Journal of Cancer Therapy
Vol. 3 No. 4 (2012) , Article ID: 21595 , 6 pages DOI:10.4236/jct.2012.34041
Cytotoxicity and Selectivity in Skin Cancer by SapC-DOPS Nanovesicles
![]()
1Division of Hematology/Oncology, Department of Internal Medicine, University of Cincinnati College of Medicine, Cincinnati, USA; 2Division of Human Genetics, Department of Pediatrics, Cincinnati Children’s Hospital Medical Center, Cincinnati, USA; 3Department of Dermatology, University of Miami, Miami, USA.
Email: *xiaoyang.qi@uc.edu
Received June 20th, 2012; revised July 25th, 2012; accepted August 9th, 2012
Keywords: Squamous Cell Carcinoma; Melanoma; SapC-DOPS Nanovesicles; Cytotoxic Effect; Cancer Selectivity
ABSTRACT
Squamous cell carcinoma (SCC) and melanoma are malignant human cancers of the skin with an annual mortality that exceeds 10,000 cases every year in the USA alone. In this study, the lysosomal protein saposin C (SapC) and the phospholipid dioloylphosphatidylserine (DOPS) were assembled into cancer-selective nanovesicles (SapC-DOPS) and successfully tested using several in vitro and in vivo skin cancer models. Using MTT assay that measures the percentage of cell death, SapC-DOPS cytotoxic effect on three skin tumor cell lines (squamous cell carcinoma, SK-MEL-28, and MeWo) was compared to two normal nontumorigenic skin cells lines, normal immortalized keratinocyte (NIK) and human fibroblast cell (HFC). We observed that the nanovesicles selectively killed the skin cancer cells by inducing apoptotic cell death whereas untransformed skin cancer cells remained unaffected. Using subcutaneous skin tumor xenografts, animals treated with SapC-DOPS by subcutaneous injection showed a 79.4% by volume tumor reduced compared to the control after 4 days of treatment. We observed that the nanovesicles killed skin cancer cells by inducing apoptotic cell death compared to the control as revealed by TUNEL staining of xenograft tumor sections.
1. Introduction
Squamous cell carcinoma (SCC) and melanoma are the two most common malignant skin cancers. SCCs often occur in the skin, lung, and head and neck is among the leading malignant cancers in the United Stated [1]. For skin SCC alone, there are more than 20,000 new cases each year and the annual mortality is estimated to be between 1300 and 2300, most of which die from metastatic lesions [1]. Melanoma is the most deadly skin cancer due to its notoriously rapid spreading. An estimated more than 10,700 persons die from skin cancers each year, 2000 from SCC and more than 8700 from melanoma [2]. One in 75 Americans has a risk of developing melanoma in his or her lifetime and the number is increasing each year [2]. Based on the above statistics, new therapeutic agents that selectively target skin cancer are needed to decrease the mortality of SCC or melanomas in patients with skin cancer.
In all normal tissues, Saposin C (SapC) acts as biological activators of lysosomal enzymes [3]. This protein is a small nonenzymatic glycoprotein with remarkably heat-stable and protease-resistant qualities and contains an N-glycosylation consensus sequence and six essential cysteines [3,4]. Several studies indicated that the degradation of glucosylceramide, sphingomyelin, and galactosylceramide to ceramide via acid β-glucosidase, acid sphingomyelinase, and acid β-galacotsylceramidase is enhanced in the presence of SapC [3,5,6]. It has been reported that SapC accumulates along with glycosphingolipids in macrophages of patients with lysosomal storage diseases [7].
A general property of the saposins family (SapA, SapB, SapC and SapD) is their lipid membrane binding activity [8] since they play important roles in reorganization of biological membranes [9-11] and lipid microdomain assembly [12] as well as in lipid transport [13]. It has been reported that SapC specifically associates with lipid membranes by partially embedding into membrane leaf-lets via the aminoand carboxyl-ends of the protein helices [8].
