
saw when the cells were treated with a specific Cdc25c inhibitor (Figures 5(c) and (f)).
4. Discussion
ATO induces the apoptosis of drug-resistant MM cell lines and primary neoplastic plasma cells from myeloma patients at clinically achievable levels by targeting many different pathways [4,22]. Furthermore, while it can normally overcome the anti-apoptotic effect of interleukin-6, a critical cytokine for the growth and progression of myeloma cells, its reduction by lenalidomide can help ATO clinically where the interaction with the tumor microenvironment hampers the killing of malignant cells specially since it is not its only target [21]. In the current study, we observed that the addition of ATO and lenalidomide to cells increased the apoptotic rate of myeloma cells in a time and concentration-dependent manner due to ATO’s effect as demonstrated by increased cleavage of PARP and caspase 3 and decreased expression of Bcl2 in these cells similar to ATO alone. ATO activates the pro-apoptotic Bcl-2 family member Bax and induces its translocation from the cytosol to the mitochondria [22,32]. Such Bax engagement plays an important role in apoptosis. Contrary to this, its activation is suppressed by the anti-apoptotic protein, Bcl-2, via inhibition of mitochondrial ROS generation while ATO is known to overcome it by generating ROS which is also part of its toxicity [21,22,32,33]. An alternative under study is the administration of ascorbic acid in combination with ATO since its can also help neutralize ROS without apparently decreasing its effectiveness which could be considered as an add-on for future studies of ATO’s combination with lenalidomide [21,34]. Importantly, a conclusion from our short study is the demonstrating that lenalidomide does not, by itself; increase the apoptosis of myeloma cells but it does not decrease the cytotoxic effect of ATO. This could help explain similar results obtained clinically with ATO’s in to treat hematologic malignancies in combination with thalidomide where they demonstrate a favorable effect by the drug combination, but a decrease in the beneficial effects of ATO when removed to leave thalidomide by itself [23, 31].
Lenalidomide is a drug known to have a broad range of biological effects, including enhancement of tumorspecific immunity. Several investigators have reported anti-proliferative and pro-apoptotic effects of lenalidomide on tumor cells in vitro, while its exact mechanisms and targets remain unknown [8,12,35,36]. We recently demonstrated that it can induce a mild level of apoptosis in some tumor cells (~10% - 20%), sensitized by allelic haplo-deficiency in del(5q) MDS cells promotes that leads to G2-M arrest after use of this agent [11,37,38]. This effect occurs specifically through a deficiency of cdc25c and PP2A phosphatases and their inhibition (which lenalidomide alone is incapable of inducing) can reinforce the effects of lenalidomide’s ability to cause G2 arrest in cancer progenitors. However, most malignant cells and their progenitors have full expression of these two phosphatases demonstrating the need to find suitable combinatorial treatments that can aid and even synergize with lenalidomide to carry out antitumoral and immune enhancing functions. Interestingly, while trying to understand the combined effects of lenalidomide and ATO, we found that ATO can downregulate Cdc25C enhancing lenalidomide’s cytostatic effects. It is likely that these results will also be linked to the induction of TRAIL since lenalidmoide was recently shown to induce the death of myeloma cells in vitro through the induction of IFN and subsequent TRAIL induction [39]. However, based on our data, we do not believe that an increase in apoptosis induction is the main mechanism that may favor a combination of these two drugs.
An important effect of lenalidomide is its ability to modulate production of various cytokines in the tumor microenvironment. Lenalidomide is reported to downregulate key prosurvival cytokines, such as interleukin-6 (IL-6) [40,41]; cytokines that enrich the malignant microenvironment favoring tumor cell survival and proliferation and resistance to therapy. Importantly, the drug combination of lemalidomide with ATO also maintained lenalidomide’s inhibitory effects on IL-6. While our research suggests that ATO itself does not modulate IL-6 production, we also find that it also does not affect lenalidomide’s inhibitory effect on IL-6 suggesting that these drugs effect on the homeostasis of the microenvironment still occur and presumably contribute to the decreased viability of myeloma cells in vivo. One explanation for the increased susceptibility is through the downregulation of Cdc25C. Our previous discovery highlighted that a reduction in Cdc25C resulted in an increased susceptibility of abnormal/malignant cells to lenalidomide [11]. Those findings suggest that a reduction of these genes through other molecular or chemical mechanisms can promote a similar sensitivity to lenalidomide, and therefore may represent a strategy for the development of tumor-selective therapies. The fact that treatment with ATO can further inhibit Cdc25c, which is involved in the regulation of the CDK1 complex and G2-M phase transition, provides a correlation to the anti-proliferative effects observed with both drugs individually or in combination. However, this combination did not inhibit or down-regulate PP2A that raises the question as to what apoptosis would be in myeloma cells if PP2A was also inhibited resulting in the hyper-phosphorylation of MDM2 destabilizing the E3 ligase’ p53 and targets it for degradation leading to G2 transition. Further, accumulation of p53 in the absence of its major negative regulator, MDM2, induces apoptosis [42]. This could explain how an effect on MDM2, but not PP2A, can cause cell cycle arrest and cytotoxicity. We could hypothesize that bypassing PP2A, and down-regulating its target MDM2, will lead to similar results.