We reported previously that SapC preferentially interacts with unsaturated, negatively charged phospholipids (such as dioleoylphosphatidylserine, DOPS), at acidic pH [8,14]. It has been reported that phosphatidylserine (PS) is relatively abundant on the surface of tumor tissues [15, 16]; it could provide a tumor-specific target for SapC. In the design of our new therapeutic agent (SapC-DOPS nanovesicles), we took advantage of two unique properties of tumor cells compared to normal cells. First, neoplastic cells are hypermetabolic and thus produce significant amounts of acid and carbon dioxide as by-products of anaerobic glycolysis and aerobic respiration, respecttively. As a result, hydrogen ions accumulate in tumor tissues making the microenvironment acidic in solid tumors (pH ~6) compared to neutral in the normal tissues (pH ~7) [17,18]. Second, cancer cells posses generalized membrane alterations [19-21] and “leakiness” of lysosomal enzymes [19]. In support to these unique properties of cancer cells, it has been found that several lysosomal hydrolases are elevated in tumor tissues [22,23]. Therefore, the unique acidic microenvironment with extracellular leakage of lysosomal enzymes makes tumor tissue an optimal target for SapC.
As reported previously, SapC-DOPS induces caspasemediated apoptotic cell death of cancer cells in culture [24]. The molecular mechanism of tumor cell entry and caspase-activation remain unknown [24]. Additional data from experiments not described here indicate that SapCDOPS has a broad, potent killing effect on several tested human cancer cells and models, including brain and pancreatic cancers (Wonjton et al. and Chu et al. unpublished data). We hypothesize that SapC-DOPS nanovesicles accumulate preferentially in cancer cells, and conesquently this may trigger acid sphingomyelinase (ASM)- derived ceramide signaling cascades to induce apoptosis of cancer cells. Additional experiments are part of the ongoing research in our lab to improve our understanding of the possible mechanism of action of SapC-DOPS cytotoxicity on tumor cells.
In a previous report, we demonstrated that SapC-DOPS nanovesicles kills cancer cells and induces apoptotic cell death using a broad range of cancer models [24]. In the current study, we extended our work and investigated the cytotoxicity and selectivity of SapC-DOPS using several in vitro and in vivo skin cancer models.
2. Materials and Methods
2.1. Cell Cultures
The human skin cancer cell lines [SCC (squamous cell carcinoma), SK-MEL-28 (skin melanoma) and MeWo (melanoma lymph node metastasis)] and normal nontumorigenic skin cells lines [normal immortalized keratinocyte (NIK) and human fibroblast cell (HFC)] were cultured with DMEM supplemented with 10% of fetal bovine serum, 100 units of penicillin/ml, and 10 mg of streptomycin/ml. All cells were cultured at 37˚C in 5% CO2. No cross-contamination was found in these cells.
2.2. Preparation of Proteins and Nanovesicles
SapC was produced as previously described [4]. Briefly, recombinant saposins were expressed using the pET system in E. coli cells, followed by purification and lyophilization steps. Then, SapC was spontaneously incorporated into the lipid bilayer of the liposomes upon sonication. Following sonication and ultracentrifugation to pellet SapC-DOPS coupled liposomes, no SapC was detected in the supernatant fraction, implicating a very high loading/coupling efficiency [25].
2.3. Cell Viability Assay
Dose-dependent killing of skin cancer and normal cells by SapC-DOPS was investigated using3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)- dye assay as previously described [24,25]. Cells seeded (1 × 104/100 µl/well) in 96-well flat-bottom tissue culture plates (Falcon, Becton Dickson Labware, Franklin Lakes, NJ) were cultured for 24 h prior to the addition of SapCDOPS or PBS vehicle to the culture medium. Three days after initiating treatment, a standard MTT-dye assay (Sigma) was carried out to measure viable cells. Experimental conditions: cells (1 × 104/100 µl/well) were cultured for 24 h prior to the treatment. Experiments were performed at least twice.
2.4. MTT Data Analysis
MTT experiments were performed in quadruplicate and data were analyzed by ANOVA. The data presented are the arithmetic mean ± SEM. T-test analysis or Two-way ANOVA Tukey test were used to determine statistical significance for experiments with two or greater than two groups, respectively. Analyses were done with SPSS 12.0.
2.5. Terminal Deoxynucleotidyl Transferase—Mediated dUTP Nick End Labeling (TUNEL) Assay for Apoptosis [24]
In vivo apoptosis in SCC cancer cells inducted by SapCDOPS was determined by TUNEL assay (terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling), using Cell Death Detection Kit, POD (Roche Applied Science, Germany) as described by the manufacturer protocol.
2.6. Subcutaneous SCC Tumor Model
SapC-DOPS nanovesicles effect on the growth of skin cancer tumors (tumor reduction comparison) in tumor bearing mice was investigated using SCC cells. Athymic nude mice (Nu/Nu) were injected on the upper-back with 5 × 106 SCC cells subcutaneously to initiate tumor growth. Single subcutaneous administration around the tumors of either SapC (10 mg/Kg)/PS (2 mg/Kg) or placebo (PBS control) was done on Day 6 (n = 9 tumors for each group). The tumor size was determined by caliper measurements [26,27].
3. Results and Discussion
3.1. SapC-DOPS Nanovesicles Are Toxic to Skin Cancer Cells but Not to Normal Cells
In a previous report, we demonstrated the cytotoxicity due to SapC-DOPS using a variety of tumor models including neuroblastoma, malignant peripheral nerve sheet tumor (MPNST) and breast cancer cells [24]. In this paper, we measured this cytotoxicity on skin cancer cells. First, we determined the optimal molar ratio of SapC and DOPS for maximal killing effect. We found that ratios between 1:5 and 1:10 had maximal in vitro cytotoxicity SCC cells (Figure 1).
Then we tested the SapC-DOPS IC50 (inhibitory concentration that induces 50% cell death) using SK-MEL- 28 melanoma cells at three different SapC-DOPS molar ratios (1:3, 1:7 and 1:10). As summarized in Table 1, the best IC50 (19.5 ± 11.0 µM SapC and 136.6 ± 78.0 µM DOPS) was obtained at a 1:7 ratio. No significant effect was observed with addition DOPS or SapC alone (Table 1). These data indicate that the observed cytotoxic effect requires coupled SapC-DOPS nanovesicles.
To demonstrate that SapC-DOPS nanovesicles specifically induce skin tumor cell killing, the effect of these nanovesicles was tested on three skin cancer cell lines (SCC, MeWo and SK-MEL-28) and untransformed normal lines (NIK keratinocyte and HFC fibroblast). As shown in Table 2 (left column), after 3 days of the starting point, the untreated samples lost only 4% - 16% of the cells for both normal (NIK and HFC) and cancerous human skin cell lines (SCC, SK-MEL-28 and MeWo). Conversely, when the cell lines were treated (Table 2, right column), only those cancerous cells induced killing in the range of 57% - 82% based on the line. In other words, when the cells, in their respective growth media, were treated with SapC-DOPS, the majority of the tumor cells died by apoptosis compared to their untreated controls, while the NIK and HFC cells remained viable compared to their untreated controls.
3.2. SapC-DOPS Shows in Vivo Anti-Skin Tumor Activity
We conducted an experiment to investigate the effectiveness of SapC-DOPS on the growth of skin tumors in a subcutaneous SCC xenograft mouse model. SCC cancer cells were used to generate tumors in athymic mice (Nu/ Nu). SCC cells (5 × 106 cells) were injected subcutaneously into the upper back of mice. The tumor masses were measured every two days using a pair of calipers
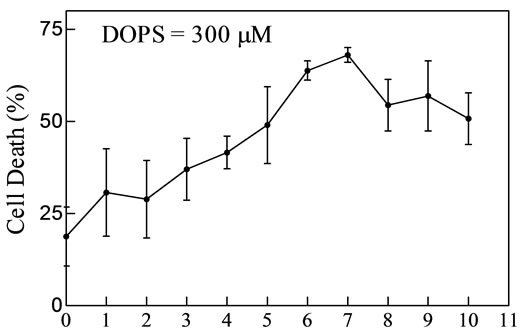 DOPS: SapC Ratio (mole:mole)
DOPS: SapC Ratio (mole:mole)
Figure 1. SapC-DOPS vesicles are cytotoxic against skin cancer cells. Squamous cell carcinoma (SCC) cells were used to determine the optimal concentration of SapC for maximum cytotoxic effect.