5. Acknowledgements
This project is partly supported by Natural Science Foundation of China (No. 81170472) and Application Bases and Advanced Technology Research Program of Tianjin (No. 09JCYBJC11200) and Clinical Research Grant of Hematologist Association of China (No. 20090109). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
REFERENCES
- R. A. Kyle and S. V. Rajkumar, “Multiple Myeloma,” New England Journal of Medicine, Vol. 351, No. 18, 2004, pp. 1860-1873. doi:10.1056/NEJMra041875
- B. Sirohi and R. Powles, “Multiple Myeloma,” Lancet, Vol. 363, No. 9412, 2004, pp. 875-887. doi:10.1016/S0140-6736(04)15736-X
- J. A. Katzel, P. Hari and D. H. Vesole, “Multiple Myeloma: Charging toward a Bright Future,” CA: A Cancer Journal for Clinicians, Vol. 57, No. 5, 2007, pp. 301-318. doi:10.3322/CA.57.5.301
- M. Cives, V. Simone, O. Brunetti, et al., “Novel Lenalidomide-Based Combinations for Treatment of Multiple Myeloma,” Critical Reviews in Oncology and Hematology, Vol. 85, No. 1, 2013, pp. 9-20.
- S. Kumar and S. V. Rajkumar, “Thalidomide and Lenalidomide in the Treatment of Multiple Myeloma,” European Journal of Cancer, Vol. 42, No. 11, 2006, pp. 1612-1622. doi:10.1016/j.ejca.2006.04.004
- M. A. Dimopoulos and E. Kastritis, “The Role of Novel Drugs in Multiple Myeloma,” Annals of Oncology, Vol. 19, No. 7, 2008, pp. vii121-vii127. doi:10.1093/annonc/mdn444
- H. Brenner, A. Gondos and D. Pulte, “Recent Major Improvement in Long-Term Survival of Younger Patients with Multiple Myeloma,” Blood, Vol. 111, No. 5, 2008, pp. 2521-2526. doi:10.1182/blood-2007-08-104984
- J. B. Bartlett, K. Dredge and A. G. Dalgleish, “The Evolution of Thalidomide and Its IMiD Derivatives as Anticancer Agents,” Nature Reviews Cancer, Vol. 4, No. 4, 2004, pp. 314-322. doi:10.1038/nrc1323
- T. Moehler and H. Goldschmidt, “Therapy of Relapsed and Refractory Multiple Myeloma,” Recent Results in Cancer Research, Vol. 183, 2011, pp. 239-271. http://www.ncbi.nlm.nih.gov/pubmed/21509688 doi:10.1007/978-3-540-85772-3_11
- A. Messori, D. Maratea, C. Nozzoli, et al., “The Role of Bortezomib, Thalidomide and Lenalidomide in the Management of Multiple Myeloma: An Overview of Clinical and Economic Information,” Pharmacoeconomics, Vol. 29, No. 4, 2011, pp. 269-285.
- S. Wei, X. Chen, K. Rocha, et al., “A Critical Role for Phosphatase Haplodeficiency in the Selective Suppression of Deletion 5q MDS by Lenalidomide,” Proceedings of National Academy of Sciences of USA, Vol. 106, No. 31, 2009, pp. 12974-12979. doi:10.1073/pnas.0811267106
- L. G. Corral, P. A. Haslett, G. W. Muller, et al., “Differential Cytokine Modulation and T Cell Activation by Two Distinct Classes of Thalidomide Analogues that Are Potent Inhibitors of TNF-Alpha,” Journal of Immunology, Vol. 163, No. 1, 1999, pp. 380-386.