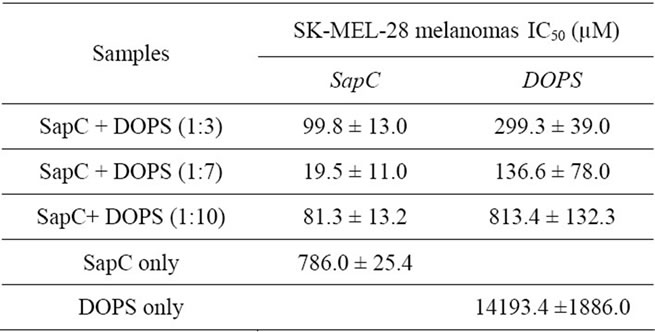
Table 1. SapC-DOPS vesicles cytotoxicity at different molar ratios (DOPS:SapC). MTT assay was carried out after culture of the treated cells for 3 days. The data are presented as the arithmetic IC50 mean ± SEM of four independent samples. The IC50 (µM) is represented using either SapC concentration or DOPS concentration in (µM).
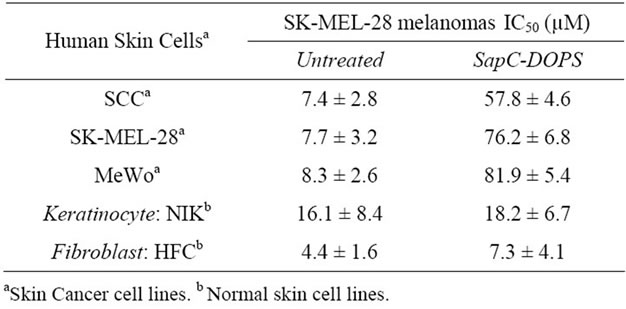
Table 2. Antineoplastic activity of SapC-DOPS in human cancer cells. MTT assay was carried out after culture of the treated cells for 3 days. The data are presented as the arithmetic IC50 mean ± SEM at least two independent samples. Experiments were performed at (SapC:DOPS) 1:3 ratio (SapC = 100 µM; DOPS = 300 µM). NIK: normal immortalized keratinocytes. HFC: human fibroblast cells. SCC: squamous cell carcinoma.
and the formula V = (π6)LW2 (V = volume, L = length, W = width) [24]. To show the success of the subcutaneous injections and the pathology of tumors for both cell lines, representation of H&E staining of xenogratfed skin cancer tumors on day 2 are shown in Figure 2. On day 6 post-implantation, the mice were randomized into two groups. One group was administered with SapC-DOPS nanovesicles and the other group was administered with the vehicle (PBS). At day 10, the SapC-DOPS treated group had a mean reduction of 79.4% in tumor by volume compared to the control (Figure 3). The difference in volumes between the treatment and control groups at post-implantation day 10 was significant (p < 0.0136). These results indicate that SapC-DOPS induces reduction of skin tumor in xenograft-bearing nude mice.
This mouse model allowed us to determine in vivo apoptotic cancer cell death induced by SapC-DOPS. To visualize apoptotic cells in tumor tissues from mice treated with SapC-DOPS, TUNEL assay was used on fixed sections of tumor tissue from animals harvested at day 10.
As shown in Figure 4, SapC-DOPS-treated sections showed abundant apoptotic cell death (pink or brown cells by TUNEL staining) for SCC (Figure 4(A)) and MeWo (Figure 4(B)) compared to the negative control (sections of tumor treated with DOPS alone for SCC (Figure 4(C)) and MeWo (Figure 4(D)). Our previous report supported the notion that SapC-DOPS kills cancer cells at least in part through the accumulation of cera-
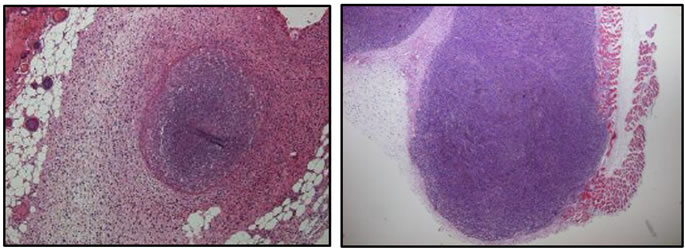 (a) (b)
(a) (b)
Figure 2. H & E staining of SCC (a) and MeWo (b) from paraffin sections of tumor tissue. The xenografts were dissected from mice skin after 48 hours of subcutaneous tumor cell injection in nude mice. Original magnification, ×40.
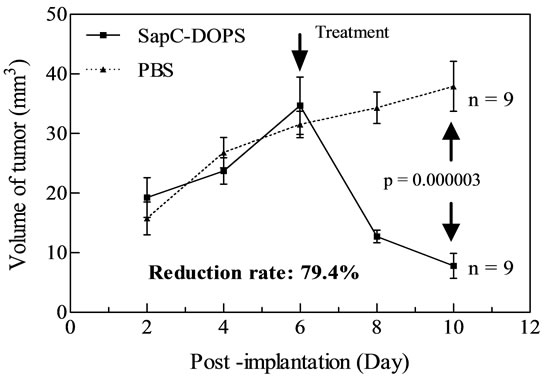
Figure 3. Treatment of nude mice bearing human squamous cell carcinoma (SCC) tumors with SapC/DOPS complex. After 10 days of treatment, the tumor reduction rate was calculated and it was reduced by 79.4% compared to the control.
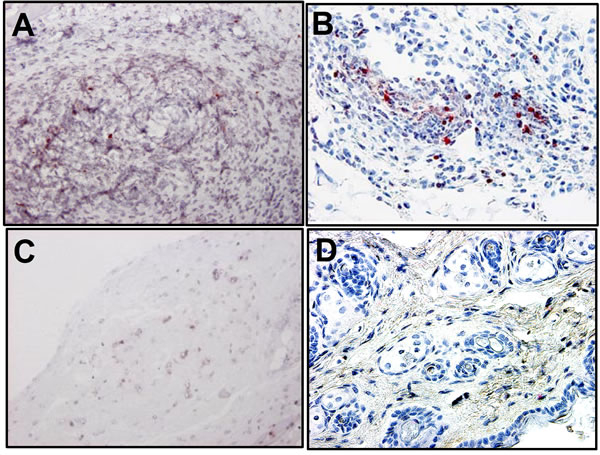
Figure 4. The tumor tissue for SapC-DOPS-treated animals contained abundant apoptotic cells by TUNEL staining (pink or brown) for SCC (A) and MeWo (B). As a negative control, animals with were treated with DOPS alone for SCC (C) and MeWo (D) tumors. Original magnification, ×200.
mides, leading to caspase activation and apoptotic cell death [24].
In this study, we tested the cytotoxicity and selectivity of a promising novel biotherapeutic agent composed of two natural cellular components that spontaneously assemble into stable nanovesicles (SapC-DOPS). Based on our current results, we demonstrated that SapC-DOPS nanovesicles can induce selective killing of skin tumor cells. In cell assays, SapC-DOPS induced the death of the skin cancer cell lines SCC, MeWo and SK-MEL-28, while untransformed skin cell lines (NIK and HFC) remained unaffected. In vivo, using a subcutaneous SCC skin tumor xenograft model in nude mice, repeated injections of SapCDOPS nanovesicles inhibited tumor growth and induced tumor apoptosis. The current report supports the notion that SapC-DOPS selectively kills skin cancer cells as indicated in previous reports on other tumor models [24].
4. Acknowledgements
This work was supported in part by Translational Research Initiative and Validation Research Grants from Cincinnati Children’s Hospital Medical Center (to X. Qi), NIH/NCI Grants Number 1R01CA158372-01 (to X. Qi), and New Drug State Key Project Grant Number 009ZX09102-205 (to X. Qi).
5. Conflict of Interest Disclosure
Xiaoyang Qi in listed as an inventor on the patent for the technology (SapC-DOPS) that is the subject of this research. Consistent with current Cincinnati Children’s Hospital Medical Center policies, the development and commercialization of this technology has been licensed to Bexion Pharmaceuticals, LLC, in which Dr. Qi holds a minor (<5%) equity interest. Dr. Qi is an inventor of scientific research that is in the pre-clinical stage for Bexion Pharmaceuticals. At this time Bexion Pharmaceuticals does not offer any marketable treatments or drugs.
REFERENCES
- S. J. Salasche, “Epidemiology of Actinic Keratoses and Squamous Cell Carcinoma,” Journal of the American Academy of Dermatology, Vol. 42, No. 1, 2000, pp. 4-7.