- H. Sartor, F. Ehlert, K. H. Grzeschik, et al., “Assignment of Two Human Cell Cycle Genes, CDC25C and CCNB1, to 5q31 and 5q12, Respectively,” Genomics, Vol. 13, No. 3, 1992, pp. 911-912. doi:10.1016/0888-7543(92)90190-4
- H. Kiyokawa and D. Ray, “In Vivo Roles of CDC25 Phosphatases: Biological Insight into the Anti-Cancer Therapeutic Targets,” Anti-Cancer Agents in Medicinal Chemistry, Vol. 8, No. 8, 2008, pp. 832-836. doi:10.2174/187152008786847693
- R. Boutros, V. Lobjois and B. Ducommun, “CDC25 Phosphatases in Cancer Cells: Key Players? Good Targets?” Nature Reviews Cancer, Vol. 7, No. 7, 2007, pp. 495-507. doi:10.1038/nrc2169
- P. G. Richardson, E. Blood, C. S. Mitsiades, et al., “A Randomized Phase 2 Study of Lenalidomide Therapy for Patients with Relapsed or Relapsed and Refractory Multiple Myeloma,” Blood, Vol. 108, No. 10, 2006, pp. 3458- 3464. doi:10.1182/blood-2006-04-015909
- M. Dimopoulos, A. Spencer, M. Attal, et al., “Lenalidomide plus Dexamethasone for Relapsed or Refractory Multiple Myeloma,” The New England Journal of Medicine, Vol. 357, No. 21, 2007, pp. 2123-2132. doi:10.1056/NEJMoa070594
- D. M. Weber, C. Chen, R. Niesvizky, et al., “Lenalidomide plus Dexamethasone for Relapsed Multiple Myeloma in North America,” The New England Journal of Medicine, Vol. 357, No. 21, 2007, pp. 2133-2142. doi:10.1056/NEJMoa070596
- N. C. Munshi, G. Tricot, R. Desikan, et al., “Clinical Activity of Arsenic Trioxide for the Treatment of Multiple Myeloma,” Leukemia, Vol. 16, No. 9, 2002, pp. 1835- 1837. doi:10.1038/sj.leu.2402599
- W. H. Miller Jr., H. M. Schipper, J. S. Lee, et al., “Mechanisms of Action of Arsenic Trioxide,” Cancer Research, Vol. 62, No. 14, 2002, pp. 3893-3903.
- J. R. Berenson, J. Matous, R. A. Swift, et al., “A Phase I/II Study of Arsenic Trioxide/Bortezomib/Ascorbic Acid Combination Therapy for the Treatment of Relapsed or Refractory Multiple Myeloma,” Clinical Cancer Research, Vol. 13, No. 6, 2007, pp. 1762-1768. doi:10.1158/1078-0432.CCR-06-1812
- A. Emadi and S. D. Gore, “Arsenic Trioxide—An Old Drug Rediscovered,” Blood Reviews, Vol. 24, No. 4, 2010, pp. 191-199. doi:10.1016/j.blre.2010.04.001
- W. Wei, F. Zhou, Y. Zhang, et al., “A Combination of Thalidomide and Arsenic Trioxide Is Effective and Well Tolerated in Patients with Myelodysplastic Syndromes,” Leukemia Research, Vol. 36, No. 6, 2012, pp. 715-719. doi:10.1016/j.leukres.2011.12.023
- X. H. Zhu, Y. L. Shen, Y. K. Jing, et al., “Apoptosis and Growth Inhibition in Malignant Lymphocytes after Treatment with Arsenic Trioxide at Clinically Achievable Concentrations,” Journal of the National Cancer Institute, Vol. 91, No. 9, 1999, pp. 772-778. doi:10.1093/jnci/91.9.772
- Y. Jing, J. Dai, R. M. Chalmers-Redman, et al., “Arsenic Trioxide Selectively Induces Acute Promyelocytic Leukemia Cell Apoptosis via a Hydrogen Peroxide-Dependent Pathway,” Blood, Vol. 94, No. 6, 1999, pp. 2102-2111.
- W. H. Park, J. G. Seol, E. S. Kim, et al., “Arsenic Trioxide-Mediated Growth Inhibition in MC/CAR Myeloma Cells via Cell Cycle Arrest in Association with Induction of Cyclin-Dependent Kinase Inhibitor, p21, and Apoptosis,” Cancer Research, Vol. 60, No. 11, 2000, pp. 3065- 3071.
- N. J. Bahlis, J. McCafferty-Grad, I. Jordan-McMurry, et al., “Feasibility and Correlates of Arsenic Trioxide Combined with Ascorbic Acid-Mediated Depletion of Intracellular Glutathione for the Treatment of Relapsed/Refractory Multiple Myeloma,” Clinical Cancer Research, Vol. 8, No. 12, 2002, pp. 3658-3668.
- M. A. Hussein, M. Saleh, F. Ravandi, et al., “Phase 2 Study of Arsenic Trioxide in Patients with Relapsed or Refractory Multiple Myeloma,” British Journal of Haematology, Vol. 125, No. 4, 2004, pp. 470-476. doi:10.1111/j.1365-2141.2004.04941.x
- K. L. Wu, M. Beksac, J. van Droogenbroeck, et al., “Phase II Multicenter Study of Arsenic Trioxide, Ascorbic Acid and Dexamethasone in Patients with Relapsed or Refractory Multiple Myeloma,” Haematologica, Vol. 91, No. 12, 2006, pp. 1722-1723.