- E. J. Lentsch and J. N. Myers, “Melanoma of the Head and Neck: Current Concepts in Diagnosis and Management,” Laryngoscope, Vol. 111, No. 7, 2001, pp. 1209- 1222. doi:10.1097/00005537-200107000-00014
- K. Sandhoff, T. Kolter and K. Harzer, “Sphingolipid Activator Proteins,” In: C. R. Scriver, A. L. Beaudet, W. S. Sly and D. Valle, Eds., The Metabolic and Molecular Bases of Inherited Disease, McGraw-Hill, Inc., New York, 2000, pp. 3371-3388.
- X. Qi, T. Leonova and G. A. Grabowski, “Functional Human Saposins Expressed in Escherichia Coli. Evidence for Binding and Activation Properties of Saposins C with Acid Beta-Glucosidase,” The Journal of Biological Chemistry, Vol. 269, No. 24, 1994, pp. 16746-16753.
- K. Harzer, B. C. Paton, H. Christomanou, M. Chatelut, T. Levade, M. Hiraiwa and J. S. O’Brien, “Saposins (Sap) A and C Activate the Degradation of Galactosylceramide in Living Cells,” FEBS Letters, Vol. 417, No. 3, 1997, pp. 270-274. doi:10.1016/S0014-5793(97)01302-1
- T. Linke, G. Wilkening, S. Lansmann, H. Moczall, O. Bartelsen, J. Weisgerber and K. Sandhoff, “Stimulation of Acid Sphingomyelinase Activity by Lysosomal Lipids and Sphingolipid Activator Proteins,” The Journal of Biological Chemistry, Vol. 382, No. 2, 2001, pp. 283-290. doi:10.1515/BC.2001.035
- S. Morimoto, Y. Yamamoto, J. S. O’Brien and Y. Kishimoto, “Distribution of Saposin Proteins (Sphingolipid Activator Proteins) in Lysosomal Storage and Other Diseases,” Proceedings of the National Academy Sciences of the United States of America, Vol. 87, No. 9, 1990, pp 3493-3497. doi:10.1073/pnas.87.9.3493
- X. Qi and G. A. Grabowski, “Differential Membrane Interactions of Saposins A and C: Implications for the Functional Specificity,” The Journal of Biological Chemistry, Vol. 276, No. 29, 2001, pp. 27010-27017. doi:10.1074/jbc.M101075200
- A. M. Vaccaro, M. Tatti, F. Ciaffoni, R. Salvioli, A. Serafino and A. Barca, “Saposin C Induces pH-Dependent Destabilization and Fusion of Phosphatidylserine-Containing Vesicles,” FEBS Letters, Vol. 349, No. 2, 1994, pp. 181- 186. doi:10.1016/0014-5793(94)00659-8
- H. X. You, X. Qi, G. A. Grabowski and L. Yu, “Phospholipid Membrane Interactions of Saposin C: In Situ Atomic Force Microscopic Study,” Biophysical Journal, Vol. 84, No. 3, 2003, pp. 2043-2057. doi:10.1016/S0006-3495(03)75012-7
- Y. Wang, G. A. Grabowski and X. Qi, “Phospholipid Vesicle Fusion Induced by Saposin C,” Arch Biochem Biophys, Vol. 415, No. 1, 2003, pp. 43-53. doi:10.1016/S0003-9861(03)00219-4
- H. X. You, L. Yu and X. Qi, “Phospholipid Membrane Restructuring Induced by Saposin C: A Topographic Study Using Atomic Force Microscopy,” FEBS Letters, Vol. 503, No. 1, 2001, pp. 97-102. doi:10.1016/S0014-5793(01)02700-4
- M. Hiraiwa, S. Soeda, Y. Kishimoto and J. S. O’Brien, “Binding and Transport of Gangliosides by Prosaposin,” Proceedings of the National Academy Sciences of the United States of America, Vol. 89, No. 23, 1992, pp. 11254-11258. doi:10.1073/pnas.89.23.11254
- X. Qi, W. Qin, Y. Sun, K. Kondoh and G. A. Grabowski, “Functional Organization of Saposin C. Definition of the Neurotrophic and Acid Beta-Glucosidase Activation Regions,” The Journal of Biological Chemistry, Vol. 271, No. 12, 1996, pp. 6874-6880.
- S. Ran and P. E. Thorpe, “Phosphatidylserine Is a Marker of Tumor Vasculature and a potential target for Cancer Imaging and Therapy,” International Journal of Radiation Oncology, Vol. 54, No. 5, 2002, pp. 1479-1484. doi:10.1016/S0360-3016(02)03928-7
- T. Utsugi, A. J. Schroit, J. Connor, C. D. Bucana and I. J. Fidler, “Elevated Expression of Phosphatidylserine in the Outer Membrane Leaflet of Human Tumor Cells and Recognition by Activated Human Blood Monocytes,” Cancer Research, Vol. 51, No. 11, 1991, pp. 3062-3066.
- P. Vaupel, F. Kallinowski and P. Okunieff, “Blood Flow, Oxygen and Nutrient Supply, and Metabolic Microenvironment of Human Tumors: A Review,” Cancer Research, Vol. 49, No. 23, 1989, pp. 6449-6465.
- J. L. Wike-Hooley, J. Haveman and H. S. Reinhold, “The Relevance of Tumour pH to the Treatment of Malignant Disease,” Radiotherapy and Oncology, Vol. 2, No. 4, 1984, pp. 343-366. doi:10.1016/S0167-8140(84)80077-8
- D. F. Wallach, “Generalized Membrane Defects in Cancer,” The New England Journal of Medicine, Vol. 280, No. 14, 1969, pp. 761-767. doi:10.1056/NEJM196904032801406
- D. F. Wallach, “Cellular Membrane Alterations in Neoplasia: A Review and a Unifying Hypothesis,” Current Topics in Microbiology and Immunology, Vol. 47, No. 1969, pp. 152-176.
- D. F. Wallach, “Membrane Biology and Cancer Therapy,” Pathobiology Annual, Vol. 8, No. 1978, pp. 189- 216.
- A. C. Allison, “Lysosomes in Cancer Cells,” Journal of Clinical Pathology. Supplement, Vol. 7, 1974, pp. 43-50.
- M. J. Boyer and I. F. Tannock, “Lysosomes, Lysosomal Enzymes, and Cancer,” Advances in Cancer Research, Vol. 60, 1993, pp. 269-291.
- X. Qi, Z. Chu, Y. Y. Mahller, K. F. Stringer, D. P. Witte, and T. P. Cripe, “Cancer-Selective Targeting and Cytotoxicity by Liposomal-Coupled Lysosomal Saposin C Protein,” Clinical Cancer Research, Vol. 15, No. 18, 2009, pp. 5840-5851. doi:10.1158/1078-0432.CCR-08-3285
- V. Kaimal, Z. Chu and Y. Y. Mahller, B. Papahadjopoulos-Sternberg, T. P. Cripe, S. K. Holland and X. Qi, “Saposin C Coupled Lipid Nanovesicles Enable Cancer-Selective Optical and Magnetic Resonance Imaging,” Molecular Imaging and Biology, Vol. 13, No. 5, 2011, pp. 886-897. doi:10.1007/s11307-010-0417-7
- F. T. Lee, A. Rigopoulos, C. Hall, K. Clarke, S. H. Cody, F. E. Smyth, Z. Liu, M. W. Brechbiel, N. Hanai, E. C. Nice, B.Catimel, A. W. Burgess, S. Welt, G. Ritter, L. J. Old and A. M. Scott, “Specific Localization, Gamma Camera Imaging, and Intracellular Trafficking of Radiolabelled Chimeric Anti-G(D3) Ganglioside Monoclonal Antibody KM871 in SK-MEL-28 Melanoma Xenografts,” Cancer Research, Vol. 61, No. 11, 2001, pp. 4474-4482.
- C. V. Hamby, R. Abbi, N. Prasad, C. Stauffer, J. Thomson, C. E. Mendola, V. Sidorov and J. M. Backer, “Expression of a Catalytically Inactive H118Y Mutant of nm23-H2 Suppresses the Metastatic Potential of Line IV Cl 1 Human Melanoma Cells, International Journal of Cancer, Vol. 88, No. 4, 2000, pp. 547-553.
NOTES
*Corresponding author.