- C. Rollig and T. Illmer, “The Efficacy of Arsenic Trioxide for the Treatment of Relapsed and Refractory Multiple Myeloma: A Systematic Review,” Cancer Treatment Reviews, Vol. 35, No. 5, 2009, pp. 425-430. doi:10.1016/j.ctrv.2009.04.007
- A. Raza, S. Buonamici, L. Lisak, et al., “Arsenic Trioxide and Thalidomide Combination Produces Multi-Lineage Hematological Responses in Myelodysplastic Syndromes Patients, Particularly in Those with High Pre-Therapy EVI1 Expression,” Leukemia Research, Vol. 28, No. 8, 2004, pp. 791-803. doi:10.1016/j.leukres.2003.11.018
- S. Y. Ho, W. J. Wu, H. W. Chiu, et al., “Arsenic Trioxide and Radiation Enhance Apoptotic Effects in HL-60 Cells through Increased ROS Generation and Regulation of JNK and p38 MAPK Signaling Pathways,” ChemicoBiological Interactions, Vol. 193, No. 2, 2011, pp. 162- 171. doi:10.1016/j.cbi.2011.06.007
- H. K. Lam, K. Li, K. W. Chik, et al., “Arsenic Trioxide Mediates Intrinsic and Extrinsic Pathways of Apoptosis and Cell Cycle Arrest in Acute Megakaryocytic Leukemia,” International Journal of Oncology, Vol. 27, No. 2, 2005, pp. 537-545.
- E. M. Ocio, M. V. Mateos, P. Maiso, et al., “New Drugs in Multiple Myeloma: Mechanisms of Action and Phase I/II Clinical Findings,” The Lancet Oncology, Vol. 9, No. 12, 2008, pp. 1157-1165. doi:10.1016/S1470-2045(08)70304-8
- K. Dredge, R. Horsfall, S. P. Robinson, et al., “Orally Administered Lenalidomide (CC-5013) Is Anti-Angiogenic in Vivo and Inhibits Endothelial Cell Migration and Akt Phosphorylation in Vitro,” Microvascular Research, Vol. 69, No. 1-2, 2005, pp. 56-63. doi:10.1016/j.mvr.2005.01.002
- K. Dredge, J. B. Marriott, C. D. Macdonald, et al., “Novel Thalidomide Analogues Display Anti-Angiogenic Activity Independently of Immunomodulatory Effects,” British Journal of Cancer, Vol. 87, No. 10, 2002, pp. 1166- 1172. doi:10.1038/sj.bjc.6600607
- A. K. Gandhi, J. Kang, S. Naziruddin, et al., “Lenalidomide Inhibits Proliferation of Namalwa CSN.70 Cells and Interferes with Gab1 Phosphorylation and Adaptor Protein Complex Assembly,” Leukemia Research, Vol. 30, No. 7, 2006, pp. 849-858. doi:10.1016/j.leukres.2006.01.010
- D. Verhelle, L. G. Corral, K. Wong, et al., “Lenalidomide and CC-4047 Inhibit the Proliferation of Malignant B Cells While Expanding Normal CD34+ Progenitor Cells,” Cancer Research, Vol. 67, No. 2, 2007, pp. 746-755. doi:10.1158/0008-5472.CAN-06-2317
- Y. Yang, A. L. Shaffer 3rd, N. C. Emre, et al., “Exploiting Synthetic Lethality for the Therapy of ABC Diffuse Large B Cell Lymphoma,” Cancer Cell, Vol. 21, No. 6, 2012, pp. 723-737. doi:10.1016/j.ccr.2012.05.024
- J. B. Marriott, K. Dredge and A. G. Dalgleish, “Thalidomide Derived Immunomodulatory Drugs (IMiDs) as Potential Therapeutic Agents,” Current Drug Targets—Immune, Endocrine & Metabolic Disorders, Vol. 3, No. 3, 2003, pp. 181-186. doi:10.2174/1568008033340207
- K. C. Anderson, “Lenalidomide and Thalidomide: Mechanisms of Action—Similarities and Differences,” Seminars in Hematology, Vol. 42, No. 4, 2005, pp. S3-S8. doi:10.1053/j.seminhematol.2005.10.001
- D. Michael and M. Oren, “The p53 and Mdm2 Families in Cancer,” Current Opinion in Genetics & Development, Vol. 12, No. 1, 2002, pp. 53-59. doi:10.1016/S0959-437X(01)00264-7